1.本发明属于药物化学和药理学技术领域,具体涉及一种灰黄霉素开环衍生物在制备抗肿瘤药物中的应用。
背景技术:
2.灰黄霉素是一种螺环苯并呋喃
‑3‑
酮类天然产物,结构如下式所示,1939年由oxford等人首次从丝状真菌分离,是具有较强抗真菌活性的非多烯类的抗真菌抗生素,它能强烈抑制真菌有丝分裂,干扰真菌dna合成,并且它与微管蛋白结合,能阻止真菌细胞分裂。目前,灰黄霉素普遍被作为抗真菌药物在临床使用。以价廉易得的灰黄霉素为原料,通过化学方法进行结构转化,对其进行化学骨架的改造,得到新的化合物,并开发其在药物化学和药理学技术领域的应用,具有重要意义。
3.
技术实现要素:
4.有鉴于此,本发明的目的在于提供一种灰黄霉素开环衍生物在制备抗肿瘤药物中的应用,此类化合物结构中具有酰胺结构,并且具有较强的抑制肿瘤细胞增殖活性,可用于制备抗肿瘤药物。
5.本发明提供了一种具有式(i)所示结构的灰黄霉素开环衍生物在制备抗肿瘤药物中的应用,
[0006][0007]
其中,r表示取代或未取代的苄胺基、c1
‑
c6的脂肪胺基、取代或未取代的芳胺基、吗啉基、羟乙基哌嗪基、n
‑
甲基哌嗪基、哌啶基、4
‑
羟基哌啶基、3
‑
羟基哌啶基、4
‑
哌啶基哌啶基、四氢吡咯基或咪唑基。
[0008]
进一步的,所述肿瘤为口腔上皮癌、胃癌、肺腺癌、子宫颈癌、结肠癌细胞和肝癌中的一种。
[0009]
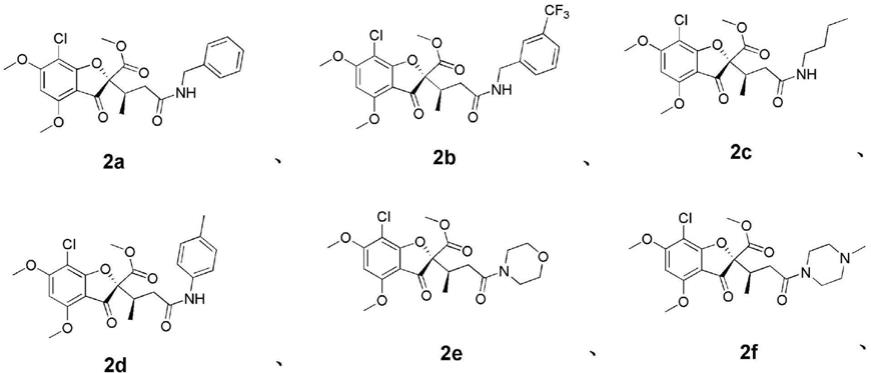
进一步的,所述灰黄霉素开环衍生物具有如式2a~2f中任意一项所示的结构:
[0010][0011]
其中,
[0012]
r为苄胺基时,该开环衍生物为式2a所示结构的化合物;
[0013]
r为3
’‑
三氟甲基苄胺基时,该开环衍生物为式2b所示结构的化合物;
[0014]
r为丁胺基时,该开环衍生物为式2c所示结构的化合物;
[0015]
r为4
’‑
甲基苯胺基时,该开环衍生物为式2d所示结构的化合物;
[0016]
r为吗啉基时,该开环衍生物为式2e所示结构的化合物;
[0017]
r为4
’‑
甲基哌嗪基时,该开环衍生物为式2f所示结构的化合物;
[0018]
本发明还提供了一种灰黄霉素开环衍生物的制备方法,包括以下步骤:
[0019]
由灰黄霉素与高碘酸钠和催化量的三氯化钌在乙腈和水的混合溶剂中发生双键氧化断裂反应得到灰黄霉素开环中间体1。然后灰黄霉素开环中间体1与胺类化合物或含氮杂环化合物在hatu和dipea的n,n
‑
二甲基甲酰胺溶液中进行酰化反应,得到相应的开环衍生物。
[0020]
其中,所述反应的反应式为:
[0021][0022]
其中,r表示取代或未取代的苄胺基、c1
‑
c6的脂肪胺基、取代或未取代的芳胺基、吗啉基、羟乙基哌嗪基、n
‑
甲基哌嗪基、哌啶基、4
‑
羟基哌啶基、3
‑
羟基哌啶基、4
‑
哌啶基哌啶基、四氢吡咯基或咪唑基。
[0023]
具体的,该制备方法包括以下步骤:
[0024]
(1)将灰黄霉素溶于乙腈和水的混合溶剂中,加入高碘酸钠和三氯化钌,60℃反应12h,得第一反应液,将所述第一反应液用饱和硫代硫酸钠水溶液淬灭以后,用有机溶剂稀释,依次经过水洗,饱和食盐水洗,mgso4干燥,减压干燥,然后柱层析得到白色固体,所述白色固体为灰黄霉素开环中间体1,其中,灰黄霉素、高碘酸钠、三氯化钌的摩尔比为1:1.5:0.05,乙腈和水的体积比为6:1;
[0025]
(2)将所述白色固体溶于n,n
‑
二甲基甲酰胺,加入dipea和hatu,搅拌15分钟后,加入胺或含氮杂环,25℃反应12h,得第二反应液,将所述第二反应液用有机溶剂稀释后,依次
经过水洗,饱和食盐水洗,mgso4干燥,减压干燥,然后柱层析得到灰黄霉素开环衍生物,其中,所述灰黄霉素开环中间体1、dipea、hatu与胺或含氮杂环的摩尔比为1:2:1.5:1.1;
[0026]
hatu名称为2
‑
(7
‑
氮杂苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸酯,dipea名称为n,n
‑
二异丙基乙胺。
[0027]
其中,所述胺为c1
‑
c6的脂肪胺、取代或未取代的苄胺、取代或未取代的芳胺中的一种;所述含氮杂环为吗啉、羟乙基哌嗪、n
‑
甲基哌嗪、哌啶、4
‑
羟基哌啶、3
‑
羟基哌啶、4
‑
哌啶基哌啶、四氢吡咯或咪唑中的一种。
[0028]
优选的,上述制备方法中,所述有机溶剂为乙酸乙酯、乙醚和苯中的至少一种。
[0029]
与现有技术相比,本技术提供了一类新的化合物灰黄霉素开环衍生物在制备抗肿瘤药物中的应用,该化合物能明显提高对肿瘤细胞的增殖抑制作用。
附图说明
[0030]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
[0031]
图1为本发明实施例2提供的灰黄霉素开环衍生物(2a)的核磁共振1h谱图;
[0032]
图2为本发明实施例2提供的灰黄霉素开环衍生物(2a)的核磁共振
13
c谱图;
[0033]
图3为本发明实施例3提供的灰黄霉素开环衍生物(2b)的核磁共振1h谱图;
[0034]
图4为本发明实施例3提供的灰黄霉素开环衍生物(2b)的核磁共振
13
c谱图;
[0035]
图5为本发明实施例4提供的灰黄霉素开环衍生物(2c)的核磁共振1h谱图;
[0036]
图6为本发明实施例4提供的灰黄霉素开环衍生物(2c)的核磁共振
13
c谱图;
[0037]
图7为本发明实施例5提供的灰黄霉素开环衍生物(2d)的核磁共振1h谱图;
[0038]
图8为本发明实施例5提供的灰黄霉素开环衍生物(2d)的核磁共振
13
c谱图;
[0039]
图9为本发明实施例6提供的灰黄霉素开环衍生物(2e)的核磁共振1h谱图;
[0040]
图10为本发明实施例6提供的灰黄霉素开环衍生物(2e)的核磁共振
13
c谱图;
[0041]
图11为本发明实施例7提供的灰黄霉素开环衍生物(2f)的核磁共振1h谱图;
[0042]
图12为本发明实施例7提供的灰黄霉素开环衍生物(2f)的核磁共振
13
c谱图。
具体实施方式
[0043]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0044]
实施例1
[0045]
将211mg(0.6mmol)的灰黄霉素溶于乙腈(3ml)和水(0.5ml)的混合溶剂,然后加入193mg(0.9mmol)的高碘酸钠和7mg(0.03mmol)的三氯化钌,60℃反应过夜。tlc检测反应结束,用饱和硫代硫酸钠水溶液淬灭,乙酸乙酯萃取(10ml
×
3),合并有机相,然后依次水洗、饱和食盐水洗,mgso4干燥,减压干燥,柱层析(石油醚:乙酸乙酯=1:1)得到185mg白色固体,即灰黄霉素开环中间体1(产率=83%)。
[0046]1h nmr(400mhz,cdcl3):δ7.78(s,1h,cooh),6.14(s,1h,arh),4.10
‑
3.99(m,3h,
och3),3.96(dd,j=4.0,1.9hz,3h,och3),3.77(dd,j=4.2,2.0hz,3h,och3),3.19(ddq,j=10.1,6.3,3.1hz,1h,ch),2.32(ddt,j=16.1,5.7,3.0hz,1h,ch2),2.13(dddd,j=15.8,13.7,6.1,3.4hz,1h,ch2),1.20(td,j=6.9,6.3,2.4hz,3h,ch3);
13
c nmr(100mhz,cdcl3):δ189.8(c=o),176.9(coo),168.8(coo),165.5(aro),164.9(aro),158.0(aro),104.4(ar),97.7(ar),95.3(ar),89.9(c),57.1(och3),56.4(och3),53.4(och3),34.8(ch),14.6(ch3);hrms(esi):m/z calcd for c
16
h
17
o8nacl:395.0510;found:395.050[m na]
.
[0047]
实施例2
[0048]
将142mg(0.38mmol)的灰黄霉素开环中间体1溶于2ml的无水dmf中,加入100mg(0.76mmol)dipea和218mg(0.57mmol)hatu,搅拌15分钟后,加入43mg(0.4mmol)苄胺,室温反应12h。加入20ml乙酸乙酯稀释,然后依次水洗、饱和食盐水洗,mgso4干燥,减压干燥,柱层析(石油醚:乙酸乙酯=1:2)得到白色固体(2a)126mg(产率=72%)。其核磁共振1h谱图如图1所示,核磁共振
13
c谱图如图2所示。
[0049]1h nmr(400mhz,cdcl3):δ7.37
‑
7.34(m,1h,arh),7.33
‑
7.27(m,2h,arh),7.26
‑
7.20(m,2h,arh),6.11(s,1h,arh),5.83(s,1h,arh),4.48
‑
4.29(m,2h,ch2),4.01(s,3h,och3),3.94(s,3h,och3),3.76(s,3h,och3),3.29
‑
3.11(m,1h,ch),2.24(dd,j=14.1,3.2hz,1h,ch2),1.95(dd,j=14.1,11.1hz,1h,ch2),1.18(d,j=6.8hz,3h,ch3);
13
c nmr(100mhz,cdcl3):δ190.3(c=o),170.3(c=on),168.9(coo),165.7(aro),164.9(aro),158.0(aro),138.0(ar),128.7(ar),127.8(ar),127.5(ar),104.5(ar),97.8(ar),95.5(ar),89.8(c),57.1(och3),56.4(och3),53.5(och3),43.7(ch2),37.0(ch),35.9(ch2),14.4(ch3);hrms(esi):m/z calcd for c
23
h
25
no7cl:462.1320;found:462.1317[m h]
.
[0050]
实施例3
[0051]
将142mg(0.38mmol)的灰黄霉素开环中间体1溶于2ml的无水dmf中,加入100mg(0.76mmol)dipea和218mg(0.57mmol)hatu,搅拌15分钟后,加入60mg(0.4mmol)3
’‑
三氟甲基苄胺,室温反应12h。加入20ml乙酸乙酯稀释,然后依次水洗、饱和食盐水洗,mgso4干燥,减压干燥,柱层析(石油醚:乙酸乙酯=1:1)得到白色固体(2b)151mg(产率=75%)。其核磁共振1h谱图如图3所示,核磁共振
13
c谱图如图4所示。
[0052]1h nmr(400mhz,cdcl3):δ7.54
‑
7.50(m,1h,arh),7.48(s,1h,arh),7.46
‑
7.39(m,2h,arh),6.12(s,1h,arh),5.99(t,j=6.0hz,1h,nh),4.54
‑
4.36(m,2h,ch2),4.01(s,3h,och3),3.95(s,3h,och3),3.76(s,3h,och3),3.32
‑
3.08(m,1h,ch),2.28(dd,j=14.1,3.3hz,1h,ch2),1.99(dd,j=14.1,10.9hz,1h,ch2),1.18(d,j=6.8hz,3h,ch3);
13
c nmr(100mhz,cdcl3):δ190.3(c=o),170.6(c=on),168.9(coo),165.6(aro),165.0(aro),158.0(aro),139.2(ar),131.2(ar),131.2(ar),129.2(ar),124.4(cf3),124.4(ar),124.3(ar),104.5(ar),97.8(ar),95.4(ar),89.8(c),57.1(och3),56.4(och3),53.5(och3),43.1(ch2),37.0(ch),35.9(ch2),14.4(ch3);hrms(esi):m/z calcd for c
24
h
24
no7clf3:530.1193;found:530.1190[m h]
.
[0053]
实施例4
[0054]
将142mg(0.38mmol)的灰黄霉素开环中间体1溶于2ml的无水dmf中,加入100mg(0.76mmol)dipea和218mg(0.57mmol)hatu,搅拌15分钟后,加入43mg(0.4mmol)正丁胺,室温反应12h。加入20ml乙酸乙酯稀释,然后依次水洗、饱和食盐水洗,mgso4干燥,减压干燥,
柱层析(石油醚:乙酸乙酯=1:1)得到白色固体(2c)151mg(产率=93%)。其核磁共振1h谱图如图5所示,核磁共振
13
c谱图如图6所示。
[0055]1h nmr(400mhz,cdcl3):δ6.13(s,1h,arh),5.49(t,j=5.3hz,1h,nh),4.02(s,3h,och3),3.97(s,3h,och3),3.77(s,3h,och3),3.24
‑
3.18(m,2h,ch2),3.16
‑
3.12(m,1h,ch),2.20(dd,j=13.9,3.3hz,1h,ch2),1.90(dd,j=13.9,11.2hz,1h,ch2),1.49
‑
1.38(m,2h,ch2),1.37
‑
1.25(m,2h,ch2),1.17(d,j=6.8hz,3h,ch3),0.90(t,j=7.3hz,3h,ch3);
13
c nmr(100mhz,cdcl3):δ190.2(c=o),170.3(c=on),168.9(coo),165.7(aro),164.8(aro),157.9(aro),104.6(=cc=o),97.8(ar),95.5(ar),89.8(c),57.1(och3),56.4(och3),53.4(och3),39.3(ch),37.1(ch2),36.0(ch2),31.6(ch2),20.0(ch2),14.3(ch3),13.7(ch3);hrms(esi):m/z calcd for c
20
h
27
no7cl:428.1476;found:428.1471[m h]
.
[0056]
实施例5
[0057]
将142mg(0.38mmol)的灰黄霉素开环中间体1溶于2ml的无水dmf中,加入100mg(0.76mmol)dipea和218mg(0.57mmol)hatu,搅拌15分钟后,加入43mg(0.4mmol)对甲基苯胺,室温反应12h。加入20ml乙酸乙酯稀释,然后依次水洗、饱和食盐水洗,mgso4干燥,减压干燥,柱层析(石油醚:乙酸乙酯=1:1)得到白色固体(2d)154mg(产率=88%)。其核磁共振1h谱图如图7所示,核磁共振
13
c谱图如图8所示。
[0058]1h nmr(400mhz,cdcl3):δ7.36(s,1h,nh),7.34(d,j=3.1hz,2h,arh),7.09(d,j=8.3hz,2h,arh),6.12(s,1h,arh),4.02(s,3h,och3),3.96(s,3h,och3),3.75(s,3h,och3),3.28(ddp,j=10.5,6.7,3.3hz,1h,ch),2.44(dd,j=14.1,3.8hz,1h,ch2),2.29(s,3h,ch3),2.11(dd,j=14.1,10.6hz,1h,ch2),1.22(d,j=6.8hz,3h,ch3);
13
c nmr(100mhz,cdcl3):δ190.5(c=o),168.9(c=on),168.5(coo),165.7(aro),164.9(aro),158.0(aro),135.2(ar),133.9(ar),129.4(ar),119.8(ar),104.7(ar),97.8(ar),95.3(ar),89.8(c),57.1(och3),56.4(och3),53.5(och3),38.2(ch),35.9(ch2),20.9(ch3),14.5(ch3);hrms(esi):m/z calcd for c
23
h
25
no7cl:462.1320;found:462.1321[m h]
.
[0059]
实施例6
[0060]
将142mg(0.38mmol)的灰黄霉素开环中间体1溶于2ml的无水dmf中,加入100mg(0.76mmol)dipea和218mg(0.57mmol)hatu,搅拌15分钟后,加入36mg(0.4mmol)吗啉,室温反应12h。加入20ml乙酸乙酯稀释,然后依次水洗、饱和食盐水洗,mgso4干燥,减压干燥,柱层析(石油醚:乙酸乙酯=1:1)得到白色固体(2e)120mg(产率=72%)。其核磁共振1h谱图如图9所示,核磁共振
13
c谱图如图10所示。
[0061]1h nmr(400mhz,cdcl3):δ6.14(s,1h,arh),4.03(s,3h,och3),3.98(s,3h,och3),3.77(s,3h,och3),3.64(d,j=4.4hz,4h,ch2),3.61
–
3.53(m,2h,ch2),3.41(d,j=4.9hz,2h,ch2),3.22
‑
3.13(m,2h,ch2),2.36(d,j=14.4hz,1h,ch),2.20
‑
2.10(m,2h,ch2),1.17(dd,j=6.8,2.0hz,3h,ch3);
13
c nmr(100mhz,cdcl3):δ190.3(c=o),169.2(c=on),168.9(coo),165.7(aro),164.9(aro),157.9(aro),104.5(ar),97.8(ar),95.5(ar),89.8(c),66.9(och2),66.7(och2),57.1(och3),56.4(och3),53.4(och3),46.1(ch2),42.0(ch2),35.0(ch2),33.5(ch2),14.4(ch3);hrms(esi):m/z calcd for c
20
h
25
no8cl:442.1269;found:442.1269[m h]
.
[0062]
实施例7
[0063]
将142mg(0.38mmol)的灰黄霉素开环中间体1溶于2ml的无水dmf中,加入100mg(0.76mmol)dipea和218mg(0.57mmol)hatu,搅拌15分钟后,加入40mg(0.4mmol)甲基哌嗪,室温反应12h。加入20ml乙酸乙酯稀释,然后依次水洗、饱和食盐水洗,mgso4干燥,减压干燥,柱层析(石油醚:乙酸乙酯=1:1)得到白色固体(2f)158mg(产率=92%)。其核磁共振1h谱图如图11所示,核磁共振
13
c谱图如图12所示。
[0064]1h nmr(400mhz,cdcl3):δ6.13(s,1h,arh),4.03(s,3h,och3),3.97(s,3h,och3),3.77(s,3h,och3),3.61(dtd,j=18.7,12.9,7.0hz,2h,ch2),3.44(tq,j=14.2,9.0,7.0hz,2h,ch2),3.18(ddd,j=11.1,8.4,2.9hz,1h,ch),2.43
‑
2.33(m,4h,ch2),2.31(s,3h,ch3),2.15(dd,j=14.3,11.3hz,2h,ch2),1.17(d,j=6.8hz,3h,ch3);
13
c nmr(100mhz,cdcl3):δ190.3(c=o),168.9(c=on),165.7(aro),164.8(aro),157.9(aro),104.6(ar),97.8(ar),95.6(ar),89.8(c),57.1(och3),56.4(och3),55.2(och3),54.7(ch2),53.4(ch2),46.0(ch2),45.6(ch2),41.6(ch2),35.1(ch),33.7(ch3),14.4(ch3);hrms(esi):m/z calcd for c
21
h
28
n2o7cl:455.1585;found:455.1586[m h]
.
[0065]
为了更好地理解本发明的实质,下面分别用本发明提供的灰黄霉素开环衍生物对六种肿瘤细胞株的生长的抑制作用的药理实验结果,说明其在抗肿瘤药物研究领域中的新用途。药理实施例给出了代表性化合物的部分活性数据。必须说明,本发明的药理实施例是用于说明本发明而不是对本发明的限制。根据本发明的实质对本发明进行的简单改进都属于本发明要求保护的范围。
[0066]
药物实验例1:化合物2a~2f和紫杉醇对人口腔上皮癌细胞(kb)细胞毒活性测试
[0067]
人口腔上皮癌细胞kb用mem培养基培养,培养基中含有10%的胎牛血清,100u/ml青霉素和100u/ml的链霉素。细胞以每孔5
×
103的浓度加入到96孔板中,在37℃含有5%co2的潮湿空气的培养箱中培养24小时。
[0068]
将化合物2a~2f溶于dmso中,配制1
×
10
‑2mol/l的母液,用完全培养基将母液稀释到相应浓度取对数生长期细胞接种于96孔板,24h贴壁后加入不同浓度的化合物溶液,每个浓度设4个平行孔,培养68h后加入四甲基偶氮唑盐(mtt)溶液,继续培养4h,弃去培养液,加入二甲亚砜150μl,振荡10min,用酶标仪测定570nm吸收度(a)值,计算半数抑制浓度(ic
50
),具体如表1所示。根据表1可知,化合物2a的ic
50
为4
×
10
‑7m,而阳性对照紫杉醇对kb细胞的ic
50
为2
×
10
‑7m。
[0069]
药物实验例2
‑
6:化合物2a~2f和紫杉醇对人胃癌细胞(mgc803),人肺腺癌细胞(a549),人子宫颈癌细胞(hela),人结肠癌细胞(hct
‑
116),人肝癌细胞(hepg2)细胞毒活性测试。
[0070]
采用药物实验例1所示方法,对人胃癌细胞(mgc803),人肺腺癌细胞(a549),人子宫颈癌细胞(hela),人结肠癌细胞(hct
‑
116),人肝癌细胞(hepg2)的生长抑制作用进行药理实验,计算半数抑制浓度(ic
50
),具体如表1所示。
[0071]
表1化合物2a~2f和紫杉醇的细胞毒活性测试结果
[0072][0073]
根据表1可知,本发明提供的灰黄霉素开环衍生物具有重要的生物活性,体外对人胃癌细胞(mgc803),人口腔上皮癌细胞(kb),人结肠癌细胞(hct
‑
116),人肺腺癌细胞(a549),人子宫颈癌细胞(hela),人肝癌细胞(hepg2)共六种肿瘤细胞的细胞毒活性试验表明:此类式(1)所示结构的灰黄霉素开环衍生物对肿瘤细胞生长具有抑制作用,有可能发展成为新的防治肿瘤药物。从以上药理实施例中我们可以看出这些化合物对这六种肿瘤细胞都显示了较强的细胞毒活性,细胞毒活性超过或与阳性对照紫杉醇相当,具有开发成抗肿瘤药物的潜力。
[0074]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。