s型t
‑
cof@cds核壳异质结复合光催化材料及其制备和在光解产氢中的应用
技术领域
1.本发明属于光解产氢领域,具体涉及光解产氢的光催化材料。
背景技术:
2.工业革命以来,随着重工业和制造业的蓬勃发展,人类社会对化石能源的需求日益增大。但地球上的化石能源十分有限,且其带来的环境污染问题日趋严重,因此,寻找一种绿色、清洁的可再生能源以替代现有的化石燃料迫在眉睫。在此情况下,太阳能作为一种取之不尽用之不竭的清洁可再生的自然资源,已成为化石燃料的一种极具前景的替代品。然而,太阳光到达地球表面的量不仅取决于地理位置、季节和时间,更难以以一种低成本的方式对其进行转换和储存。氢能凭借其高热值、环境友好、易于运输等特点成为公认的清洁能源,是太阳能潜在的能量载体。氢气可以通过直接燃烧或氢燃料电池释放能量,其唯一的副产物是清洁无污染的水。因此,利用太阳能光解水产氢是一种极具前景的光能转化方式。半导体光催化剂在这项技术中至关重要,通过设计、调控和改性,大多数半导体可以满足宽范围光吸收,有效电荷分离和迁移等要求。在过去的几十年中,已经对各种半导体(例如tio2,cds,g
‑
c3n4等)进行了充分的研究。
3.cds作为带隙为2.4ev的无机半导体,被认为是利用太阳能产氢最有前途的材料之一。但cds作为光催化剂有两个主要缺点,首先是cds中光生电子/空穴对很容易复合,导致可见光催化活性低。其次cds在光照条件下,价带上的空穴易与s
2
反应,导致cds被腐蚀,限制了其在实际中的大规模应用。如果能将cds与合适的材料复合,使电子集中在cds上发生还原反应生,而空穴集中在另一种材料上发生氧化反应,这不仅可以避免cds被腐蚀,而且在一定程度上可以抑制光生电子和空穴的复合。
4.共价有机框架材料(cofs)作为一种有结晶性的多孔有机聚合物材料,不仅具有刚性规整的孔道结构,而且有超高的比表面积,为光催化反应提供了更多活性位点,同时有序规整的孔道结构可加快传质速率,缩短反应时间;(2)此外通过调整构筑单元,可以设计和调控cofs的拓扑结构、孔道大小和能带结构,以满足不同应用的需求;(3)cofs易于功能化,可通过“自上而下法”或后修饰法将特定功能的官能团嵌入到材料的骨架中,从而设计出适用性强、性能优异的cofs光催化剂;(4)cofs由共价键连接而成,具较高的热力学和化学稳定性,易于循环利用。但目前cofs作为光催化剂还面临着不少挑战,尤其是内部光生电子空穴对的复合率较高,降低了cofs材料的催化性能量子效率。
5.目前,cds型材料主要是mofs复合材料,cofs复合材料较少,为数不多的cds
‑
cofs复合材料也仅是常规物理混合的ii
‑
type异质结复合材料;该类材料的稳定性较差,材料的氧化还原能力较低。
技术实现要素:
6.针对现有的技术中的不足,本发明的第一目的在于提供一种s型t
‑
cof@cds核壳异
质结复合光催化材料(本发明也称为s型核壳异质结光催化剂),旨在提供一种具有光吸收范围宽,具有大量孔道结构、且结合力强,稳定性好的s型核壳异质结光催化剂。
7.本发明的第二个目的是提供一个简易、温和、经济的制备方法用于上述s型核壳异质结光催化剂的制备。
8.本发明的第三个目的是将所所述的s型核壳异质结复合材料应用于光驱动下水分解产氢,旨在改善光催化产氢的稳定性以及产氢活性。
9.为了实现上述技术目的,本发明提供了一种s型t
‑
cof@cds核壳异质结复合光催化材料,具有核
‑
壳结构,其中,核为cds纳米颗粒,壳为t
‑
cof多孔材料;所述的核
‑
壳之间形成s型异质结结构;
10.所述的t
‑
cof多孔材料由式1和式2的单体聚合形成;
[0011][0012][0013]
本发明创新地发现,通过式1和式2单体聚合构建的t
‑
cof和cds存在特殊的物理、化学双适配协同性,其能够意外地形成s型核壳异质结。研究发现,该全新结构的材料具有良好的稳定性以及光驱动下的产氢活性。
[0014]
cds纳米颗粒为球形纳米颗粒;优选地,其颗粒度为50~70nm。
[0015]
优选的方案,所述t
‑
cof具有多孔结构,其孔道为均一的球形结构,孔径为~1.1nm,比表面积为1000~1605m2/g。丰富的孔结构有利于水分子进入孔道,进而到达cds表面。
[0016]
作为优选,式1、式2的摩尔比为1~1.5:1;优选为1:1。
[0017]
作为优选,所述的t
‑
cof中的式2单体、cds的质量比为0.5~3:1;优选为0.7~1.5:1;进一步优选为1~1.5:1;最优选为1~1.2:1。研究发现,控制在该优选的比例下,有助于进一步改善核
‑
壳之间物理
‑
化学的适配协同性,有助于进一步改善材料的稳定性以及产氢性能。
[0018]
本发明还提供了一种所述的s型t
‑
cof@cds核壳异质结复合光催化材料的制备方法,将cds纳米颗粒、式1、式2用乙腈分散均匀,随后再和催化剂混合,进行聚合反应,在cds纳米颗粒的表面原位聚合形成t
‑
cof多孔材料,制得所述的s型t
‑
cof@cds核壳异质结复合光催化材料;
[0019]
所述的催化剂为醋酸。
[0020]
本发明研究发现,如何诱导t
‑
cof在cds表面原位聚合以及如何促使t
‑
cof和cds表面的物理
‑
化学相适配是合成本发明所述材料的关键难点。本发明通过研究发现,创新地基于cds表面和式1、式2单体之间特殊的诱导机制以及聚合过程的条件(如溶剂、催化剂等)的
协同控制,能够意外地诱导单体在cds表面原位聚合,从而能意外地构建所述的s型t
‑
cof@cds核壳异质结复合光催化材料。研究发现,该方法构建得到的特殊异质结结构的材料能够意外地表现出更优的稳定性以及催化产氢活性。
[0021]
本发明的s型核壳异质结复合材料关键在cofs能够自动迁移到cds周围,在cds表面生长,且cds的存在,不能影响cofs的有序生长。本发明通过研究意外地发现,cds和本发明的t
‑
cof之间存在特殊的诱导电荷诱导机制,可利用静电荷相互作用,使t
‑
cof在生长的过程中自发地迁移到cds表面,并配合所述的乙腈溶剂等条件的联合控制,可以实现t
‑
cof和cds之间特殊的物理
‑
化学双适应,从而利于在cds表面原位形成t
‑
cof,构建特殊的s型异质结。
[0022]
本方案制得的s型核壳异质结复合光催化材料,光吸收范围宽,且光生电子和空穴分别集中在导带更负和价带更正的半导体上,提高了材料的氧化还原能力,抑制了光生载流子的快速复合。
[0023]
本发明中,式1、式2的摩尔比为1~1.5:1;优选为1:1。
[0024]
优选地,式2单体、cds的质量比为0.5~3:1;优选为0.7~1.5:1;进一步优选为1~1.5:1;最优选为1~1.2:1。
[0025]
作为优选,所述的乙腈相对于cds纳米颗粒的重量比为1000:1~5;优选为180~800:1。
[0026]
作为优选,所述的催化剂以水熔液形式添加;优选地,催化剂的水溶液中,催化剂的摩尔浓度大于或等于9m;优选为12m至饱和浓度;进一步优选为12m~15m;
[0027]
优选地,催化剂相对于式1单体摩尔比为50~250:1;优选为100~150:1。
[0028]
优选地,聚合反应过程在室温下进行,所述的室温温度例如为20~45℃;进一步优选为20~30℃。
[0029]
作为优选,聚合反应过程在静置条件下进行。
[0030]
作为优选,静置反应的时间为72
‑
120h;进一步优选为90~100h。
[0031]
本发明中,所述的cds可以采用现有手段制备。本发明优选采用以下方法制备:
[0032]
将硫源、镉源、pvp和乙二醇进行水热反应,随后经过洗涤、干燥即得。
[0033]
所述的硫源例如为硫脲。所述的镉源例如为能电离出cd
2
的水溶性盐,例如为cd
2
的硝酸盐。
[0034]
所述的硫源、镉源中的s:cd的摩尔比为1:1。所述的pvd和cd的摩尔比为0.5~1.5:1;乙二醇作为溶剂,其中cd的浓度为0.01~0.2m。水热温度例如为100~160℃。水热时间例如为6~20h。
[0035]
本发明优选的t
‑
cof@cds核壳复合光催化材料的合成方法:将一定量的球形cds和式2分散在乙腈溶液中,超声后,加入式1的乙腈溶液,然后再次超声,最后加入催化剂醋酸水溶液,室温下静置反应。
[0036]
本发明还包括采用所述的制备方法构建得到的s型t
‑
cof@cds核壳异质结复合光催化材料,其具有所述的制备方法构建的特殊的物理
‑
化学作用,且能够表现出更优的稳定性以及光驱动产氢性能。
[0037]
本发明还提供了一种所述的s型t
‑
cof@cds核壳异质结复合光催化材料的应用,作用光催化剂,用于催化水光解产氢。
[0038]
本发明所述的复合材料,其具有优异的稳定性,还具有优异的光驱动水解产氢性能。本发明所述的材料,在无需任何助剂辅助下,即可实现良好的光驱动水解产氢。
[0039]
作为优选,将所述的s型t
‑
cof@cds核壳异质结复合光催化材料和水、牺牲剂混合,在光照下进行光解产氢反应,获得氢气;
[0040]
优选地,所述的牺牲剂为三乙醇胺、乳酸、甲醇,优选的为三乙醇胺;
[0041]
优选地,光照的功率没有特别的要求,例如,可以大于或等于100w,优选为200~400w。
[0042]
相对于现有技术,本发明的技术方案带来的有效成果是:
[0043]
(1)本发明提供了一种全新的具有s型t
‑
cof@cds核壳异质结复合光催化材料。该全新结构的材料可以形成特殊的s型异质结,可以表现出更优的稳定性以及光驱动下的产氢性能。
[0044]
(2)本发明还提供了所述的材料的制备方法,其创新地基于cds和所述的单体成分以及制备过程的条件(如溶剂)的联合控制,能够产生协同性,能够意外地在cds表面诱导t
‑
cof形成,并促使核
‑
壳之间物理、化学相适配,从而成功构建所述的s型t
‑
cof@cds核壳异质结材料。
[0045]
本发明优选的方案,以式1和式2为构筑单体,醋酸水溶液为催化剂,乙腈为溶剂,在室温下即可让t
‑
cof原位生长在cds表面,形成形貌均一的核壳结构,告别了传统高温合成cofs的方法,适用于规模化制备。
[0046]
本发明所制备的t
‑
cof@cds复合材料孔道结构有序,比表面积较高。丰富的孔结构有利于提高传质速率,使反应物进入孔道到达cds表面,为催化反应奠定了基础。
[0047]
(3)本发明以t
‑
cof@cds作为光催化剂,在光照的条件下,可以分解水产生氢气,且其性能远超于单一的cds和t
‑
cof,具有良好的催化活性、及稳定性。
[0048]
(4)本发明制备的t
‑
cof@cds复合光催化剂制备方法简单,容易操作,采用无毒,价廉的原料,无需大型复杂器件,环保,可用于工业化。
附图说明
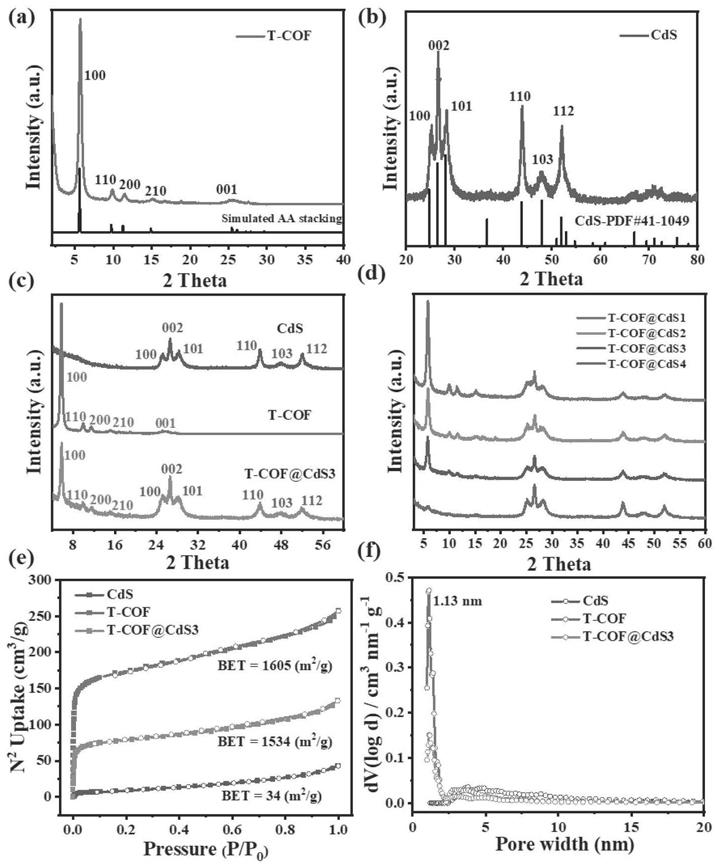
[0049]
【图1】为本发明实施案例1、2、3、4、5、6制备的样品的x射线衍射(xrd)图、氮气吸/脱附曲线图和孔径分布图:(a)实施案例1制备的t
‑
cof样品的x射线衍射(xrd)图;(b)实施案例2所制备的球形cds的x射线衍射(xrd)图;(c)实施案例1、2、5、制备的t
‑
cof、cds、t
‑
cof@cds3样品的x射线衍射(xrd)对比图;(d)实施案例3、4、5、6制备的t
‑
cof@cds样品的x射线衍射(xrd)对比图;(e)实施案例1、2、5、制备的t
‑
cof、cds、t
‑
cof@cds3样品的的氮气吸/脱附曲线图;(f)实施案例1、2、5、制备的t
‑
cof、cds、t
‑
cof@cds3样品的的孔径分布图。
[0050]
【图2】为本发明实施案例1、2、5、制备的t
‑
cof、cds、t
‑
cof@cds3的扫描电子显微镜(sem)、透射电子显微镜(tem)和mapping图像。(a)实施案例2制备的cds样品的sem图;(b)实施案例1制备的t
‑
cof样品的sem图;(c)实施案例5制备的t
‑
cof@cds3样品的sem图;(d)实施案例2制备的cds样品的tem图(e)实施案例1制备的t
‑
cof样品的tem图;(f)实施案例5制备的t
‑
cof@cds3样品的tem图;(g)实施例5所制备的t
‑
cof@cds3样品的tem图;(h)实施例5所制备的t
‑
cof@cds3样品的c元素的mapping图;(i)实施例5所制备的t
‑
cof@cds3样品的n元素的mapping图;(j)实施例5所制备的t
‑
cof@cds3样品的s元素的mapping图;(k)实施例5所
制备的t
‑
cof@cds3样品的cd元素的mapping图;
[0051]
【图3】为本发明实施案例1、2、3、4、5、6制备的样品的紫外光电子能谱(ups)、x
‑
射线光电子能谱(vb
‑
xps)、紫外可见漫反射光谱(drs):(a)为本发明实施案例1所制备的cds样品的ups图谱;(b)为本发明实施案例2所制备的t
‑
cof样品的ups图谱;(c)为本发明实施案例1所制备的cds样品的vb
‑
xps图谱;(d)为本发明实施案例2所制备的t
‑
cof样品的vb
‑
xps图谱;(e)为本发明实施案例1、2、5、制备的t
‑
cof、cds、t
‑
cof@cds3样品的drs光谱图;(f)为本发明实施案例1、2所制备的cds、t
‑
cof样品的能带结构图。
[0052]
【图4】为本发明实施案例1、2、5制备的样品的能带偏移图、界面电荷性质、电荷转移图以及zeta电位谱图:(a)为本发明实施案例1、2、5所制备的cds、t
‑
cof、t
‑
cof@cds样品的能带结构图;(b)为本发明实施案例5所制备的t
‑
cof@cds在光照条件下的电荷转移图;(c)为本发明实施案例1、2所制备的cds、t
‑
cof样品的zeta电位图。
[0053]
【图5】为本发明实施案例1、2、3、4、5、6、7(对比例1)制备的样品的稳态固体荧光(pl)谱图和光催化性能图:(a)为本发明实施案例1、2、5制备的t
‑
cof、cds、t
‑
cof@cds3样品的pl图谱;(b)为本发明实施案例1、2、3、4、5、6、7制备的样品的光解水产氢动力学图;(c)为本发明实施案例1、2、3、4、5、6、7制备的样品的光解水产氢性能对比图;(d)为本发明实施案例5所制备的t
‑
cof@cds3的光解水产氢循环实验图。
[0054]
【图6】为本发明实施案例的制备示意图;
具体实施方式
[0055]
下面通过具体实施例对本发明技术方案进行进一步的说明,但本发明的保护范围不限于下述实施例。
[0056]
以下案例,所述的室温的温度例如为25~45℃。
[0057]
实施例1
[0058]
t
‑
cof的制备:称取式1(0.04mmol,6.5mg)和式2(0.04mmol,14.2mg)置于15ml玻璃小瓶中,然后加入5ml乙腈,超声30min,最后缓慢滴加12m的醋酸水溶液(0.4ml)。立即封口静置,在室温下反应96h。待反应结束后,用n
‑
n二甲基甲酰胺洗涤三次,收集到的固体在70℃真空干燥烘箱干燥12h。获得的样品命名为t
‑
cof。
[0059]
实施例2
[0060]
球形cds的制备:称取硫脲(0.53g,7.0mmol),cd(no3)2·
4h2o(2.16g,7.0mmol),聚乙烯吡咯烷酮pvp(0.78g,7.0mmol)于150ml反应釜内胆中,加入70ml乙二醇,超声至固体完全溶解,然后将反应密封放入120℃烘箱中反应12h。待反应冷却后,用甲醇多次洗涤,收集到的固体在70℃真空干燥烘箱干燥12h。
[0061]
实施例3
[0062]
t
‑
cof@cds1的制备:称取式2(0.04mmol,14.2mg)和cds(实施例2制备;5mg)置于15ml玻璃小瓶中,然后加入3ml乙腈,超声60min,再加入式1(0.04mmol,6.5mg)的乙腈溶液(2ml),再次超声10min,然后缓慢滴加12m的醋酸水溶液(0.4ml)。立即封口静置,在室温下反应96h。待反应结束后,用n
‑
n二甲基甲酰胺洗涤三次,收集到的固体在70℃真空干燥烘箱干燥12h。,获得的样品命名t
‑
cof@cds1。
[0063]
实施例4
[0064]
t
‑
cof@cds2的制备:称取式2(0.04mmol,14.2mg)和cds(10mg)置于15ml玻璃小瓶中,然后加入3ml乙腈,超声60min,再加入式1(0.04mmol,6.5mg)的乙腈溶液(2ml),再次超声10min,然后缓慢滴加12m的醋酸水溶液(0.4ml)。立即封口静置,在室温下反应96h。待反应结束后,用n
‑
n二甲基甲酰胺洗涤三次,收集到的固体在70℃真空干燥烘箱干燥12h。,获得的样品命名t
‑
cof@cds2。
[0065]
实施例5
[0066]
t
‑
cof@cds3的制备:称取式2(0.04mmol,14.2mg)和cds(14mg)置于15ml玻璃小瓶中,然后加入3ml乙腈,超声60min,再加入式1(0.04mmol,6.5mg)的乙腈溶液(2ml),再次超声10min,然后缓慢滴加12m的醋酸水溶液(0.4ml)。立即封口静置,在室温下反应96h。待反应结束后,用n
‑
n二甲基甲酰胺洗涤三次,收集到的固体在70℃真空干燥烘箱干燥12h。获得的样品命名t
‑
cof@cds3。
[0067]
实施例6
[0068]
t
‑
cof@cds4的制备:称取式2(0.04mmol,14.2mg)和cds(20mg)置于15ml玻璃小瓶中,然后加入3ml乙腈,超声60min,再加入式1(0.04mmol,6.5mg)的乙腈溶液(2ml),再次超声10min,然后缓慢滴加12m的醋酸水溶液(0.4ml)。立即封口静置,在室温下反应96h。待反应结束后,用n
‑
n二甲基甲酰胺洗涤三次,收集到的固体在70℃真空干燥烘箱干燥12h。,获得的样品命名t
‑
cof@cds4。
[0069]
实施例7
[0070]
调控单体比例,分别为:(a):称取式1(0.04mmol,6.5mg)和式2(0.04mmol,14.2mg)置于15ml玻璃小瓶1中;(b):称取式1(0.05mmol,8.1mg)和式2(0.04mmol,14.2mg)置于15ml玻璃小瓶2中;(c):称取式1(0.06mmol,9.7mg)和式2(0.04mmol,14.2mg)置于15ml玻璃小瓶3中。
[0071]
然后分别加入5ml乙腈,超声30min,最后再分别缓慢滴加12m的醋酸水溶液(0.4ml)。立即封口静置,在室温下反应96h。待反应结束后,用n
‑
n二甲基甲酰胺洗涤三次,收集到的固体在70℃真空干燥烘箱干燥12h。获得的样品命名为t
‑
cof1(也即是实施例1的t
‑
cof),t
‑
cof2,t
‑
cof3。
[0072]
对比例1
[0073]
和实施例5相比,区别仅在于,将得到的t
‑
cof、cds按相同比例混合,步骤为:
[0074]
t
‑
cof/cds3的制备:称取20.7mg t
‑
cof(实施例1制备)和14mg cds(实施例2制备)于干净的研钵中,将两种样品充分研磨,通过物理共混制得t
‑
cof/cds3。
[0075]
对比例2
[0076]
改变单体种类:将式2改为1,3,5
‑
三(4
‑
氨苯基)苯。
[0077]
称取1,3,5
‑
三(4
‑
氨苯基)苯(0.04mmol,14.1mg),不同量的cds(4mg,10mg,14mg,20mg)置于15ml玻璃小瓶中,然后加入3ml乙腈,超声60min,再加入式1(0.04mmol,6.5mg)的乙腈溶液(2ml),再次超声10min,然后缓慢滴加12m的醋酸水溶液(0.4ml)。立即封口静置,在室温下反应96h。待反应结束后,用n
‑
n二甲基甲酰胺洗涤三次,收集到的固体在70℃真空干燥烘箱干燥12h,获得的样品分别命名为b
‑
cof@cds1、b
‑
cof@cds2、b
‑
cof@cds3、b
‑
cof@cds4。研究发现,采用1,3,5
‑
三(4
‑
氨苯基)苯作为单体,并没有形成和本发明所述的t
‑
cof@cds类似的包覆s
‑
型异质结,且其产氢性能相较于单独b
‑
cof和cds没有显著提升。
[0078]
对比例3
[0079]
和实施例1相比,区别主要在于,改变溶剂,例如,将乙腈分别换成二氧六环或均三甲苯。
[0080]
称取式1(0.04mmol,6.5mg)和式2(0.04mmol,14.2mg)置于15ml玻璃小瓶1中;称取式1(0.04mmol,6.5mg)和式2(0.04mmol,14.2mg)置于15ml玻璃小瓶2中,然后瓶1和瓶2中分别加入5ml二氧六环和均三甲苯,超声30min,最后缓慢滴加12m的醋酸水溶液(0.4ml)。立即封口静置,在室温下反应96h。待反应结束后,用n
‑
n二甲基甲酰胺洗涤三次,收集到的固体在70℃真空干燥烘箱干燥12h。获得的样品分别命名为t
‑
cof
‑
d,t
‑
cof
‑
m。在这两种溶剂中得到的t
‑
cof的微观形貌无序且不规则。在此类溶剂中合成t
‑
cof@cds时,t
‑
cof无法有序的长在cds上,无法合成核壳型s型异质结。
[0081]
对比例4
[0082]
和实施例1相比,区别主要在于,改变催化剂种类:将醋酸换成hcl或h2so4。
[0083]
分别称取式1(0.04mmol,6.5mg)和式2(0.04mmol,14.2mg)各五份,分别置于5个15ml玻璃小瓶中,然后分别加入5ml乙腈,超声30min,最后分别缓慢滴加0.5m,1m,2m,3m,6m的hcl 0.4ml。立即封口静置,在室温下反应96h。待反应结束后,用n
‑
n二甲基甲酰胺洗涤三次,收集到的固体在70℃真空干燥烘箱干燥12h。所得样品都为无定形材料,即在此条件下无法合成t
‑
cof。
[0084]
分别称取式1(0.04mmol,6.5mg)和式2(0.04mmol,14.2mg)各五份,分别置于5个15ml玻璃小瓶中,然后分别加入5ml乙腈,超声30min,最后分别缓慢滴加0.5m,1m,2m,3m,6m的h2so4 0.4ml。立即封口静置,在室温下反应96h。待反应结束后,用n
‑
n二甲基甲酰胺洗涤三次,收集到的固体在70℃真空干燥烘箱干燥12h。所得样品都为无定形材料,即在此条件下无法合成t
‑
cof。
[0085]
实施例8
[0086]
和实施例1相比,区别主要在于,改变醋酸浓度,步骤为:
[0087]
分别称取式1(0.04mmol,6.5mg)和式2(0.04mmol,14.2mg)各五份,分别置于5个15ml玻璃小瓶中,然后分别加入5ml乙腈,超声30min,最后分别缓慢滴加3m,6m,9m,12m,15m的醋酸水溶液0.4ml。立即封口静置,在室温下反应96h。待反应结束后,用n
‑
n二甲基甲酰胺洗涤三次,收集到的固体在70℃真空干燥烘箱干燥12h。获得的样品命名为t
‑
cof
‑
3m,t
‑
cof
‑
6m,t
‑
cof
‑
9m,t
‑
cof
‑
12m,t
‑
cof
‑
15m。3m醋酸作催化剂时,得到的固体样品为无定形材料,即在此条件无法合成t
‑
cof。6m醋酸作催化剂时,得到的固体样品有微弱的晶型。9m,12m,15m醋酸作催化剂时,都可以得到有一定晶型的cofs材料,但是醋酸为12m时,得到的材料晶型最好。
[0088]
光驱动产氢步骤:光催化产氢实验是在室温下,25ml的光反应器中使用300w的氙灯(>380nm)照射完成的。具体操作过程如下:
①
将6mg光催化剂分散在6ml去离子水和0.5ml三乙醇胺(牺牲剂)中,不停搅拌并通n2约40min以排净其内空气;
②
组装好反应体系,将光反应器固定后用氙灯照射;
③
体系中产生的h2可通过气相色谱进行检测,即用进样针抽取200μl的上层气体注射到气相色谱仪中,计算体系的产氢量。
[0089]
图1(a)所示为t
‑
cof样品的xrd图谱。100、110、200、210等晶面对应的峰与模拟计算得出的a
‑
a堆积型cof的图谱基本一致,表明a
‑
a堆积的t
‑
cof成功合成。图1(b)所示为cds
的xrd图谱。100、002、101、110等晶面对应的峰与cds标准卡片上的峰基本一致,表明cds成功合成。图1(c)所示为t
‑
cof、cds、t
‑
cof@cds3样品的xrd对比图。从图中可以看出由t
‑
cof包覆cds形成的核壳复合材料的xrd峰位移与单一t
‑
cof和cds的完全一致,说明cds的存在并没有影响t
‑
cof的有序生长,t
‑
cof@cds3样品成功合成。图1(d)所示为包覆不同含量cds的t
‑
cof@cds复合材料的xrd对比图。可以看出随着cds含量的增加,t
‑
cof的有序生长会收到影响,当cds含量增加到20mg,t
‑
cof各晶面对应的峰已经消失,说明此条件下cof已经无法有序生长,无法合成t
‑
cof@cds核壳结构。图1(e)所示为t
‑
cof、cds、t
‑
cof@cds3样品的氮气吸/脱附曲线图,t
‑
cof的比表面积为1605m2/g,包覆cds后,比表面积仍可达1534m2/g。图1(f)所示为t
‑
cof、cds、t
‑
cof@cds3样品的孔径分布图,t
‑
cof的孔径约为1.56nm,而水分子的直径为0.4nm,丰富的孔结构有利于水分子传输到cds表面,进而发生还原水产氢反应。
[0090]
【图2】图2(a)所示为cds样品的sem图,从图中可以看出cds为形貌均一的球形颗粒。图2(b)所示为t
‑
cof的sem图,t
‑
cof也是形貌均一的球形颗粒。图2(c)所示为t
‑
cof@cds3的sem图,可以看出复合材料的形貌仍然呈均一、有序的球形。图2(d)所示为cds样品的tem图,与其sem一致,为规整的球形。图2(e)所示为t
‑
cof的tem图,与其sem一致,为规整的球形。图2(f)和(g)所示为t
‑
cof@cds3的tem图,可以看出cds成功被t
‑
cof包裹。图2(h)、(i)、(j)、(k)分别为t
‑
cof@cds3的c、n、s、cd元素的mapping图,进一步可以证实中间深色球形为cds,外围为t
‑
cof,t
‑
cof包裹cds的核壳结构成功合成。
[0091]
图3(a)所示为cds的紫外光电子能谱,通过标定二次电子截止边(e
cutoff
)和费米边(e
fermi
),根据公式(wf=21.22
‑
e
cutoff
e
fermi
)可以求出cds的功函数(wf)为4.68ev。图3(b)所示为t
‑
cof的紫外光电子能谱,通过标定二次电子截止边(e
cutoff
)和费米边(e
fermi
),根据公式(wf=21.22
‑
e
cutoff
e
fermi
)可以求出t
‑
cof的功函数(wf)为4.98ev。图3(c)所示为cds的vb
‑
xps谱,从图中可知cds的vb
‑
xps值为1.42ev,根据公式vb(vs.rhe)=vb xps 功函数(wf)
‑
4.44ev,可以求出cds的价带为1.42vvs rhe。图3(d)所示为t
‑
cof的vb
‑
xps谱,从图中可知t
‑
cof的vb
‑
xps值为1.59ev,根据公式vb(vs.rhe)=vb xps 功函数(wf)
‑
4.44ev,可以求出t
‑
cof的价带为2.04vvs rhe。图3(e)所示为t
‑
cof、cds、t
‑
cof@cds3样品的drs图谱,从图中可以看出三种材料对可见光都有一定的吸收能力,且通过drs数据可以求出t
‑
cof、cds的带隙分别为2.65ev、2.4ev。已知带隙和价带的数值,可求出t
‑
cof、cds的导带分别为
‑
0.61v、
‑
0.71vvs rhe。图3(f)所示为t
‑
cof、cds的带隙结构图。
[0092]
图4(a)为cds、t
‑
cof和t
‑
cof@cds的能带偏移图、界面电荷性质图。cds、t
‑
cof复合前,作为独立的半导体,其功函数分别为4.68ev、4.89ev,即cds的费米能级高于t
‑
cof的费米能级,因此当两种材料接触时,cds上的电子流向t
‑
cof,直至两种半导体材料的费米能级相平。接触后形成的内建电场如图所示,由cds指向t
‑
cof,形成s
‑
型异质结。图4(b)为接触后t
‑
cof@cds异质结在光照条件下的电子流向图。光照条件下,cds和t
‑
cof价带上的电子会被激发到导带,导带上留下空穴。因为光生电场的方向与内建电场的方向相反,因此t
‑
cof导带上的电子将被驱动,自发流向cds的价带,保证电场的平衡。最后电子集中在cds的导带上,发生还原反应,而空穴集中在t
‑
cof的价带上发生氧化反应,这是典型的s
‑
型异质结的优势,增强了材料的氧化还原能力。图4(c)为cds、t
‑
cof的zeta电位图,由实验数据可知,cds的表面电位比t
‑
cof更负,进一步说明当两种材料形成异质结时,电子将从cds流向t
‑
cof,与上述由功函数分析电子流向的结果相一致。
[0093]
图5(a)为cds、t
‑
cof、t
‑
cof@cds3的光致发光光谱图,从图中可以看出,t
‑
cof@cds3异质结的发光强度远低于cds和t
‑
cof,说明在光照条件下t
‑
cof@cds3的光生载流子寿命更长。图5(b)为cds、t
‑
cof、系列t
‑
cof@cds和物理共混的t
‑
cof/cds3的光解水产氢动力学图。随着反应时间的延长,各催化剂的产氢量在不断增加。图5(c)为cds、t
‑
cof、系列t
‑
cof@cds和物理共混的t
‑
cof/cds3的光解水产氢的性能对比图。从图中可以看出核壳结构的t
‑
cof@cds异质结催化剂,其催化性能远高于单一的cds、t
‑
cof以及物理共混的t
‑
cof/cds3,说明s型核壳异质结的形成确实可以提升材料的光催化性能。图5(d)为t
‑
cof@cds3的产氢循环性能图,可以看出t
‑
cof@cds3的稳定性较好,连续循环5次,产氢性能仍没有明显下降。
[0094]
通过以上实施实例,申请人以举例的方式证明了s型核壳异质结光催化剂的制备方法以及其光解水产氢性能的影响。以上所述仅为本发明的较佳实施例,本发明的保护范围不限于上述的实施案例,凡依本发明申请专利范围所做的均等变化和修饰,皆应属本发明的涵盖范围,本技术所要求的保护范围如本技术权利要求书所示。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
