一种具有高口服生物利用度的jak抑制剂
技术领域
1.本发明属于小分子化合物领域,具体地,涉及一种具有高口服生物利用度的jak抑制剂,该jak抑制剂能够用于预防或治疗自身免疫性疾病或相关的炎症性皮肤病,特别是在口服施用时具有明显的优势。
背景技术:
2.自从辉瑞的jak3抑制剂tofacitinib和incyte的jak1/jak2抑制剂ruxolitinib 上市以来,jak抑制剂已经逐渐成为治疗变态反应和自身免疫相关的炎症性疾病、骨髓增值性疾病(myeloproliferative diseases,mpd)、移植物抗宿主疾病 (graft
‑
versus
‑
host disease,gvhd)等疾病的重要治疗手段和药物开发方向。
3.至今世界范围内已经批准了8种jak抑制剂通过用于口服或外用治疗人类疾病。由于jak家族成员存在四种结构上非常近似、功能既分化又重叠的亚型,而现有jak抑制剂多集中在对jak1、jak2和jak3亚型表现出高效抑制。这类jak抑制剂在临床应用中遇到疗效不佳或有明显副作用等问题,如已经批准上市的几款口服jak抑制剂长期使用后的安全风险(癌症及感染)逐渐暴露, fda对jak上市药物提出安全性黑盒警示。
4.目前看来jak抑制剂存在这样几个亟待解决的痛点:1)pan
‑
jak抑制剂由于抑制3种以上的jak亚型,特别是抑制jak2造成贫血和凝血障碍,口服治疗非恶性的炎症性疾病前景不乐观;2)抑制1
‑
2种jak亚型,如jak1和tyk2,应能做到安全性更高,但是特异性存在的前提下抑制效应不够;3)抑制1
‑
2种 jak亚型的jak抑制剂生物利用度不够高,无法通过口服进行系统治疗,极大地限制了其临床应用;4)同时具有高特异性抑制tyk2和jak1亚型的小分子化合物比较稀缺,缺乏这类化合物在自身免疫性疾病上的临床应用数据。
技术实现要素:
5.本发明的目的在于克服如上所述的现有技术中的不足,提供一种具有高活性、高选择性和高口服生物利用度的jak抑制剂。
6.具体地,本发明的发明人发现现有的jak抑制剂可适用于外用涂抹给药,在炎症性皮肤病动物模型上,该类jak抑制剂药物通过被动/主动吸收进入皮肤局部病变组织控制炎症、治疗炎症性皮肤疾病并显示很好疗效。然而,该类小分子jak抑制剂的口服生物利用度极低,从而不适用作为口服药物进一步开发。
7.为解决口服生物利用度低的障碍,本发明通过进一步化合物修饰以改变一类或几类小分子化合物的物理化学特性,从而提高该类化合物的体内细胞吸收性能,极大提高药物的生物利用度,对该类化合物药物的给药方式及使用提供了新的可能。因此,对在体外和体内细胞水平对jak激酶抑制很强的jak抑制剂的应用治疗相关疾病拓展了临床使用途径,是新药研发领域解决未满足的临床需求的一个重要的创新性手段。本发明也是基于上述发现而完成的。
8.为了实现上述目的,在一方面,本发明提供了一种具有高口服生物利用度的jak抑
制剂,其特征在于,所述jak抑制剂包含如下的式i化合物,或其立体异构体、几何异构体、互变异构体、水合物、溶剂化物、以及药学上可接受的盐作为活性成分:
9.[式i]
[0010][0011]
其中,
[0012]
r1为c或n;
[0013]
r2选自其中r4选自卤素或氰基;
[0014]
r3选自未取代的、或任选地被卤素、c1‑
c3烷基、羟基、c1‑
c3羟烷基、氨基、酰胺基和c1‑
c3烷基酰胺基中的至少一种取代的五元或六元芳基或杂芳基。
[0015]
在本发明的一个实施方式中,所述r4选自氟或氰基。
[0016]
在本发明的一个实施方式中,所述五元或六元芳基或杂芳基选自苯基、吡啶基、嘧啶基、吡唑基、吡咯基、咪唑基、吡嗪基或哒嗪基。
[0017]
在本发明的一个实施方式中,所述c1‑
c3烷基为甲基、乙基或丙基;所述 c1‑
c3羟烷基为羟甲基、羟乙基或羟丙基;所述c1‑
c3烷基酰胺基为甲基甲酰胺基、二甲基甲酰胺基、乙基甲酰胺基或甲基乙酰胺基。
[0018]
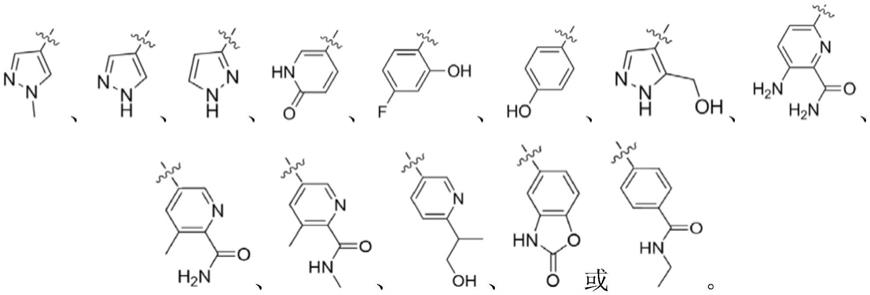
在本发明的一个实施方式中,所述r3选自以下基团中的任一种:
[0019][0020]
在另一方面,本发明还提供了如上所述的式(i)化合物,或其立体异构体、几何异构体、互变异构体、水合物、溶剂化物、以及药学上可接受的盐在制备用于预防或治疗自身免疫性疾病、以及相关的炎症性皮肤病的药物中的用途。
[0021]
在本发明的一个实施方式中,所述自身免疫性疾病选自类风湿性关节炎、炎症性肠病、系统性红斑狼疮、干燥综合征、皮肌炎、强直性脊柱炎、多发性硬化、白塞氏病、covid
‑
19重症肺炎、reiter综合征和葡萄膜炎中的至少一种。
[0022]
在本发明的一个实施方式中,所述相关的炎症性皮肤疾病选自银屑病、自身免疫相关血管炎、硬皮病、皮肌炎、肠病性肢端皮炎、化脓性汗腺炎、扁平苔藓、白癜风、皮肤型红斑狼疮和硬化萎缩性苔藓中的至少一种。
[0023]
在本发明的一个实施方式中,所述药物为口服施用的药物。
[0024]
在本发明的一个实施方式中,所述口服施用的药物为片剂、丸剂、颗粒剂、胶囊剂、锭剂或液体剂。
[0025]
本发明的作用和效果:
[0026]
本发明提供的具有高口服生物利用度的jak抑制剂能够克服现有的jak抑制剂口服生物利用度低的障碍,通过进一步化合物修饰以改变一类或几类小分子化合物的物理化学特性,从而提高该类化合物的体内细胞吸收性能,极大提高药物的生物利用度,也对该类化合物药物的给药方式及使用提供了新的可能。
[0027]
具体地,本发明的jak抑制剂通过增加可水解代谢的基团以降低化合物的极性,提高水溶性,以其通过延迟原药的半衰期,延缓原药的清除,提高原药的血药浓度来显著提升原药的生物利用度,将一类高效jak抑制剂由无法口服施用改造成可以口服施用,拓展药物使用途径,从而相应地拓展药物的治疗适应症。
具体实施方式
[0028]
以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
[0029]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
[0030]
在详细描述本发明前,应了解,在此使用的术语只在于描述特定的实施方式,而不希望限制本发明的范围,本发明的范围仅由所附权利要求书限定。为了更完全地了解在此描述的本发明,采用以下术语,它们的定义如下所示。除非另外定义,在此使用的所有技术和科学术语具有与本发明所属领域的普通技术人员所理解的相同的含义。
[0031]
在一方面,本发明提供了一种具有高口服生物利用度的jak抑制剂,其特征在于,所述jak抑制剂包含如下的式i化合物,或其立体异构体、几何异构体、互变异构体、水合物、溶剂化物、以及药学上可接受的盐作为活性成分:
[0032]
[式i]
[0033][0034]
其中,
[0035]
r1为c或n;
[0036]
r2选自其中r4选自卤素(例如氟、氯或溴,特别是氟)或氰基;
[0037]
r3选自未取代的、或任选地被卤素、c1‑
c3烷基、羟基、c1‑
c3羟烷基、氨基、酰胺基和c1‑
c3烷基酰胺基中的至少一种取代的五元或六元芳基或杂芳基。
[0038]
在本发明中,所述未取代的、或被取代基取代的五元或六元芳基或杂芳基可以本领域熟知的各种五元或六元芳基或杂芳基。另外,对于所述杂芳基,其中的杂原子可以如本领域技术人员所熟知地为n、o或s等杂原子,优选为n 原子,并且其数量可以为例如1、2或3个。在本发明的一个优选实施方式中,所述五元或六元芳基或杂芳基可以选自例如苯基、吡啶基、嘧啶基、吡唑基、吡咯基、咪唑基、吡嗪基或哒嗪基,但不限于此。
[0039]
对于上述取代基,所述c1‑
c3烷基可以为例如甲基、乙基或丙基;所述c1‑
c3羟烷基可以为例如羟甲基、羟乙基或羟丙基;所述c1‑
c3烷基酰胺基可以为例如甲基甲酰胺基、二甲基甲酰胺基、乙基甲酰胺基或甲基乙酰胺基,但不限于此。
[0040]
进一步地,在本发明的一个更优选实施方式中,所述r3可以选自以下基团中的任一种:
[0041][0042]
如本文所用,术语“药学上可接受的”是指不影响本发明化合物的生物活性或性质的物质,并且相对无毒,即该物质可施用于个体而不造成不良的生物反应或以不良方式与组合物中包含的任意组分相互作用。在本发明中,“药学上可接受的盐”可以包括无机盐和有机盐,其中,所述有机盐可以包括但不限于铵、锂、钠、钾、铯、钙、镁、铜、铝、锌、钡或季铵盐,并且所述无机盐可以包括但不限于精氨酸、叔丁胺、二甲胺、二乙醇胺、乙醇胺、乙二胺、咪唑、赖氨酸、甲胺、吡啶、吡啶甲酸酯、哌嗪、三乙胺、三乙醇胺、三甲胺或脲盐。
[0043]
在另一方面,本发明还提供了上述式(i)化合物,或其立体异构体、几何异构体、互变异构体、水合物、溶剂化物、以及药学上可接受的盐在制备用于预防或治疗自身免疫性疾病、以及相关的炎症性皮肤病的药物中的用途。
[0044]
如本文所用,术语“治疗”是指根据治疗性方案的治疗性试剂的任何施用,所述治疗性方案达到所需效果,即部分或完全减轻、改善、缓解、抑制、延迟发作、降低严重程度和/
或降低特定疾病、障碍和/或病症的一种或多种症状或特征的发生率;在一些实施方式中,根据治疗性方案的治疗性试剂的施用与所需效果的实现相关。这种治疗可以针对没有表现出相关疾病、障碍和/或病症的受试者和/或针对仅表现出疾病、障碍和/或病症的早期迹象的受试者。替代地或另外地,这种治疗可以针对表现出相关疾病、障碍和/或病症的一种或多种所确定迹象的受试者。在一些实施方式中,治疗可以针对已被诊断患有相关疾病、障碍和/或病症的受试者。在一些实施方式中,治疗可以针对已知具有一种或多种易感因素的受试者,所述易感因素在统计学上与相关疾病、障碍和/或病症发展的风险增加相关。
[0045]
根据本发明,上述用途中制得的药物可以包含有效量的本发明的式(i)化合物,以及药学上可接受的赋形剂、载体或稀释剂。
[0046]
如本文所用,术语“有效量”、“治疗有效量”或“药学有效量”是指对于治疗的受试者以适用于任何药物治疗的合理受益/风险比赋予治疗效果的治疗性试剂的量。这样的治疗效果可以是客观的(即可以通过某种测试或标记测量) 或主观的(即受试者给出指示或感觉到效果)。在一些实施方式中,“治疗有效量”是指诸如通过改善与疾病有关的症状、预防或延迟疾病发作和/或还减轻疾病症状的严重性或频率来有效治疗、改善或预防(例如延迟发作)相关疾病或病症和/或表现出可检测的治疗或预防效果的治疗性试剂或组合物的量。
[0047]
本领域的技术人员将认识到,待施用的所述式(i)化合物的治疗有效量将根据以下各项而变化:受试者和疾病的性质和严重程度、受试者的身体状况、治疗方案(例如是否使用第二治疗剂)、以及所选择的施用途径;合适的剂量可以由本领域的技术人员容易地确定。另外,该药物的个体剂量的最佳数量和间隔将通过所治疗的病状的性质和程度、施用的形式、途径和位置、以及所治疗的特定受试者的年龄和病状确定,并且医师将最终确定待施用的合适剂量。此剂量可以视需要重复多次。如果出现副作用,则可以根据正常临床实践改变或减少剂量的量和/或频率。
[0048]
在本发明中,“药学上可接受的赋形剂、载体或稀释剂”包括但不限于任何被相关的政府管理部门许可为可接受供人类或家畜使用的佐剂、载体、赋形剂、助流剂、增甜剂、稀释剂、防腐剂、染料/着色剂、矫味剂、表面活性剂、润湿剂、分散剂、助悬剂、稳定剂、等渗剂、溶剂或乳化剂等。
[0049]
根据本发明,进一步地,上述用途中制得的药物除了可以包含本发明的式 (i)化合物作为有效成分之外,还可以包含其他可用于预防或治疗自身免疫性疾病、以及与免疫有关的炎症性皮肤疾病的药剂作为另一种有效成分。所述药剂的实例包括但不限于维生素d衍生物、维生素a衍生物、糖皮质激素、钙调神经磷酸酶抑制剂或非甾体类抗炎药等。当该药物包含多种有效成分时,各有效成分可以根据医师的判断同时、依次或分开施用。
[0050]
另外,本发明的式(i)化合物可以通过多种途径施用于患者,这些途径诸如口服、透皮、皮下、鼻内、静脉内、肌内、鞘内、区域或局部(例如粘膜)。在任何给定情况下最适合的施用途径将取决于受试者和疾病的性质和严重程度、以及受试者的身体状况等。在本发明的一个实施方式中,本发明的式(i) 化合物制得的药物可以经口服施用。在此情况下,所述口服施用的药物可以为片剂、丸剂、颗粒剂、胶囊剂、锭剂或液体剂。
[0051]
经发明人研究发现,本发明的式(i)化合物或其制得的药物在用于预防或治疗jak相关的自身免疫性疾病、以及相关的炎症性皮肤病时(特别是即便以口服方式施用时)能够
发挥优异的效果。具体地,所述自身免疫性疾病选自类风湿性关节炎、炎症性肠病、系统性红斑狼疮、干燥综合征、皮肌炎、强直性脊柱炎、多发性硬化、白塞氏病、covid
‑
19重症肺炎、reiter综合征和葡萄膜炎等;而所述相关的炎症性皮肤疾病选自银屑病、自身免疫相关血管炎、硬皮病、皮肌炎、肠病性肢端皮炎、化脓性汗腺炎、扁平苔藓、白癜风、皮肤型红斑狼疮和硬化萎缩性苔藓等。
[0052]
以下,将通过实施例对本发明的特定化合物的效果进行详细描述。
[0053]
实施例
[0054]
实施例1:合成化合物1的一般方法(tdm
‑
181055)
[0055][0056]
步骤1:化合物1(tdm
‑
181055)的制备((s)
‑
(2,2
‑
二氟
‑
n
‑
(5
‑
(2
‑
(((1
‑
甲基
ꢀ‑
1h
‑
吡唑
‑4‑
基)氨基)嘧啶
‑4‑
基)吡啶
‑2‑
基)环丙烷
‑1‑
羧酰胺基)新戊酸甲酯)
[0057]
向化合物1a(500mg,1.346mmol)的n,n
‑
二甲基甲酰胺(15ml)溶液中加入碳酸钾(372mg,2.692mmol)和化合物1b(608mg,4.039mmol),将混合物用氩气吹扫几次并加热至40℃,搅拌过夜。然后将混合物在减压下浓缩,向残余物中加入水,并通过过滤收集固体,将固体通过硅胶色谱法(二氯甲烷: 10%甲醇的二氯甲烷=0
‑
30%)纯化得到黄色固体产物(tdm
‑
181055,化合物 1,19.8mg,收率3%)。lcms[m 1]
=486.2。
[0058]1h nmr(400mhz,dmso)δ9.60(s,1h),9.24(d,j=2.1hz,1h),8.63(d,j =7.3hz,1h),8.55(d,j=5.1hz,1h),7.93(s,1h),7.63(d,j=8.5hz,1h),7.56(s, 1h),7.41(d,j=5.2hz,1h),6.03
–
5.92(m,2h),3.83(s,3h),3.04(s,1h),2.17
–ꢀ
1.88(m,2h),1.10(s,9h)。
[0059]
实施例2:合成化合物2的一般方法(tdm
‑
181065)
[0060][0061]
步骤1:化合物2(tdm
‑
181065)的制备(((1
‑
(3
‑
氰基苯基)
‑
n
‑
(4
‑
(2
‑
(((1
‑ꢀ
甲基
‑
1h
‑
吡唑
‑4‑
基)氨基)嘧啶
‑4‑
基)苯基)甲基)磺酰胺基)新戊酸甲酯)
[0062]
向化合物2a(150mg,0.337mmol)的n,n
‑
二甲基甲酰胺(10ml)溶液中加入碳酸铯(219.4mg,0.673mmol),反应液升温至50℃搅拌1小时。自然冷却至室温,然后加入化合物2b(122.2mg,0.505mmol),反应液在室温下搅拌10 分钟,lcms[m h]
=560,检测反应完全。后处理:将反应液倒入70ml水中,水相用乙酸乙酯ea(3*150ml)萃取三次,合并有机相,并用饱
和食盐水洗,无水硫酸钠干燥,抽滤,滤液拉干,得到的粗品过柱纯化[洗脱剂:(d/m=10:1) /dcm=0
‑
15%],得到黄色固体目标化合物(tdm
‑
181065,化合物2,60mg,收率31.8%)。lcms[m 1]
=560.2。
[0063]1h nmr(400mhz,dmso)δ9.54(s,1h),8.51(d,j=5.1hz,1h),8.18(d,j =8.6hz,2h),7.94
–
7.86(m,3h),7.79(d,j=8.1hz,1h),7.63(t,j=7.8hz,1h), 7.58(s,1h),7.50(d,j=8.3hz,2h),7.31(d,j=5.2hz,1h),5.63(s,2h),4.86(s, 2h),3.84(s,3h),1.17(s,9h)。
[0064]
实施例3:合成化合物3的一般方法(tdm
‑
181058)
[0065][0066]
以与实施例2类似的方法得到黄色固体化合物(tdm
‑
181058,化合物3, 90.5mg,收率41.13%)。lcms[m 1]
=553.3。
[0067]1h nmr(400mhz,dmso)δ9.53(s,1h),8.51(d,j=5.1hz,1h),8.18(d,j =8.6hz,2h),7.91(s,1h),7.57(s,1h),7.53
–
7.41(m,3h),7.27(ddd,j=17.1, 11.2,3.5hz,4h),5.60(s,2h),4.79(s,2h),3.83(s,3h),1.17(s,9h)。
[0068]
实施例4:合成化合物4的一般方法(tdm
‑
181059)
[0069][0070]
以与实施例2类似的方法得到类白色固体(tdm
‑
181059,化合物4,67mg,收率23%)。lcms[m 1]
=566.3。
[0071]1h nmr(400mhz,dmso)δ9.81(s,1h),8.62(d,j=5.2hz,1h),8.32(d,j =7.4hz,2h),8.09(t,j=5.6hz,1h),7.71(d,j=6.6hz,2h),7.51(d,j=5.2hz, 3h),7.32(d,j=9.0hz,1h),5.74(d,j=10.4hz,2h),3.28
–
3.18(m,2h),2.55
–ꢀ
2.52(m,1h),2.37(s,3h),2.03(dt,j=13.3,6.6hz,1h),1.87(s,1h),1.19
–
1.06 (m,12h)。
[0072]
实施例5:合成化合物5的一般方法(tdm
‑
181060)
[0073][0074]
以与实施例2类似的方法得到类白色固体(tdm
‑
181060,化合物5,89.4mg,收率22%)。lcms[m 1]
=552.2。
[0075]1h nmr(400mhz,dmso)δ10.00(s,1h),8.65(d,j=5.2hz,1h),8.41
–
8.24(m,3h),7.92(d,j=8.9hz,2h),7.83(d,j=8.9hz,2h),7.59
–
7.37(m,3h), 5.75(d,j=10.4hz,2h),3.31
–
3.24(m,2h),2.52(d,j=1.9hz,1h),2.13
–
1.78 (m,2h),1.19
–
1.06(m,12h)。
[0076]
对比例1:合成化合物6(tdm
‑
180935)的一般方法
[0077][0078]
步骤1:化合物6(tdm
‑
180935)的制备
[0079]
在室温下向化合物6a(80mg,0.299mmol)即4
‑
(6
‑
氨基吡啶
‑3‑
基)
‑
n
‑
(1
‑
甲基
ꢀ‑
1h
‑
吡唑
‑4‑
基)嘧啶
‑2‑
胺的n,n
‑
二甲基甲酰胺(5ml)溶液中加入n,n
‑
二异丙基乙胺(72.9mg,0.564mmol),将混合物搅拌5分钟,然后加入2
‑
(7
‑
氧化苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸盐(170mg,0.449mmol)和化合物6b(54mg, 0.449mmol)即(s)
‑
2,2
‑
二氟环丙烷
‑1‑
羧酸。将混合物加热到90℃并搅拌16小时。减压浓缩混合物以除去一些溶剂,向剩余物中加入水,然后将溶液用乙酸乙酯 (60ml*3)萃取。有机层用饱和盐水(50ml*5)洗涤,并用无水硫酸钠干燥。减压浓缩滤液,残余物通过甲酸制备纯化,得到黄色固体化合物,即(s)
‑
2,2
‑
二氟
‑
n
‑
(5
‑
(2
‑
((1
‑
甲基
‑
1h
‑
吡唑
‑4‑
基)氨基)嘧啶
‑4‑
基)吡啶
‑2‑
基)环丙烷
‑1‑
甲酰胺 (化合物6,21.6mg,收率19.5%)。lcms[m 1]
=372.1。
[0080]1h nmr(400mhz,dmso
‑
d6)δ11.29(s,1h),9.54(s,1h),9.11(d,j=2.0hz, 1h),8.51(dd,j=14.3,6.9hz,2h),8.22(d,j=8.7hz,1h),7.93(s,1h),7.54(s, 1h),7.34(d,j=5.2hz,1h),3.83(s,3h),3.04(dd,j=22.4,9.9hz,1h),2.05(dt,j =11.8,8.0hz,2h)。
[0081]
对比例2:合成化合物7的一般方法(tdm
‑
180958)
[0082][0083]
步骤1:化合物7c(3
‑
氰基苄基氨基硫代氨基甲酸酯)的制备
[0084]
向化合物7a(1.78g,9.08mmol)的乙醇(13ml)溶液中加入化合物7b (690mg,9.08mmol),反应液升温至80℃搅拌1小时,lcms[m h]
=192,检测反应完全。后处理:反应液浓缩至干得到白色目标化合物(化合物3c,1.7g,产率97.7%),lcms[m 1]
=192。
[0085]
步骤2:化合物7d((3
‑
氰基苯基)甲磺酰氯)的制备
[0086]
向n
‑
氯代丁二酰亚胺(4.85g,36.32mmol)的乙腈(20ml)溶液中加入2n 的盐酸溶液(2.5ml)和化合物7c(1.74g,9.08mmol),反应液室温搅拌30分钟。后处理:反应完成后,将乙腈浓缩除去,加入水(15ml),有白色固体析出,过滤,固体用油泵拉干得到白色目标化合物(化合物7d,1.776g,收率90.7%)。
[0087]
步骤3:化合物7(tdm
‑
180958)的制备
[0088]
向化合物7e(500mg,1.878mmol)的无水吡啶(20ml)溶液中加入化合物7d(1g,4.64mmol),反应液升温至80℃反应30分钟,lcms[m h]
=446,检测反应完全。后处理:反应液浓缩至干,粗品过柱[洗脱剂:(d:m=10:1)/dcm= 0~50%],粗品用dcm/meoh=30/1打浆得到黄色固体,再通过制备型hplc纯化得到黄色目标化合物,即1
‑
(3
‑
氰基苯基)
‑
n
‑
(4
‑
(2
‑
(((1
‑
甲基
‑
1h
‑
吡唑
‑4‑
基)氨基) 嘧啶
‑4‑
基)苯基)甲磺酰胺(化合物7,155.4mg,收率18.6%)。
[0089]1h nmr(400mhz,dmso)δ10.27(s,1h),9.46(s,1h),8.46(d,j=5.2hz, 1h),8.12(d,j=8.7hz,2h),7.93(s,1h),7.84(dt,j=7.2,1.6hz,1h),7.71(s,1h), 7.61(ddd,j=17.3,10.8,4.8hz,3h),7.32(d,j=8.6hz,2h),7.24(d,j=5.2hz, 1h),4.71(s,2h),3.85(s,3h)。lcms[m h]
=446。
[0090]
对比例3:合成化合物8的一般方法(tdm
‑
180945)
[0091][0092]
步骤1:化合物8(tdm
‑
180945)的制备
[0093]
在室温下向化合物8a(60mg,0.225mmol)的吡啶(5ml)溶液中加入化合物8b(65.8mg,0.315mmol),将混合物加热至70℃并搅拌6h。然后将混合物在减压下浓缩,残余物通过硅胶色谱(二氯甲烷:含10%甲醇的二氯甲烷=70: 30)和甲酸制备纯化,得到黄色目标固体化合物8,tdm
‑
180945,即1
‑
(3
‑
氟苯基)
‑
n
‑
(4
‑
(2
‑
(((1
‑
甲基
‑
1h
‑
吡唑
‑4‑
基)氨基)嘧啶
‑4‑
基)苯基)甲磺酰胺(26.1mg,收率19.8%)。
[0094]1h nmr(400mhz,dmso
‑
d6)δ10.25(s,1h),9.45(s,1h),8.45(d,j=5.2hz, 1h),8.12(d,j=8.6hz,2h),7.93(s,1h),7.56(s,1h),7.40(dd,j=14.3,7.6hz, 1h),7.32(d,j=8.6hz,2h),7.26
–
7.15(m,2h),7.11(d,j=7.3hz,2h),4.63(s, 2h),3.84(s,3h)。lcms[m 1]
=439.2。
[0095]
对比例4:合成化合物9的一般方法(tdm
‑
180977)
[0096][0097]
步骤1:化合物9b(4
‑
氨基
‑
n
‑
乙基
‑2‑
甲基苯甲酰胺)的制备
[0098]
向化合物9a(1.8g,11.91mmol)的n,n
‑
二甲基甲酰胺(80ml)溶液中加入2
‑
(7
‑
氮杂苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸酯(5.4g,14.289mmol) 和n,n
‑
二异丙基乙胺(3.8g,29.775mmol),将混合物搅拌5分钟,然后加入乙胺的四氢呋喃(2m)(9ml,18mmol)溶液,将混合物在室温搅拌过夜。减压浓缩混合物除去一些溶剂,向残余物中加入水并用乙酸乙酯(100ml*3)萃取,合并有机相,用水(150ml*3)和饱和盐水(150ml)洗涤,用硫酸钠干燥,减压浓缩滤液,通过硅胶色谱法纯化(石油醚/乙酸乙酯=0%
‑
50%),以得到黄色油状目标化合物(化合物9b,1.32g,收率62.2%)。lcms[m 1]
=179。
[0099]
步骤2:化合物9((s)
‑4‑
((4
‑
(4
‑
(4
‑
(2,2
‑
二氟环丙烷
‑1‑
羧酰胺基)苯基)嘧啶
‑2‑ꢀ
基)氨基)
‑
n
‑
乙基
‑2‑
甲基苯甲酰胺)的制备
[0100]
向化合物9c(80mg,0.258mmol)的正丁醇(8ml)溶液加入化合物9b(92mg, 0.517mmol)和对甲苯磺酸一水合物(98mg,0.517mmol)。将所得混合物加热至110℃并搅拌3小时。反应结束将混合物在减压下浓缩,残余物通过制备型 hplc(甲酸)纯化,以得到白色固体目标化合物tdm
‑
180977(化合物9,19.4mg,收率13.3%。lcms[m h]
=425.2。
[0101]1h nmr(400mhz,dmso)δ10.70(s,1h),9.72(s,1h),8.54(d,j=5.3hz, 1h),8.18(d,j=8.8hz,2h),8.08(d,j=5.6hz,1h),7.81
–
7.67(m,4h),7.40(d,j =5.3hz,1h),7.32(d,j=8.4hz,1h),3.28
–
3.17(m,2h),2.86(ddd,j=13.6,10.8, 8.1hz,1h),2.37(s,3h),2.14
–
1.93(m,2h),1.11(t,j=7.2hz,3h)。
[0102]
对比例5:合成化合物10的一般方法(tdm
‑
180972)
[0103][0104]
步骤1:化合物10c(4
‑
(2
‑
氯嘧啶
‑4‑
基)苯胺)的制备
[0105]
向三口烧瓶加入化合物10a(2g,9.129mmol),化合物10b(1.36g, 9.129mmol),四三苯基膦钯(527g,0.45mmol),碳酸钾(2.5g,18.258mmol),二氧六环(20ml)和水(20ml),用氮气置换数次,然后将混合物加热至80℃并搅拌45分钟。反应结束减压浓缩反应物,残余物
通过硅胶色谱法进行纯化(石油醚/乙酸乙酯=0
‑
60%),以得到淡黄色固体目标化合物(化合物10c,394mg,收率21%)。lcms[m 1]
=206。
[0106]
步骤2:化合物10e((s)
‑
n
‑
(4
‑
(2
‑
氯嘧啶
‑4‑
基)苯基)
‑
2,2
‑
二氟环丙烷
‑1‑
羧酰胺)的制备
[0107]
向三口烧瓶中加入化合物10c(300mg,1.459mmol)和化合物10d(187mg, 1.531mmol),将混合物用氮气置换几次,然后在0℃下加入吡啶(10ml)和三氯氧磷(335.6mg,2.189mmol)。将混合物在室温搅拌1h,然后减压浓缩,残余物用乙酸乙酯(30ml*3)萃取,合并有机层,并用水(50ml*3)和饱和盐水 (50ml*2)洗涤,用硫酸钠干燥,将滤液减压浓缩,残余物通过硅胶色谱法进行纯化(石油醚/乙酸乙酯=0
‑
12%),以得到黄色固体目标化合物(化合物10e, 327.7mg,收率72.5%)。lcms[m h]
=310。
[0108]
步骤3:化合物1((s)
‑4‑
((4
‑
(4
‑
(4
‑
(2,2
‑
二氟环丙烷
‑1‑
羧酰胺基)苯基)嘧啶
‑2‑ꢀ
基)氨基)
‑
n
‑
乙基苯甲酰胺)的制备
[0109]
向化合物10e(80mg,0.258mmol)的正丁醇(8ml)溶液加入化合物1f(85mg, 0.517mmol)和对甲苯磺酸一水合物(98mg,0.517mmol)。将所得混合物加热至110℃并搅拌3小时。反应结束将混合物在减压下浓缩,残余物通过制备型hplc(甲酸)纯化,以得到黄色固体目标化合物tdm
‑
180972(化合物10,19.7mg,收率17.5%)。lcms[m h]
=438.2。
[0110]1h nmr(400mhz,dmso)δ10.70(s,1h),9.91(s,1h),8.57(d,j=5.3hz, 1h),8.28(t,j=5.5hz,1h),8.19(d,j=8.8hz,2h),7.92(d,j=8.9hz,2h),7.83 (d,j=8.9hz,2h),7.78(d,j=8.8hz,2h),7.44(d,j=5.3hz,1h),3.28(dt,j= 12.7,6.4hz,2h),2.86(ddd,j=13.6,10.8,8.0hz,1h),2.12
–
1.94(m,2h),1.13 (t,j=7.2hz,3h)。
[0111]
大鼠口服pk生物学描述:
[0112]
为验证本发明实施例的化合物1
‑
5是否比与其具有类似结构的对比例的化合物6
‑
10具有显著提高的口服生物利用度,将合成的化合物1
‑
10在6
‑
8周左右的sprague dawley(sd)大鼠(体重大约250
‑
300克)体内进行了单次口服给药的药代动力学(pk)的测试,以便将化合物1
‑
10的各项pk参数进行对比。简言之,化合物在溶媒[dma:30%solutol hs 15:saline=10:10:80(v/v/v)]中配制成透明溶液制剂,浓度为1mg/ml,每个测试化合物使用3只大鼠(n=3)进行单次口腔灌胃给药(5mg/kg)。给药前以及给药后0.25,0.5,1,2,4,8,12,24小时分别从每只实验大鼠经锁骨下静脉取大约150μl的全血,收集到含有 edta
‑
k2的eppendorf管中,随后取100μl全血加入含有300μl的氰基甲烷(acn)的eppendorf管中,混合后离心分离血浆,确保样品没有溶血后将血浆样品在零下90到零下60度冰箱保存,直到通过lc
‑
ms/ms对样品中的药物浓度进行分析。所有口服pk试验均未发现大鼠对任何测试化合物产生不良反应。
[0113]
血浆样品中的化合物浓度通过lc
‑
ms/ms系统(api4000:lc
‑
ms
‑
ms
‑
001) 进行检测分析,取10μl的样品和100μl含有标准品的acn进行混合,随后加入110μl的水并进行震荡混合,最后取2μl的混合溶液注射进lc
‑
ms/ms系统。
[0114]
测试结果如下所示:
[0115]
1)对比例1化合物:c
max
=116ng/ml,auc0‑
t
=891ng
·
h/ml,进一步合成为实施例1化合物后,c
max
=668ng/ml,auc0‑
t
=1670ng
·
h/ml。
[0116]
2)对比例2化合物:c
max
=20.2ng/ml,auc0‑
t
=47.4ng
·
h/ml,进一步合成为实施例
2化合物后,c
max
=791ng/ml,auc0‑
t
=1580ng
·
h/ml。
[0117]
3)对比例3化合物:c
max
=85.5ng/ml,auc0‑
t
=381ng
·
h/ml,进一步合成为实施例3化合物后,c
max
=1860ng/ml,auc0‑
t
=2890ng
·
h/ml。
[0118]
4)对比例4化合物:c
max
=5.02ng/ml,auc0‑
t
=30.7ng
·
h/ml,进一步合成为实施例4化合物后,c
max
=635ng/ml,auc0‑
t
=3090ng
·
h/ml。
[0119]
5)对比例5化合物:c
max
=16.5ng/ml,auc0‑
t
=141ng
·
h/ml,进一步合成为实施例5化合物后,c
max
=689ng/ml,auc0‑
t
=2710ng
·
h/ml。
[0120]
根据大鼠口服pk生物学数据可以看出,在进一步合成后,化合物的系统暴露得到了极大提升,表现为血浆药物浓度峰值比原药提高5
‑
120倍,auc提高 2
‑
100倍,证明了本发明实施例的化合物的口服生物利用度明显提高,同时体内清除速率则明显降低。
[0121]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
[0122]
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
[0123]
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。

