中性粒细胞弹性蛋白酶抑制剂在肝病中的用途
1.相关申请的交叉引用
2.本技术是非临时申请,根据美国法典第35篇第119条(e)款要求2018年4月24日提交的临时申请序列号62/662,074的优先权,其内容以引用的方式整体并入本文。
技术领域
3.本发明涉及使用中性粒细胞弹性蛋白酶抑制剂治疗慢性肝病、特别是非酒精性脂肪肝病(nafld)和非酒精性脂肪性肝炎(nash)的方法。本发明还涉及包含中性粒细胞弹性蛋白酶抑制剂的药物组合物。
背景技术:
4.非酒精性脂肪肝病(nafld)是指从肝细胞脂肪变性经由非酒精性脂肪性肝炎(nash)到纤维化以及不可逆的肝硬化的一组越来越严重的肝脏病症。同肥胖和糖尿病流行一样,nafld/nash现在是西方世界慢性肝病的最常见原因,并且有可能在未来一至二十年内成为肝移植的主要原因(staels,b.等,hepatology,2013,58:1941
‑
1952;charlton,m.r.等,gastroenterology,2011,141:1249
‑
1253)。胰岛素抵抗和由此产生的肝脂质积累(脂肪变性)被广泛认为是nafld/nash的主要病理生理性伤害。导致肝细胞损伤(脂肪性肝炎)和纤维化的炎症被认为是重要的额外病理生理性伤害,其特征是疾病从nafld进展到nash并最终到肝硬化(chalasani,n.等,hepatology,2012,55(6):2005
‑
2023;anderson,n.和borlak,j.,pharmaological reviews,2008,60:311
‑
357)。
5.目前,还没有获得批准的用于nafld/nash的疗法。鉴于这些病状是在35年前首次描述的,并且全世界的研究人员一直认真研究这些疾病的病理生理学和诊断已超十五年,因此这种情况令人惊讶(ratzui,v.等,j.hepatology,2015,62:s65
‑
s75)。ratzui等推测,从历史上看,肝脂肪变性被认为是在糖尿病患者中观察到的良性病状,没有临床相关性,并且因此药物研究集中在抗糖尿病药物上(ratzui 2015)。现已认识到nafld/nash的更严重影响,并且鉴于这些疾病的发病率增加,存在对安全且有效的治疗的需求。
技术实现要素:
6.在一方面,本发明提供了一种治疗慢性肝病的方法,其包括向需要治疗的患者施用治疗有效量的(4s)
‑4‑
[4
‑
氰基
‑2‑
(甲基磺酰基)苯基]
‑
3,6
‑
二甲基
‑2‑
氧代
‑1‑
[3
‑
(三氟甲基)苯基]
‑
1,2,3,4
‑
四氢嘧啶
‑5‑
甲腈或其药学上可接受的盐、多晶型物、溶剂化物或盐的溶剂化物。在一个实施方案中,所述慢性肝病是非酒精性脂肪肝病(nafld)。在另一个实施方案中,所述慢性肝病是非酒精性脂肪性肝炎(nash)。在又一个实施方案中,治疗有效量包括每天一次1mg、2mg、5mg、10mg或20mg的剂量。在另一个实施方案中,治疗慢性肝病的方法还包括施用一种或多种额外的治疗剂。在一个实施方案中,所述额外的治疗剂是中性粒细胞弹性蛋白酶抑制剂。在又一个实施方案中,所述中性粒细胞弹性蛋白酶抑制剂是西利司他(silevestat)或阿韦司他(avelestat)。在另一个实施方案中,所述额外的治疗剂通过
除抑制中性粒细胞弹性蛋白酶以外的机制来治疗或改善nafld或nash。在又一个实施方案中,所述额外的治疗剂是gft505、塞拉德帕(seladelpar)、森昔洛韦(cenecriviroc)、gs
‑
0976、gs
‑
9674、塞隆替尼(selonsertib)或奥贝胆酸(obeticholic acid)。在另一个实施方案中,所述额外的治疗剂是抗糖尿病剂。在又一个实施方案中,所述抗糖尿病剂是吡格列酮(pioglitazone)、罗格列酮(rosiglitazone)、利拉鲁肽(liraglutide)、杜拉鲁肽(dulaglutide)、索马鲁肽(semaglutide)、卡格列净(canagliflozin)、恩格列净(empagliflozin)、鲁格列净(luseogliflozin)或伊格列净(ipragliflozin)。
[0007]
在另一方面,本发明提供了一种用于治疗慢性肝病的药物组合物,其包含(4s)
‑4‑
[4
‑
氰基
‑2‑
(甲基磺酰基)苯基]
‑
3,6
‑
二甲基
‑2‑
氧代
‑1‑
[3
‑
(三氟甲基)苯基]
‑
1,2,3,4
‑
四氢嘧啶
‑5‑
甲腈或其药学上可接受的盐、多晶型物、溶剂化物或盐的溶剂化物以及药学上可接受的载体。在一个实施方案中,所述药物组合物被配制成片剂。在另一个实施方案中,所述片剂包含(4s)
‑4‑
[4
‑
氰基
‑2‑
(甲基磺酰基)苯基]
‑
3,6
‑
二甲基
‑2‑
氧代
‑1‑
[3
‑
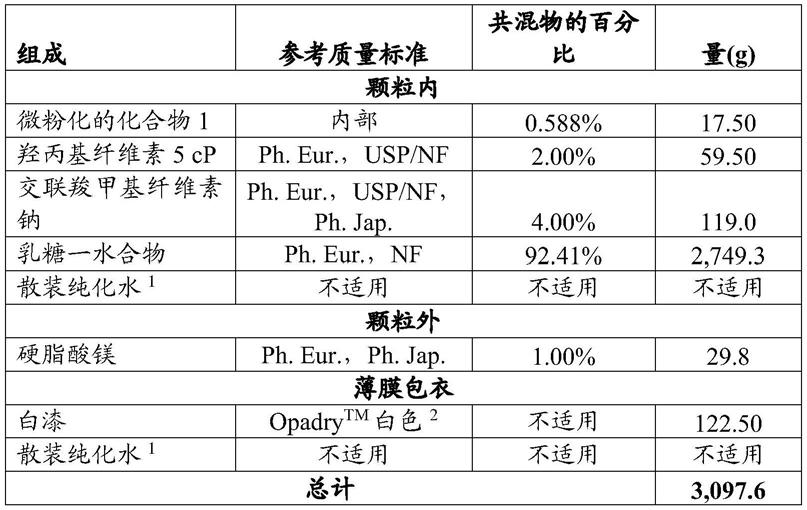
(三氟甲基)苯基]
‑
1,2,3,4
‑
四氢嘧啶
‑5‑
甲腈或其药学上可接受的盐、多晶型物、溶剂化物或盐的溶剂化物以及一种或多种稀释剂、崩解剂、表面活性剂或润滑剂。在又一个实施方案中,所述药物组合物包含1mg、2mg、5mg、10mg或20mg的(4s)
‑4‑
[4
‑
氰基
‑2‑
(甲基磺酰基)苯基]
‑
3,6
‑
二甲基
‑2‑
氧代
‑1‑
[3
‑
(三氟甲基)苯基]
‑
1,2,3,4
‑
四氢嘧啶
‑5‑
甲腈或其药学上可接受的盐、多晶型物、溶剂化物或盐的溶剂化物。
[0008]
在另一方面,本发明提供了一种在需要此类治疗的患者中治疗慢性肝病的方法,其包括向所述患者施用治疗有效量的药物组合物,所述药物组合物包含(4s)
‑4‑
[4
‑
氰基
‑2‑
(甲基磺酰基)苯基]
‑
3,6
‑
二甲基
‑2‑
氧代
‑1‑
[3
‑
(三氟甲基)苯基]
‑
1,2,3,4
‑
四氢嘧啶
‑5‑
甲腈或其药学上可接受的盐、多晶型物、溶剂化物或盐的溶剂化物以及药学上可接受的载体。在一个实施方案中,所述慢性肝病是nafld。在另一个实施方案中,所述慢性肝病是nash。在又一个实施方案中,所述药物组合物包含1mg、2mg、5mg、10mg或20mg的(4s)
‑4‑
[4
‑
氰基
‑2‑
(甲基磺酰基)苯基]
‑
3,6
‑
二甲基
‑2‑
氧代
‑1‑
[3
‑
(三氟甲基)苯基]
‑
1,2,3,4
‑
四氢嘧啶
‑5‑
甲腈或其药学上可接受的盐、多晶型物、溶剂化物或盐的溶剂化物。
[0009]
在阅读以下说明书和权利要求书之后,本发明的其他目的对于本领域技术人员而言可为显而易见的。
附图说明
[0010]
本发明的新型特征在所附权利要求书中具体阐述。参考以下阐述说明性实施方案的具体实施方式可获得对本发明的特征和优点更好的理解,其中利用了本发明的原理,并且其中附图为:
[0011]
图1示出了如美国专利号8,288,402(von nussbaum)所述的(4s)
‑4‑
[4
‑
氰基
‑2‑
(甲基磺酰基)苯基]
‑
3,6
‑
二甲基
‑2‑
氧代
‑1‑
[3
‑
(三氟甲基)苯基]
‑
1,2,3,4
‑
四氢嘧啶
‑5‑
甲腈的总合成。反应方案如下:从式(ii)的化合物经由式(iii)、(iv)和(v)的化合物到式(vi)的化合物的反应顺序在von nussbaum专利的方案6和实施例1a、实施例2a方法b和实施例3a方法b以及实施例4a方法b中;从式(vi)的化合物经由式(ix)的化合物到式(x)的化合物的反应顺序在von nussbaum专利的方案1和实施例3与实施例4中;并且从式(x)的化合物经由式(xi)和(xii)的化合物到式(xiii)的化合物的反应顺序在von nussbaum专利的方案
2和实施例5a、实施例5与实施例6中。式(i)的化合物(本文化合物1)的合成描述于von nussbaum专利的实施例33方法b中。
[0012]
图2示出了在使用罗格列酮、3mg/kg化合物1、15mg/kg化合物1或媒介物治疗的6周期间,dio(饮食诱导的肥胖)小鼠的体重变化:a)针对四个治疗组的治疗天数绘制的体重克数;b)四个治疗组的终末体重的图形描绘;c)针对四个治疗组的治疗天数绘制的体重(百分比);d)四个治疗组的体重增长率百分比的图形描绘。
[0013]
图3示出了在使用罗格列酮、3mg/kg化合物1、15mg/kg化合物1或媒介物治疗的6周期间,dio小鼠的葡萄糖处置曲线:a)在四个治疗组的治疗前一天(第
‑
1天)测定的葡萄糖激发口服剂量后的葡萄糖处置曲线;b)四个治疗组在治疗四周后(第28天)的葡萄糖处置曲线;c)四个治疗组在治疗六周后(第42天)的葡萄糖处置曲线;d)四个治疗组在治疗之前(第
‑
1天)和之后(第28天和第42天)的增量葡萄糖auc的图形描绘。
[0014]
图4示出了在使用罗格列酮、3mg/kg化合物1、15mg/kg化合物1或媒介物治疗的6周期间,dio小鼠的空腹血清胰岛素水平的变化:a)针对四个治疗组的治疗天数绘制的血清胰岛素(ng/ml);b)在研究结束时四个治疗组的空腹血清胰岛素水平的图形描绘。
[0015]
图5示出了在使用罗格列酮、3mg/kg化合物1、15mg/kg化合物1或媒介物治疗的6周期间,dio小鼠的终末肝脏重量和附睾脂肪重量的比较:a)四个治疗组的以体重百分比表示的终末肝脏重量的图形描绘;b)四个治疗组的以体重百分比表示的终末附睾脂肪重量的图形描绘。
[0016]
图6示出了在使用15mg/kg化合物1、30mg/kg化合物1或媒介物治疗的4周期间,dio小鼠的体重变化:a)三个治疗组的针对治疗天数绘制的体重克数;b)三个治疗组的针对治疗天数绘制的体重变化(百分比)。
[0017]
图7示出了在使用15mg/kg化合物1、30mg/kg化合物1或媒介物治疗的4周后,dio小鼠的口服葡萄糖耐量试验(ogtt)的葡萄糖处置曲线:a)三个治疗组在治疗四周后第28天的葡萄糖处置曲线;b)三个治疗组在治疗前第
‑
1天和治疗4周后第28天的葡萄糖auc的图形描绘。
[0018]
图8示出了在使用15mg/kg化合物1、30mg/kg化合物1或媒介物治疗的4周后第29天,dio小鼠的空腹血清胰岛素水平(ng/ml)。
具体实施方式
[0019]
本技术不限于所述的特定方法学或具体组合物,因为本技术的范围将仅由所附权利要求书及其等效物所限制。还应当理解,本文所用的术语仅出于描述特定实施方案的目的,而不旨在是限制性的。
[0020]
除非另外定义,否则本文中所用的所有技术和科学术语均具有与本技术所属领域的普通技术人员通常所理解的相同的含义。必须注意,除非上下文另外清楚地指出,否则如本文和所附权利要求书中所用,单数形式“一个”、“一种”以及“所述”包括复数个指示物。
[0021]
现在将详细参考某些优选的治疗方法、化合物和施用这些化合物的方法。本发明不限于那些优选的化合物和方法,而是由其发出的权利要求书所限定。
[0022]
简介
[0023]
本发明提供了一种使用化合物1,(4s)
‑4‑
[4
‑
氰基
‑2‑
(甲基磺酰基)苯基]
‑
3,6
‑
二
甲基
‑2‑
氧代
‑1‑
[3
‑
(三氟甲基)苯基]
‑
1,2,3,4
‑
四氢嘧啶
‑5‑
甲腈或其药学上可接受的盐、多晶型物、溶剂化物或盐的溶剂化物来治疗慢性肝病、特别是非酒精性脂肪肝病(nafld)和非酒精性脂肪性肝炎(nash)的方法。本发明还提供了适用于治疗nafld和nash的化合物1的药物组合物。
[0024]
先前已经公开了将化合物1作为强效的中性粒细胞弹性蛋白酶(ne)抑制剂(nagelschmitz,j.等,european respiratory j.,2014,44,增刊58,摘要号3416)。它对人中性粒细胞弹性蛋白酶(k
i
[m]=8.0x10
‑
11
)的选择性是对鼠中性粒细胞弹性蛋白酶(k
i
[m]=8.0
×
10
‑
11
)约100倍(von nussbaum,f.等,chemmedchem.,2015,10:1163
‑
1173)。人中性粒细胞弹性蛋白酶(hne,ne)是在炎症过程中由中性粒细胞分泌的一种非常活跃的丝氨酸蛋白酶。它也被称为人白细胞弹性蛋白酶(hle,ec 3.4.21.37)。这种蛋白水解酶经发现于多形核白细胞(pmn白细胞)的嗜天青颗粒中。细胞内的弹性蛋白酶通过分解经由吞噬作用吸收的外来颗粒,来在针对病原体的防御中起着重要的作用(nagelschmitz,2014)。高活性蛋白水解酶能够分解多种结缔组织蛋白,诸如蛋白弹性蛋白、胶原蛋白和纤连蛋白。弹性蛋白以高浓度出现在所有表现出高弹性的组织类型中,诸如在肺和动脉中。ne也是炎性过程的重要调节因子。hne活性过高与炎性肺病,像支气管扩张、copd和肺动脉高压的发病机制有关。
[0025]
在许多专利和申请中已经公开了将化合物1作为各种肺病的治疗和对慢性伤口的治疗(美国专利号8,288,402;美国专利号8,889,700;美国专利号9,174,997;pct公开wo 2017/081044,其公开内容以引用的方式并入本文)。
[0026]
化合物1(也称为bay 85
‑
8501)的安全性和耐受性已在若干人类临床试验中进行了评估。四项临床研究,包括健康受试者中的两项1期单剂量研究、健康受试者中的1期多剂量研究以及患有非囊性纤维化支气管扩张(ncf be)的受试者中的2a期多剂量研究,已经评估了化合物1作为口服溶液和/或速释(ir)片剂施用的安全性、药代动力学(pk)和药效学(pd)。在参与三项1期研究的健康受试者中,以高达1mg的剂量施用长达14天的单一和重复的化合物1治疗是安全且耐受性良好的。在1期研究中报告的不良事件(ae)总体较轻,与研究治疗无关,并且没有报告严重的ae(sae)。没有观察到关于研究药物诱导的实验室或ecg异常的安全信号。(参见:nagleschmitz,j.等,european respiratory j.,2014,44:3416;nagelschmitz,j.等,european respiratory j.,2014,44:p1511)。
[0027]
使用化合物1的28天口服施用,对患有非cf be的受试者进行了多中心、2a期、随机、双盲、安慰剂对照研究(www.clinicaltrials.gov;识别码:nct01818544)。将九十四位患者(平均年龄66岁,53%男性)随机化以接受治疗,其中45位患者接受1.0mg口服剂量的作为ir片剂施用的化合物1。所述药物在28天内总体安全且耐受性良好。接受化合物1和安慰剂的受试者的安全性结果总体类似。ae的严重程度总体为轻度或中度,与研究治疗无关,并且在研究药物与安慰剂之间没有差异。sae和由于ae而退出研究治疗的发生率很低,并且研究者没有将sae归因于所述药物。没有观察到关于研究药物诱导的实验室参数或ecg影响的安全信号。(参见:watz,h.等,european respiratory j.,2016,48:pa4088)。
[0028]
化学描述
[0029]
化合物1,(4s)
‑4‑
[4
‑
氰基
‑2‑
(甲基磺酰基)苯基]
‑
3,6
‑
二甲基
‑2‑
氧代
‑1‑
[3
‑
(三氟甲基)苯基]
‑
1,2,3,4
‑
四氢嘧啶
‑5‑
甲腈,具有以下化学结构:
[0030][0031]
可替代地,化合物1可以命名为(4s)
‑4‑
[4
‑
氰基
‑2‑
(甲基磺酰基)苯基]
‑
1,2,3,4
‑
四氢
‑
3,6
‑
二甲基
‑2‑
氧代
‑1‑
[3
‑
(三氟甲基)苯基]
‑5‑
嘧啶甲腈。化合物1在文献中通常称为bay 85
‑
8501。应当理解,化合物1的任何这些名称都可以互换使用并且具有相同的含义。
[0032]
化合物1及其盐、多晶型物、溶剂化物或盐的溶剂化物可以各种立体异构形式存在,即以构型异构体的形式,或者,如果适当的话,还作为构象异构体(对映体和/或非对映体,包括阻转异构体)。因此,化合物1也指对映体和非对映体以及它们各自的混合物。可以已知方式从对映体和/或非对映体的此类混合物中分离出立体异构纯组分。化合物1还涵盖任何可能的互变异构形式。
[0033]
化合物1可以多种物理形式存在,包括但不限于多种结晶形式、非结晶态无定形形式和多晶型物。一般来说,所有物理形式对于本文所预期的用途而言都是等同的并且旨在处于本公开的范围内。多晶型是指分子以两种或更多种结晶形式存在的能力,其中具有晶格的分子在结构排列和/或构象上可能不同。多晶型结构具有相同的化学组成,虽然它们的晶格结构和/或构象不同可导致不同的物理、化学或药理特性,诸如溶解度、稳定性、熔点、密度和生物利用度。无定形形式不具有确定的晶体结构。化合物1的所有多晶型物和其他物理形式对于本文所预期的用途而言都是等同的并且旨在处于本发明的范围内。
[0034]
出于本发明的目的所优选的盐是化合物1的生理上可接受的盐。还涵盖本身不适合用于药物用途但可以例如用于分离或纯化根据本发明的化合物的盐。盐可以多种物理形式存在,包括但不限于多种结晶形式、非结晶态无定形形式和多晶型物。
[0035]
化合物1的生理上可接受的盐包括无机酸、羧酸和磺酸的酸加成盐,例如盐酸、氢溴酸、硫酸、磷酸、甲磺酸、乙磺酸、甲苯磺酸、苯磺酸、萘二磺酸、乙酸、三氟乙酸、丙酸、乳酸、酒石酸、苹果酸、柠檬酸、甲酸、富马酸、马来酸和苯甲酸的盐。化合物1的生理学上可接受的盐还包括常规碱的盐,诸如,举例并优选地,碱金属盐(例如钠盐和钾盐)、碱土金属盐(例如钙盐和镁盐)和源自氨或具有1至16个碳原子的有机胺的铵盐,举例并优选地所述有机胺为诸如乙胺、二乙胺、三乙胺、乙基二异丙胺、单乙醇胺、二乙醇胺、三乙醇胺、二环己胺、二甲氨基乙醇、普鲁卡因、二苄胺、n
‑
甲基吗啉、精氨酸、赖氨酸、乙二胺和n
‑
甲基哌啶。
[0036]
出于本发明的目的,溶剂化物是指根据本发明的化合物1的那些形式,其以固态或液态通过与溶剂分子配位来形成络合物。溶剂化物可以多种物理形式存在,包括但不限于多种结晶形式、非结晶态无定形形式和多晶型物。溶剂化物也可以与化合物1的药学上可接受的盐一起形成。水合物是与水发生配位的特定形式的溶剂化物。各种有机溶剂可与化合物1形成溶剂化物,包括但不限于1,4
‑
二噁烷、1
‑
丙醇、1
‑
丁醇、1,2
‑
二甲氧基乙烷、2
‑
乙氧
基乙醇、2
‑
甲氧基乙醇、2
‑
甲基
‑1‑
丙醇、2
‑
甲基四氢呋喃、3
‑
甲基
‑1‑
丁醇、乙酸、丙酮、乙腈、苯甲醚、乙酸丁酯、氯苯、异丙苯、二甲基亚砜、乙醇、乙酸乙酯、乙醚、甲酸乙酯、乙二醇、甲酸、庚烷、乙酸异丁酯、异丙醚、乙酸异丙酯、甲醇、乙酸甲酯、甲基乙基酮、甲基异丁基酮、n
‑
甲基吡咯烷酮、叔丁醇、叔丁基甲基醚、四氢呋喃和甲苯。
[0037]
化学合成
[0038]
化合物1,(4s)
‑4‑
[4
‑
氰基
‑2‑
(甲基磺酰基)苯基]
‑
3,6
‑
二甲基
‑2‑
氧代
‑1‑
[3
‑
(三氟甲基)苯基]
‑
1,2,3,4
‑
四氢嘧啶
‑5‑
甲腈,可如von nussbaum等(美国专利号8,288,402)所述来制备,所述文献以引用的方式整体并入本文。可替代地,可以使用如美国公开申请号2018/0072685所述的schirmer等的方法,所述文献以引用的方式整体并入本文。
[0039]
von nussbaum等的方法描述于美国专利号8,288,402。从3
‑
氟
‑4‑
甲基苄腈开始,化合物1以10个步骤产生,其中总产率为理论的4.45%。图1详细示出合成中的中间步骤。最后一步是n
‑
甲基化,然后进行柱色谱法。通过将色谱级分浓缩成无定形固体来获得s
‑
对映体。合成的更多细节可见于von nussbaum等的专利的实施例33。
[0040]
schrimer等提供了如美国公开申请号2018/0072685中提供的方案所描述的化合物1的改进合成。改进的方法有两种变体,其中方法变体(a)以8个步骤(参见美国2018/0072685的方案7、2和3)来提供化合物1,获得超过理论的17%的总体产率,无需对中间体进行色谱纯化。方法变体(b)(参见美国2018/0072685的方案7、4、5和6)以9个步骤提供化合物1,同样无需对中间体进行色谱纯化,其中总体产率取决于反应管理,如美国2018/0072685中所详细描述。
[0041]
化合物1为白色至黄色固体,其中熔点为232℃。它被认为是中性的并且不易形成盐。化合物1在正常储存条件下不吸湿。化合物1几乎不溶于水,极微溶于乙醇,并且溶于丙酮。
[0042]
药物组合物
[0043]
包含(4s)
‑4‑
[4
‑
氰基
‑2‑
(甲基磺酰基)苯基]
‑
3,6
‑
二甲基
‑2‑
氧代
‑1‑
[3
‑
(三氟甲基)苯基]
‑
1,2,3,4
‑
四氢嘧啶
‑5‑
甲腈(化合物1)或其药学上可接受的盐、多晶型物、溶剂化物或盐的溶剂化物作为活性成分的组合物可以有利地用于治疗慢性肝病。虽然化合物1或其药学上可接受的盐、多晶型物、溶剂化物或盐的溶剂化物可以单独施用,但优选将其呈现为制剂。所述组合物或剂型可以单独施用或施加,或与其他试剂,包括一种或多种稀释剂、崩解剂、表面活性剂或润滑剂组合。所述制剂还可以将化合物1与另一种药物活性剂组合递送至患者。
[0044]
如本文所用,术语“组合物”旨在涵盖含有预定量或比例的指定成分的产物,以及由指定量的指定成分的组合直接或间接产生的任何产物。和药物组合物有关的此术语旨在涵盖包含一种或多种活性成分和含有惰性成分的任选的药学上可接受的载体的产物,以及由所述成分中任何两种或更多种成分的组合、络合或聚合,或由所述成分中一种或多种成分的解离,或由所述成分中一种或多种成分的其他类型反应或相互作用而直接或间接产生的任何产物。一般来说,通过使活性成分与液体载体或微细的固体载体或两者均匀且紧密地结合,并且然后在需要时使产物成型为所需制剂来制备药物组合物。在药物组合物中,活性目标化合物以足以对疾病的过程或病状产生所需作用的量而含入。因此,本发明的药物组合物涵盖通过混合本发明的化合物和药学上可接受的载体而制备的任何组合物。所述组
合物分别根据常规混合、制粒或包衣方法来制备,并且含有0.1%至75%、优选1%至50%的活性成分。
[0045]“药学上可接受的”意指载体、稀释剂或赋形剂必须与制剂的其他成分相容并且对于其受体无害。意图用于口服使用的药物组合物可根据本领域中已知用于制造药物组合物的任何方法来制备,并且此类组合物可含有一种或多种选自由甜味剂、调味剂、着色剂以及防腐剂组成的组的试剂,以便提供药学上优良且可口的制剂。
[0046]
片剂含有与无毒的药学上可接受的赋形剂混合的活性成分,所述赋形剂适用于制造片剂,包括但不限于稀释剂、崩解剂、表面活性剂和润滑剂。这些赋形剂可以是例如惰性稀释剂,诸如碳酸钙、碳酸钠、乳糖、磷酸钙或磷酸钠;成粒剂和崩解剂,例如玉米淀粉或海藻酸;粘合剂,例如淀粉、明胶或阿拉伯胶;以及润滑剂,例如硬脂酸镁、硬脂酸或滑石。片剂可以是未包衣的,或者它们可以通过已知技术包衣以延迟在胃肠道中的崩解和吸收,并从而在更长的时间内提供持久的作用。可以通过任选地与一种或多种药学上可接受的成分一起压制或模制活性成分来制备片剂。可以通过在适合的机器中压制任选地与粘合剂、润滑剂、惰性稀释剂、表面活性剂或分散剂混合的呈自由流动形式(诸如粉末或颗粒)的活性成分来制备压制片剂。可以通过在适合的机器中模制用惰性液体稀释剂润湿的粉状活性成分和适合的载体的混合物来制备模制片剂。片剂可以如以下实施例所述或如pct申请wo 2017/081044(may等)所述来制备,所述申请以引用的方式整体并入本文。
[0047]
用于口服使用的组合物还可呈现为硬明胶胶囊剂,其中活性成分与惰性固体稀释剂(例如碳酸钙、磷酸钙或高岭土)混合,或呈现为软明胶胶囊剂,其中活性成分与水或油介质(例如花生油、液体石蜡或橄榄油)混合。具体地说,本发明的药物组合物可包括液体填充的胶囊剂型,其中活性成分以液体和半固体赋形剂的某些组合存在于溶液中。
[0048]
用于口服施用的组合物也可配制为水性混悬液,其含有与适合制造水性混悬液的赋形剂混合的活性成分。可以通过将活性成分悬浮在适合的油中来配制油性混悬液。也可以使用水包油乳剂。适用于通过添加水来制备水性混悬液的可分散粉末和颗粒提供了与分散剂或润湿剂、悬浮剂以及一种或多种防腐剂混合的活性成分。化合物1的口服混悬液可以如pct申请wo 2017/081044(may等)所述来制备。
[0049]
本发明的活性成分可以口服速释(ir)制剂或缓释制剂来施用。“缓释”是指相对于通过口服施用活性剂的常规制剂所达到的治疗量,在全身血液循环中在长时间段内活性剂以有效达到治疗量的活性剂或其活性代谢物的速率从剂型中释放。活性剂的释放发生在长时间段内,例如,在至少6小时内、在至少8小时内、在至少12小时内或在至少24小时内。
[0050]
化合物1可以通过静脉内(i.v.)输注来施用。适用于静脉内施用的化合物1的溶液可以如pct公开申请号wo 2017/081044(may等)所述来制备。
[0051]
适合的局部制剂和剂型包括软膏、乳膏、凝胶、洗剂、糊剂等,如remington:the science and practice of pharmacy(第21版,university of the sciences in philadelphia,2005)所述。软膏是半固体制剂,通常基于矿脂或其他石油衍生物。如本领域技术人员将理解的,要使用的特定软膏基质是一种将提供最佳的药物递送,并且优选地还将提供其他期望的特性(例如润肤性等)的软膏基质。乳膏为水包油型或油包水型的粘性液体或半固体乳剂。乳膏基质是可水洗的,并包含油相、乳化剂和水相。油相,也称为“内部”相,一般包含矿脂和脂肪醇诸如鲸蜡醇或硬脂醇。水相经常(虽然不一定)在体积上超过油
相,并且一般包含保湿剂。乳膏制剂中的乳化剂一般是非离子、阴离子、阳离子或两性表面活性剂。凝胶是半固体、混悬型系统。单相凝胶包含基本上均匀地分布在整个载体液体中的有机大分子(聚合物),所述载体液体通常是水性的,但是优选还包含醇诸如乙醇或异丙醇,以及任选地油。为了制备均匀的凝胶,可以添加分散剂诸如醇或甘油,或者可以通过磨碎、机械混合或搅拌或其组合来分散胶凝剂。洗剂是无需摩擦即可施加到皮肤表面的制剂,并且通常是液体或半液体制剂,其中包括活性剂的固体颗粒存在于水或醇基中。洗剂经常是微细固体的混悬液,并且通常包含悬浮剂以产生更好的分散体,以及可用于使活性剂定位并保持与皮肤接触的化合物。糊剂是半固体剂型,其中活性剂悬浮在适合的基质中。根据基质的性质,将糊剂分为脂肪糊剂或由单相水性凝胶制成的糊剂。
[0052]
本领域技术人员已知的各种添加剂可以包括在局部制剂中。例如,可以使用包括相对少量的醇的溶剂来增溶某些原料药。其他任选的添加剂包括遮光剂、抗氧化剂、香料、着色剂、胶凝剂、增稠剂、稳定剂、表面活性剂等。也可以添加其他试剂诸如抗菌剂,以防止储存后变质,即抑制微生物诸如酵母和霉菌的生长。对于那些具有异常低的透过皮肤或粘膜组织的渗透率的药物,可期望的是在制剂中包含渗透促进剂。所述制剂还可包含刺激缓和添加剂,以最小化或消除由药物、促进剂或剂型的其他组分引起的皮肤刺激或皮肤损害的可能性。所述制剂还可包含醚生理上可接受的赋形剂或其他次要添加剂,诸如香料、染料、乳化剂、缓冲剂、凉味剂(例如薄荷醇)、抗生素、稳定剂等。在一些情况下,一种组分可以提供不止一种功能。
[0053]
局部制剂中活性剂的浓度可以有很大不同,并且取决于多种因素,包括待治疗的疾病或病状、活性剂的性质和活性、所需的效果、可能的不良反应、活性剂达到其预期目标的能力和速度,以及患者和医师的特定知识范围内的其他因素。所述制剂通常包含约0.1重量%至50重量%的活性剂,优选0.1重量%至5重量%的活性剂,最佳5重量%至20重量%的活性剂。
[0054]
本发明的药物组合物可以配制成通过肌肉内或皮下注射来施用的贮库制剂。贮库制剂是高效的、耐受性良好的、缓释或延迟释放的活性成分组合物,其有效治疗数周,诸如至少一周、至少两周、至少三周、至少四周、至少五周或至少六周或更长。除活性剂之外,可在本发明的贮库制剂中使用额外的成分,包括表面活性剂、增溶剂、乳化剂、防腐剂、等渗剂、分散剂、润湿剂、填充剂、溶剂、缓冲剂、稳定剂、润滑剂和增稠剂。也可以使用额外成分的组合。贮库制剂中活性成分的量将取决于所治疗的慢性肝病的严重程度。
[0055]
本发明的组合物可以单位剂型呈现并且可通过药学领域中众所周知的任何方法来制备。术语“单位剂型”意指单剂量,其中所有活性和非活性成分在适合的系统中组合,使得患者或向患者施用药物的人可以打开具有包含在其中的全部剂量的单个容器或包装,并且不必将两个或更多个容器或包装中的任何组分混合在一起。单位剂型的典型实例是用于口服施用的片剂或胶囊。单位剂型的这些实例不旨在以任何方式进行限制,而仅仅是代表单位剂型的药学领域中的典型实例。
[0056]
本发明的组合物还可以呈现为试剂盒,由此可以提供两种或更多种组分,其可以是活性或非活性成分、载体、稀释剂等,并带有由患者或向患者施用药物的人制备实际剂型的说明书。可以提供具有包含在其中的所有必需的材料和成分的此类试剂盒,或者它们可以包含使用或制备必须由患者或向患者施用药物的人独立获得的材料或组分的说明书。
[0057]
nafld和nash
[0058]
本质上,非酒精性脂肪肝病(nafld)是脂肪堆积在肝脏中(脂肪变性)的病状。所述术语包括从单纯性脂肪变性到非酒精性脂肪性肝炎、纤维化和肝硬化的进行性的疾病谱。非酒精性脂肪性肝炎(nash)被定义为脂肪变性(肝脂质积累)的存在与肝脏炎症和肝细胞损伤(脂肪性肝炎)并存。nash是nafld的一种更晚期形式,其中已发生肝损伤,并且单纯的良性脂肪变性可演变为纤维化和肝硬化,导致终末期肝病,包括肝细胞癌的发生(staels,b.等,hepatology,2013,58:1941
‑
1952)。
[0059]
nafld是全世界最常见的肝病形式,其中在西方人群中的患病率为15%
‑
30%,并且是由甘油三酯在肝脏内积累引起的。然而,超重人群中的患病率增加到58%,并且肥胖人群中的患病率增加到98%。基于血清转氨酶水平升高以及没有其他可识别的肝损伤原因,四分之一以上患有nafld的成人被推测患有nash。目前,nash的明确诊断是基于不仅有肝细胞中的脂肪积累(脂肪变性),而且有肝细胞损伤以及炎性细胞死亡和积累的组织学证据。因为具有nash的肝脏比具有孤立性脂肪变性的肝脏受损更大,因此nash比孤立性脂肪变性更可能导致进行性肝纤维化以及最终的肝脏相关疾病和死亡(diehl,a.m.和day,c.,n.engl.j.med.,2017,377:2063
‑
72)。
[0060]
若干项研究表明,肥胖与慢性脂肪组织炎症有关,并且在肥胖的早期阶段,已显示脂肪组织被促炎细胞诸如淋巴细胞、肥大细胞、自然杀伤细胞和中性粒细胞浸润(elgazar
‑
carmon,v.等,j.lipid research,2008,49:1894
‑
1903;feuerer,m.等,nature medicine,2009,15:930
‑
939;liu,j.等,nature medicine,2009,17(8):940
‑
946;nishimura,s.等,nature medicine,2009,15(8):914
‑
921;weisberg,s.p.等,j.clin.invest.,2003,112:1796
–
1808;winer,d.a.等,nature medicine,2011,17(5):610
‑
618;以及xu,j.等,j.clin.invest.,2003,112:1821
–
1830)。脂肪炎症也与胰岛素抵抗的发生有关(ouchi,n.等,nature reviews immunol.,2011,11:85
‑
97;osborn,o.和olefsky,j.m.,nature medicine,2012,18(3):363
‑
374;以及sun,s.等,annul.rev nutr.,2012,32:261
‑
286)。胰岛素抵抗可能反过来独立地导致促炎和促纤维化状态并导致nash/nafld。中性粒细胞参与以肥胖为特征的炎症,并且ne对于调节中性粒细胞介导的脂肪组织和肝脏炎症的调控至关重要。作为ne抑制剂,化合物1有望同时显示出可用于治疗nash/nafld的胰岛素增敏剂和抗炎特性(talukdar,s.,olefsky,j.m.等,nature medicine,2012,18(9):1407
‑
1412)。
[0061]
使用重组鼠ne治疗的小鼠表现出葡萄糖耐受不良,而用高脂饮食(hfd)喂养的ne基因敲除小鼠(neko)与野生型对照相比具有受保护的表型,也就是说,体重增加减少、白色脂肪组织和肝脏重量降低、葡萄糖耐量增加、空腹胰岛素浓度降低、肝脏和脂肪组织胰岛素敏感性增加并且脂肪和肝脏的中性粒细胞浸润减少(talukdar等,2012)。在相同的研究中,使用ne抑制剂gw311616a治疗hfd小鼠也保护了正常表型,改善了葡萄糖耐量,并在葡萄糖钳夹/示踪剂研究中表现出外周和肝脏胰岛素敏感性增加。类似地,mansuy
‑
aubert等发现无ne(ela2
‑
/
‑
)小鼠和a1at转基因小鼠对高脂饮食(hfd)诱导的体重增加、胰岛素抵抗、炎症和脂肪肝具有抵抗力。在hfd喂养的小鼠中,ne抑制剂gw311616a逆转了胰岛素抵抗和体重增加(mansuy
‑
aubert,v.等,cell metabolism,2013,17:534
‑
548)。
[0062]
此外,已经阐明了ne介导胰岛素抵抗的机制。在喂养hfd的小鼠中,肝脏中性粒细胞浸润和由此产生的ne活性升高(talukdar,2012),这与患有nash的肥胖患者肝脏中的观
察结果一致(rensen,s.等,am.j.pathol.,2009,175:1473
‑
1482)。更具体地说,中性粒细胞在门静脉炎性浸润物中的存在与人类疾病发展为nash相关(gadd,v.l.等,hepatology,2014,59(4):1393
‑
1405)。与此观察结果一致,中性粒细胞对淋巴细胞的比率已被提出作为潜在的新型生物标志物来预测患有nafld的患者中的nash和晚期纤维化(alkhouri,n.等,liver international,2012,32(2):297
‑
302)。使用ne治疗肝细胞导致细胞胰岛素抵抗。已显示细胞外ne进入细胞内空间并介导胰岛素受体底物1(irs1)的降解(houghton,a.m.等.,nature med.,2010,16(2):219
‑
223;houghton,a.m.,cell cycle,2010,9(9):1732
‑
1737)。在一项胰岛素反应研究中,已显示neko动物具有与wt对照相比更高的胰岛素信号传导测量值,如akt磷酸化增加所证明(talukdar 2012)。相反,施用重组ne导致小鼠肝脏和脂肪组织中的基础和胰岛素刺激的irs1和p
‑
akt信号传导减少,并且在原代小鼠和人类肝细胞中,防止了胰岛素抑制胰高血糖素刺激的肝细胞葡萄糖输出的能力。
[0063]
迄今为止,尚无人类临床试验证明任何提议的nafld/nash治疗的功效。然而,已经开发出许多模仿这些疾病的动物模型。肥胖与nafld之间的紧密联系已经刺激了各种饮食诱导肥胖(dio)啮齿动物模型的开发,所述模型模仿了nash的病因和自然史。当喂养高脂、高胆固醇(致肥胖的)饮食时,不同品系的小鼠对nash显示出不同的易感性。最常用的品系是c57bl/6小鼠,其对致肥胖饮食显示出高易感性。与常用的balb/c和c3h/hen小鼠相比,这些小鼠也容易发生饮食诱导的肝脏坏死性炎症和纤维化。参见hansen,h.h.等(drug discovery today,2017,22(11):1707
‑
1718)。
[0064]
zang等已经研究了中性粒细胞和ne抑制剂西维来司他(sivelestat)在喂养高脂、高胆固醇饮食的c57bl/6j apoe
‑
/
‑
小鼠中的作用(zang,s.f.等,cell biochemistry biophysics,2015,73(2):479
‑
487;zang,s.f.等,chinese j.hepatology,2017,25(5):371
‑
376))。同一研究小组已经研究了中性粒细胞弹性蛋白酶与其天然抑制剂α1
‑
抗胰蛋白酶(a1at)之间的失衡,以及受影响患者相对于正常患者的nafld的组织学进展。他们发现ne:a1at比率的增加与患有nash的患者中的肝脏炎症密切相关(zang,s.f.等,clinical exper.pharm.physiology,2016,43(1):13
‑
21)。
[0065]
治疗施用和剂量
[0066]
术语化合物1的“施用”或“施用”化合物1应理解为意指向需要治疗的个体提供(4s)
‑4‑
[4
‑
氰基
‑2‑
(甲基磺酰基)苯基]
‑
3,6
‑
二甲基
‑2‑
氧代
‑1‑
[3
‑
(三氟甲基)苯基]
‑
1,2,3,4
‑
四氢嘧啶
‑5‑
甲腈或盐、溶剂化物、盐的溶剂化物或多晶型物,其以治疗有用的形式和治疗有效量的形式引入该个体体内,包括但不限于口服剂型,诸如片剂、胶囊、糖浆、混悬液等。
[0067]
术语“治疗(treat)”、“治疗(treating)”和“治疗(treatment)”慢性肝病均是指降低慢性肝病症状或体征的频率(包括完全消除它们),避免慢性肝病的发生和/或降低慢性肝病症状或体征的严重程度。术语“慢性肝病”包括但不限于非酒精性脂肪肝病(nafld)和非酒精性脂肪性肝炎(nash)。
[0068]
术语“治疗有效量”是指适合的组合物中和适合的剂型中用于治疗所述疾病病状的足够数量的化合物1。“治疗有效量”将根据化合物、引起慢性肝病的病状的严重程度以及待治疗患者的年龄、体重等而变化。
[0069]
本发明用于治疗慢性肝病的方法要求向需要此类治疗的患者施用化合物1或包含
inc.),一种法尼醇x受体(fxr)激动剂;tern
‑
201(terns pharmaceuticals inc.),一种氨基脲敏感型胺氧化酶(ssao)抑制剂;泰鲁司特(tipelukast)(mn
‑
001;medicinova,inc.),一种白三烯(lt)受体拮抗剂;伏昔巴特(volixibat)(shp
‑
626;shire),一种回肠钠胆汁酸共转运抑制剂;和(奥利司他(orlistat),roche),一种降脂剂。
[0074]
慢性肝病nafld和nash常常发生在糖尿病患者中。因此,将化合物1与旨在治疗或改善糖尿病影响的药物组合施用可能是特别适当的,其中所述药物不是胰岛素或其衍生物,特别是那些剂还显示出在治疗nafld或nash中的功效。在一个实施方案中,适当的抗糖尿病剂包括但不限于利拉鲁肽(例如,来自novo nordisk的来自novo nordisk的);杜拉鲁肽(eli lilly);阿卡波糖(acarbose)(bayer);阿必鲁肽(albiglutide)(glaxosmithkline);阿格列汀(alogliptin)(takeda);甲磺酸溴隐亭(bromocriptine mesylate)(salix pharmaceuticals);卡格列净(canaglifozin)(janssen pharmaceuticals);达格列净(dapagliflozin)(astrazeneca);恩格列净(boehringer ingelheim);伊格列净(astellas pharma);格列美脲(glimepiride)(sanofi
‑
aventis);格列美脲(glyburide)(diasanofi
‑
adventis);鲁格列净(taisho pharmaceutical);二甲双胍;米格列醇(miglitol)(pfizer);普罗布考(probucol);瑞格列奈(repaglinide)(novo nordisk);吡格列酮罗格列酮沙格列汀(saxagliptin)(astrazeneca);索马鲁肽(novo nordisk);和西他列汀(sitagliptin)(merck)。
[0075]
可以根据在一定的施用频率上施用的药物总量来陈述用于口服施用的化合物1的剂量范围。根据上述因素,可以适当地一天一次或多次给予一定量的活性成分。例如,可以一天一次、一天两次、一天三次、一天四次或更多次服用剂量。适合的剂量范围为0.1mg至100mg,并且优选1mg至20mg,一天一次或多次。适合的剂量通常为0.10mg、0.15mg、0.20mg、0.25mg、0.5mg、0.75mg、1mg、2mg、2.5mg、3mg、4mg、5mg、6mg、7mg、8mg、9mg、10mg、15mg、20mg、25mg、30mg、40mg、50mg或100mg,每天一次或多次。优选地,每天一次施用1mg、2mg、5mg、10mg或20mg的剂量。
[0076]
可替代地,可以根据体重依赖性剂量来陈述用于口服施用的化合物1的剂量范围。适合的剂量一般是每公斤体重0.001mg至1mg药物(mg/kg),一天一次或多次。适合的体重依赖性剂量通常为0.001mg/kg、0.0015mg/kg、0.002mg/kg、0.0025mg/kg、0.005mg/kg、0.0075mg/kg、0.01mg/kg、0.02mg/kg、0.025mg/kg、0.03mg/kg、0.04mg/kg、0.05mg/kg、0.06mg/kg、0.07mg/kg、0.08mg/kg、0.09mg/kg、0.1mg/kg、0.15mg/kg、0.2mg/kg、0.25mg/kg、0.3mg/kg、0.4mg/kg、0.5mg/kg或1mg/kg,每天一次或多次,并且优选0.01mg/kg、0.02mg/kg、0.05mg/kg、0.1mg/kg或0.2mg/kg,每天一次。通过技术人员已知的方法可以容易地确定剂量范围。可与例如载体物质组合以产生单剂型的活性成分的量将根据所治疗的
患者和特定的施用方式而变化。
[0077]
治疗有效性的确定
[0078]
可以在本领域技术人员已知的慢性肝病实验动物模型中测试本发明的方法和组合物的有效性。具体地说,上述nafld和nash的饮食诱导肥胖(dio)啮齿动物模型是适当的。这些模型通常利用c57bl/6小鼠,其对致肥胖饮食显示出高易感性。这些小鼠也容易发生饮食诱导的肝脏坏死性炎症和纤维化,类似于人类中的相同情况。参见hansen,h.h.等(drug discovery today,2017,22(11):1707
‑
1718)。
[0079]
例如,在一种模型中,每天通过口服管饲法(qd或bid)给dio小鼠投加化合物1。匹配的对照dio小鼠仅接受媒介物。观察动物数周至数月的时间,包括监测它们的体重和其他参数。将小鼠人道地处死,收集终末血样,并检查两组小鼠之间肝脏和其他器官的差异。可以比较许多参数,包括肝酶、血浆脂质、各种标志基因的表达、炎症的标志物以及包括纤维化形成程度的nafld活性评分。
[0080]
本发明的方法和组合物在治疗慢性肝病中的功效也可以在由美国食品药品管理局(fda)和其他国际机构列出的适当标准和道德准则下进行的人类临床试验中进行评估。在通常在健康志愿者中进行的1期临床试验中确定了药物的一般安全性和药代动力学后,就进行评估药物在患有待治疗病状或目标疾病的患者中的安全性和功效的2期试验。通常,此类试验是双盲和对照的,并且可能是剂量范围研究。3期研究收集了关于安全性的更多信息,并试图通过在特定剂量下研究目标人群以及任选地将所述药物与其他药物组合使用来证明有效性。
[0081]
提供以下实施例以作说明而非限制。
[0082]
实施例
[0083]
实施例1.片剂的制备
[0084]
化合物1,(4s)
‑4‑
[4
‑
氰基
‑2‑
(甲基磺酰基)苯基]
‑
3,6
‑
二甲基
‑2‑
氧代
‑1‑
[3
‑
(三氟甲基)苯基]
‑
1,2,3,4
‑
四氢嘧啶
‑5‑
甲腈,可以被配制为用于口服使用的片剂。这些片剂的制造利用标准的制药工艺技术。如前所述,以下实施例中的所有非活性药物成分均符合美国药典(usp)、国家处方集(nf)、欧洲药典(ph.eur.)和/或日本药典(ph.jap.)的要求,并根据各论对指定标准中规定的每种成分进行测试和释放。批次大小根据特定临床目的所需的量而变化。以下两个实施例展示了示例性剂量的定性/定量组成,并且用于说明目的。应当理解,本发明考虑了额外的剂量大小和批次量。
[0085]
实施例1a.0.5mg片剂的制备。表1中示出了0.5mg口服片剂的批次组成。
[0086]
表1
[0087][0088]1散装纯化水用作制造过程中去除的溶剂。
[0089]2包含:羟丙甲纤维素(hypromellose)15cp,ph.eur.,nf,ph.jap.;聚乙二醇(macrogol),ph.eur.,usp,ph.jap.;二氧化钛,ph.eur.,指令95/45/ec,usp,ph.jap.
[0090]
使用表1中规定的量,将微粉化的化合物1、交联羧甲基纤维素钠和乳糖一水合物在流化床制粒机中混合。添加羟丙基纤维素水溶液作为制粒液体。在制粒、干燥、研磨和筛选后,添加颗粒外的硬脂酸镁。将最终共混物压制成片剂,对片剂进行质量均匀性、厚度和抗破碎性测试。片剂用opadry
tm
水溶液包衣。目视检查包衣的片剂是否有缺陷。丢弃具有明显包衣缺陷的片剂。
[0091]
实施例1b.1mg和5mg片剂的制备。表2和表3中分别示出了1mg和5mg口服片剂的批次组成。
[0092]
表2
[0093][0094]1散装纯化水用作制造过程中去除的溶剂。
[0095]2包含:聚乙烯醇,ph.eur.,usp,fcc,ph.jap.;聚乙二醇,ph.eur.,usp,fcc,jecfa,ph.jap.;二氧化钛,ph.eur.,usp,fcc,ph.jap.,chp,gb;滑石,usp,fcc,ph.eur.,ph.jap.,jecfa。
[0096]
表3
[0097][0098]1散装纯化水用作制造过程中去除的溶剂。
[0099]2包含:聚乙烯醇,ph.eur.,usp,fcc,ph.jap.;聚乙二醇,ph.eur.,usp,fcc,
jecfa,ph.jap.;二氧化钛,ph.eur.,usp,fcc,ph.jap.,chp,gb;滑石,usp,fcc,ph.eur.,ph.jap.,jecfa。
[0100]
使用表2和表3中规定的量,将微粉化的化合物1、交联羧甲基纤维素钠和乳糖一水合物在高剪切制粒机中混合。添加羟丙基纤维素水溶液作为制粒液体。在制粒、干燥、研磨和筛选后,添加颗粒外的微晶纤维素和硬脂酸镁,在添加硬脂酸镁之前测试共混物的均匀性。将最终共混物压制成片剂,对片剂进行质量均匀性、厚度和抗破碎性测试。片剂用opadry
tm
ii水溶液包衣。目视检查包衣的片剂是否有缺陷。丢弃具有明显包衣缺陷的片剂。
[0101]
实施例2.化合物1对dio小鼠中葡萄糖耐量的影响
[0102]
方法。雄性dio(饮食诱导的肥胖)小鼠(c57bl/6ntac dio mpf,n=35)购自taconic biosciences(rensselaer,ny)。通过从6
‑
16周龄开始给雄性c57bl/6小鼠喂养高脂饮食(hfd,d12492;research diets,inc.,new brunswick,nj)来准备dio小鼠。在整个研究期间维持十二小时的光照周期。每天监测室温并使其保持在22
‑
25℃。将dio小鼠单独饲养,并在7天的设施适应期和整个研究期期间保持于hfd。在适应期期间每天一次向所有小鼠施用媒介物(5%dmso/3%乙醇/92%花生油)。
[0103]
在适应设施7天后,根据体重和空腹(6小时)血糖将dio小鼠随机分为4组,每组8只,用于分配来接受媒介物、30mg/kg/天的罗格列酮或3.0mg/kg/天或15mg/kg/天的化合物1。每天早上通过口服管饲施用化合物一次,持续6周。在整个研究中剂量体积保持在5ml/kg。以粉末形式提供化合物1,并且每周进行新鲜配制。罗格列酮(批号foj407)由cbin提供;罗格列酮也是每周新鲜配制。罗格列酮是噻唑烷二酮类的抗糖尿病药物。
[0104]
每周通过尾夹从进食动物中获得全血,用于首次给药后第7天、第14天、第21天和第35天测量非空腹血糖(statnova biomedical,waltham,ma)。在基线处葡萄糖测量之前记录体重,并且此后每周记录一次。在基线(第
‑
1天)、此后的第28天和第42天对空腹小鼠进行口服葡萄糖耐量测试。在进入空腹期1小时后施用罗格列酮或化合物1。在6小时空腹期结束时,使用葡萄糖(2g/kg,10ml/kg,通过口服管饲)对小鼠进行激发。在葡萄糖负荷后0、20、40、60、90和120分钟时,通过尾夹获得血样,用于通过statstrip来评估血糖。使用小鼠/大鼠胰岛素试剂盒(msd#k152bzc;meso scale diagnostics,rockville,md)从ogtt的0时血样中测量血清胰岛素。
[0105]
使用co2吸入和气胸诱导终止动物。来自每个治疗组的四只小鼠在给药后0.5小时处死,而四只在给药后4小时处死。通过心脏穿刺来获得终末血样(k2edta),并处理成血浆用于测定化合物浓度。记录器官重量(肝脏,附睾脂肪)。
[0106]
结果。所有数据均表示为组平均值
±
sem。使用jmp(sas软件)分析数据。所有归一化均使用终末体重计算。所有分配的动物均完成了研究。接受3mg/kg的化合物1的一只动物被鉴定为若干个测量参数(mahalanobis,t2)的离群值,并从所有分析中去除。将每只动物对应的0、30、60、90和120分钟时间点之间的梯形面积的总和相加,用于从ogtt数据获得葡萄糖的曲线下面积(auc)。使用因素方差分析(oneway anova)确定与媒介物相比的治疗效果(*,p<0.05),然后适当时进行邓尼特检验(dunnett’s test)。
[0107]
体重。18周龄dio小鼠的体重平均为42.5
±
0.4g,并且在基线时任何治疗组与媒介物相比均没有显著差异(媒介物、罗格列酮以及3mg/kg和15mg/kg的化合物1分别为42.2
±
0.7、42.8
±
1.0、42.8
±
1.1和42.8
±
1.0)。如图2a所示,在6周的研究期间,所有治疗组的体重与基线相比均增加。施用罗格列酮的小鼠在治疗6周后明显重于媒介物治疗的动物(49.4
±
0.7对55.0
±
1.4g)。在研究结束时,如图2b所示,虽然注意到与媒介物相比,化合物1对体重没有统计学上的显著影响(媒介物以及3mg/kg和15mg/kg的化合物1分别为49.4
±
0.7、49.8
±
0.9和47.3
±
1.1g),但与媒介物相比,15mg/kg的化合物1显示出显著较低的体重增长率(媒介物和化合物1分别为14
±
1.8%和6.9
±
1.8%)。参见图2c和图2d。
[0108]
血糖。18周龄小鼠的血糖(空腹)平均为163.1
±
3.9mg/dl并且在研究开始时与媒介物相比没有显著差异(媒介物、罗格列酮以及3mg/kg和15mg/kg的化合物1分别为163.0
±
12.7、164.0
±
4.0、160.3
±
5.7和164.6
±
7.3mg/dl)。随着研究的进展,在所有治疗组(包括媒介物治疗的小鼠)中,空腹血糖与基线值相比均降低。在治疗6周后,罗格列酮治疗的小鼠的空腹血糖与基线相比降低了23%(164.0
±
4至125.8
±
4.8mg/dl);然而,与媒介物相比,在第35天的进食葡萄糖没有显著差异(134.5
±
6.9对125.5
±
4.5mg/dl)。在治疗6周后,对于15mg/kg的剂量,化合物1治疗的小鼠中的空腹血糖相对于基线降低了11%。类似地,与媒介物相比,任何化合物1治疗的组在第35天的进食葡萄糖均没有显著差异(媒介物以及3mg/kg和15mg/kg的化合物1分别为134.5
±
6.9、128.3
±
3.4和134.9
±
4.7mg/dl)。
[0109]
ogtt。媒介物、罗格列酮和化合物1治疗的小鼠的葡萄糖处置曲线在基线处是可叠加的,如图3a所示,使得注意到组之间的葡萄糖auc没有显著差异(媒介物、罗格列酮以及3mg/kg和15mg/kg的化合物1分别为34.12
±
3.3、37.6
±
2.6、36.3
±
2.0和34.4
±
2.2mg/dl
·
min x103),如图3d所示。在治疗28天后,与媒介物相比,罗格列酮和两个化合物1治疗的组的葡萄糖处置曲线的变化是明显的,如图3b所示;与媒介物相比,罗格列酮和两个化合物1治疗的组在第28天的葡萄糖auc显著较低(媒介物、罗格列酮以及3mg/kg和15mg/kg的化合物1分别为27.9
±
1.1、20.7
±
0.4、24.0
±
0.9和22.7
±
0.7mg/dl
·
min x103),如图3d所示。虽然罗格列酮对葡萄糖处置曲线的影响在第42天仍然明显,但对化合物1的反应似乎是短暂的,因为在此时间点,那些曲线接近媒介物,如图3c所示。事实上,与媒介物相比,罗格列酮的葡萄糖auc仍然显著较低(27.8
±
1.2对20.1
±
0.5mg/dl/min x103),而与媒介物相比,化合物1治疗的显著性丧失(媒介物以及3mg/kg/天和15mg/kg/天的化合物1分别为27.8
±
1.2、28.6
±
0.7和26.3
±
1.1mg/dl/min x103),如图3d所示。
[0110]
与基线值相比,罗格列酮(44.9
±
3.9%)、3mg/kg的化合物1(20.0
±
4.5%)和15mg/kg的化合物1(
‑
21.9
±
5.0%)在第42天的葡萄糖auc表现出降低。在媒介物治疗的动物中,auc相对于基线的降低为12.6
±
10.3%。与媒介物相比,仅用罗格列酮治疗的动物的这种作用是显著的。
[0111]
空腹血清胰岛素。在基线时,18周龄dio小鼠的空腹血清胰岛素平均为4.7
±
0.4ng/ml;注意到在基线时,治疗组之间的空腹胰岛素没有差异(媒介物、罗格列酮以及3mg/kg和15mg/kg的化合物1分别为4.44
±
0.8、5.2
±
1.1、3.5
±
0.5和5.4
±
1.0ng/ml),如图4a所示。在第42天,与媒介物相比,施用罗格列酮的小鼠的空腹胰岛素水平显著较低(8.2
±
1.5对1.3
±
0.1ng/ml)。与媒介物相比,低剂量化合物1的胰岛素水平较高(8.2
±
1.5对11.8
±
1.3ng/ml),而与媒介物相比,使用高剂量化合物1治疗的动物胰岛素水平较低(8.2
±
1.5对5.9
±
1.5ng/ml),但与媒介物相比,这些作用不显著,如图4b所示。
[0112]
器官重量。与媒介物相比,施用3mg/kg的化合物1的小鼠肝脏重量(体重%)显著较
高(5.6
±
0.2对6.8
±
0.3%)。注意到与媒介物相比,对肝脏重量没有其他显著影响(媒介物、罗格列酮和15mg/kg的化合物1分别为5.6
±
0.2、5.0
±
0.2和6.4
±
0.2%),如图5a所示。注意到与媒介物相比,对附睾脂肪重量没有显著影响(媒介物、罗格列酮以及3mg/kg和15mg/kg的化合物1分别为3.4
±
0.3、3.7
±
0.2、3.1
±
0.3和3.5
±
0.2%),如图5b所示。
[0113]
概述。基于临床观察,化合物1(3
‑
15mg/kg/天)总体上耐受性良好。在治疗6周后,与媒介物治疗的小鼠相比,施用罗格列酮的动物更重、胰岛素水平更低,并且葡萄糖处置得到改善,这与其已知的对体重增加和胰岛素增敏的作用一致。虽然在整个研究期期间,罗格列酮和15mg/kg的化合物1降低了空腹血糖,但进食血糖在所有组中均没有显示出显著差异。化合物1在高剂量时降低了体重增加,并且在施用3mg/kg和15mg/kg的化合物1后,葡萄糖的处理和处置得到了短暂的改善。
[0114]
实施例3.化合物1在肥胖的dio小鼠模型中的作用
[0115]
方法。雄性dio小鼠(c57bl/6ntac dio mpf,n=24)购自taconic biosciences(rensselaer,ny)。在taconic通过从6周龄开始给雄性c57bl/6小鼠喂养高脂饮食(hfd,research diets d12492)来准备dio小鼠。它们在17周龄的时候到达研究机构,并继续接受hfd直到研究结束。在整个研究期间,从上午6点到下午6点,维持十二小时的光照周期。每天监测室温并使其保持在22
‑
25℃。将dio小鼠单独饲养,并在7天的设施适应期和整个研究期期间保持于hfd。在适应期期间每天一次向所有小鼠施用媒介物(5%dmso/3%乙醇/92%花生油)。
[0116]
在适应设施7天后,根据体重和空腹(6小时)血糖将dio小鼠随机分为3组,每组8只,用于分配来接受媒介物、15mg/kg或30mg/kg的化合物1。如表4所示,每天早上(上午6点至上午8点)以及给药后至少8小时通过口服管饲施用化合物两次,持续4周。在整个研究中,剂量体积保持在5ml/kg。以粉末形式提供化合物1,并且每周的周一、周三和周五使用新鲜媒介物溶液进行新鲜配制。
[0117]
表4
[0118][0119]
每周通过尾夹从进食动物中获得全血,用于第7天、第14天和第21天就在首次早晨给药之前测量血糖(statstrip)。在基线处葡萄糖测量之前记录体重,并且此后每周记录一次。
[0120]
在基线(第
‑
1天)和第28天对空腹(0600
‑
1200)小鼠进行口服葡萄糖耐量测试(ogtt)。在进入空腹期1小时后(上午7点左右)施用化合物1。在6小时空腹期结束时,使用葡萄糖(2g/kg,10ml/kg,通过口服管饲)对小鼠进行激发,并且在葡萄糖负荷后0、20、40、60、90和120分钟时,通过尾夹获得血样,用于通过statstrip来评估血糖。使用小鼠/大鼠胰岛素试剂盒(msd#k152bzc)在第
‑
1天和第29天从6h空腹血样中测量血清胰岛素。
[0121]
使用co2吸入和气胸诱导终止动物。在给药后4小时处死所有动物用于测定化合物
浓度。通过心脏穿刺来获得终末血样(k2edta),并处理成血浆。
[0122]
结果。所有数据均表示为组平均值
±
sem。使用prism 7.0(graphpad软件)分析数据。所有归一化均使用终末体重计算。所有分配的动物均完成了研究。将每只动物对应的0、20、40、60、90和120分钟时间点之间的梯形面积的总和相加,用于从ogtt数据获得葡萄糖的曲线下面积(auc)。使用oneway anova确定与媒介物相比的治疗效果(*,p<0.05;**,p<0.01),然后适当时进行邓尼特检验。
[0123]
体重。18周龄dio小鼠的体重平均为44.8
±
0.4g,并且在基线时任何治疗组与媒介物相比均没有显著差异(媒介物、15mg/kg和30mg/kg的化合物1分别为44.7
±
0.9、44.8
±
0.7和44.8
±
0.7)。如图6a所示,在4周的研究期间,所有治疗组的体重与基线相比均增加。在研究结束时,虽然注意到与媒介物相比,化合物1对体重没有统计学上的显著影响(媒介物以及15mg/kg和30mg/kg的化合物1分别为48.8
±
0.9、47.5
±
1.3和46.0
±
1.2g),但在给药4周后,与媒介物相比,30mg/kg的化合物1显示出显著较低的体重增长率(在研究结束时,媒介物、15mg/kg和30mg/kg的化合物分别为9.2
±
2.0%、5.9
±
1.6%和2.8
±
2.3%),如图6b所示。
[0124]
血糖。18周龄小鼠的血糖(空腹)平均为178.4
±
4.2mg/dl并且在研究开始时与媒介物相比没有显著差异(媒介物以及15mg/kg和30mg/kg的化合物1分别为182.9
±
9.1、185.1
±
8.3和180.1
±
7.0)。随着研究的进展,在所有治疗组(包括媒介物治疗的小鼠)中,血糖与基线值相比均降低。在整个研究期期间,发现所有治疗组的血糖均没有显著差异(在第28天,媒介物以及15mg/kg和30mg/kg的化合物1分别为143.0
±
7.1、151.9
±
6.1和137.3
±
4.1mg/dl)。
[0125]
ogtt。媒介物和化合物1治疗的小鼠的葡萄糖处置曲线在基线处是可叠加的,使得注意到组之间的葡萄糖auc没有显著差异(媒介物以及15mg/kg和30mg/kg的化合物1分别为33.0
±
2.5、33.8
±
0.9和32.8
±
1.8mg/dl
·
min x103)。在治疗28天后,与媒介物相比,两个化合物1治疗的组的葡萄糖处置曲线的峰值变化是明显的,如图7a所示。然而,与媒介物相比,两个化合物1治疗的组在第28天的葡萄糖auc没有显著差异(媒介物以及15mg/kg和30mg/kg的化合物1分别为25.5
±
1.4、24.7
±
1.9和22.4
±
1.0mg/dl
·
min x103)。
[0126]
空腹血清胰岛素。在基线时,18周龄dio小鼠的空腹血清胰岛素平均为5.3
±
0.5ng/ml;注意到在基线时,治疗组之间的空腹胰岛素没有差异(媒介物以及15mg/kg和30mg/kg的化合物1分别为6.1
±
0.6、7.0
±
1.1和4.9
±
0.6ng/ml),如图8所示。在第28天,当媒介物组显示空腹胰岛素水平增加时,使用两个化合物1组治疗的小鼠的空腹胰岛素水平降低(媒介物以及15mg/kg和30mg/kg的化合物1分别为7.7
±
1.6、5.8
±
1.4和4.0
±
0.3ng/ml)。与使用媒介物治疗的小鼠相比,使用30mg/kg的化合物1治疗的小鼠的空腹血清胰岛素显著较低。
[0127]
概述。在研究中,化合物1(15
‑
30mg/kg/天)总体上耐受性良好。在4周的治疗期间,与使用媒介物治疗的小鼠相比,使用15mg/kg和30mg/kg的化合物1治疗的小鼠体重增加均较少。这种趋势在用30mg/kg的化合物1治疗3周或更长时间的小鼠中具有统计学意义。
[0128]
在整个研究期期间,所有组的血糖均降低,并且这种降低在所有组之间均没有显示出显著差异。治疗4周后的ogtt处置曲线显示,使用15mg/kg和30mg/kg的化合物1治疗的小鼠中的峰值均降低。因此,与媒介物治疗的小鼠相比,这两个治疗组中存在稍微降低的
ogtt auc。然而,由于与基线相比,媒介物组中的ogtt auc存在类似降低,因此ogtt auc的这些降低均没有统计学意义。与基线相比,所有组的ogtt auc均显著降低。
[0129]
与媒介物治疗的小鼠相比,化合物1倾向于减少空腹血清胰岛素。在治疗的第28天,使用30mg/kg的化合物1治疗的小鼠的血清胰岛素水平显著低于媒介物治疗的小鼠。
[0130]
实施例4.化合物1在健康患者中的1期临床研究
[0131]
研究描述。根据良好临床实践(gcp)、起源于赫尔辛基宣言(declaration of helsinki)的道德原则以及所有其他适用的法律、规则和法规,进行了一项旨在评估化合物1在健康受试者中的安全性、耐受性和药代动力学(pk)的1期、单中心、随机化、双盲、安慰剂对照的单次递增剂量(sad)研究。
[0132]
在每个剂量队列内,将受试者以3:1的比率(6个活性和2个安慰剂)随机化来接受化合物1或安慰剂。筛选后,使受试者接受单剂量的研究药物,并在诊所期(in
‑
clinic period)期间和门诊随访期期间进行监测。在研究的第2天至第7天,将受试者限制在研究地点,以收集pk和安全性评估结果。在研究的第7天从研究地点离开后,使受试者在研究的第14、21、28和35天返回研究地点。
[0133]
结果。共有36位受试者接受了化合物1(剂量范围为1至40mg),并且有12位受试者接受了安慰剂。在最初随机化的48位受试者中,有三位因行政原因而中止。剂量递增审查委员会(dose escalation review committee)评估了每个队列的所有可用安全性和pk数据,并同意在每种情况下的剂量递增是适当的(计划最大剂量高达40mg)。
[0134]
在每个治疗组中,治疗紧急不良事件(teae)的总体分布具有可比性,其中29%(48位中的14位)的受试者经历一种或多种不良事件(ae)。身体系统发生ae的情况没有明显的差异,并且没有明确的剂量关系。化合物1受试者的ae率略低于对照(安慰剂)受试者的ae率。比起其他任何ae,更多受试者经历的头痛仅在一位化合物1受试者中观察到。没有严重的ae,并且没有中止或死亡。化合物1对实验室安全性评估(临床化学和血液学)没有明显影响。
[0135]
随着暴露量(auc和cmax)随剂量成比例增加,化合物1的pk似乎表现良好。观察到的pk支持每天一次施用化合物1。
[0136]
实施例5.化合物1在肥胖患者中的1期临床研究
[0137]
根据良好临床实践(gcp)、起源于赫尔辛基宣言的道德原则、美国联邦法规(us cfr)第21条第50部分(人类受试者的保护)和第56部分(机构审查委员会)和第312部分(研究性新药申请)和国际协调会议e6(良好临床实践指南)(ich e6 r2)以及所有其他适用的法律、规则和法规,进行了一项旨在评估化合物1在超重或肥胖但其他方面健康的受试者中的安全性、耐受性和药代动力学(pk)的1期、单中心、随机化、双盲、安慰剂对照的多次递增剂量(mad)研究。
[0138]
在每个递增剂量队列内以及总体上,将受试者以4:1的比率(每个队列8个活性和2个安慰剂)随机化来接受化合物1或安慰剂。在筛选和随机化后,受试者将每天接受研究药物治疗持续4周,其中在参与过程中进行部分诊所内限制。在研究的第2天至第7天,受试者将被限制在研究地点,以收集pk和安全性评估结果。在研究的第7天从研究地点离开后,受试者将在家中继续施用研究药物,并在研究的第11、14、18、21、23天(
±
1天)返回研究地点进行门诊研究访视。然后受试者将从研究的第27天到研究的第33天被限制,包括在研究的
第28天的最终剂量施用。随访访视将在研究的第42天(
±
2天)和第56天(
±
2天)进行。
[0139]
计划了五个递增剂量队列(1mg至20mg),每组10位受试者。除了确保起始剂量的可接受安全倍数外,剂量递增将不会进行至在28天重复剂量的狗毒理学研究中预计导致低于5倍安全倍数的人类暴露量到中枢神经系统(cns)血管周围炎/血管炎不良发现的noael暴露量的任何剂量(基于对来自先前队列的所有可用pk数据的分析)。
[0140]
队列将独立纳入,并按顺序进行,其中连续队列的给药至少间隔五周开始。至少包含首要研究员或副研究员和赞助方医学监查员的剂量递增审查委员会将以盲法方式评估来自每个队列的所有可用中期安全性和pk数据,以确定剂量递增的适当性,之后如有必要,可以对个别受试者治疗分配进行非盲评估以确认任何剂量限制性毒性(dlt)。如果同一队列内在接受活性药物的受试者中出现两例或更多例dlt,则可以停止剂量递增或可以测试中间剂量。如果由于dlt而停止了剂量递增,则最大耐受剂量(mtd)将由先前队列中的剂量来定义,除非随后通过相同算法测试了中间剂量。也可以基于对药物pk的审查(例如,如果连续剂量队列之间的暴露量没有增加)来停止剂量递增。值得注意的是,剂量递增将不会进行至/超过在28天重复剂量的狗毒理学研究中预计导致低于5倍安全倍数的暴露量到noael暴露量(发现cns血管周围炎/血管炎)的任何剂量(基于对所有可用先前pk的分析)。
[0141]
适用时,安全性和耐受性的解释(包括mtd)将基于所有可用的安全数据,包括ae/sae、体格检查结果、临床实验室参数、生命体征和心电图(ecg)的收集进行评估。
[0142]
表5中总结了研究参数。
[0143]
表5
[0144]
[0145][0146]
可以进行具有针对临床开发阶段的适当设计的额外临床试验,以测试化合物1在治疗nafld或nash患者中的功效。可以进行利用不同剂量水平的活性成分或用于区分最佳剂量或给药方案的进一步试验。此外,可以在以类似方式进行的额外临床试验中,确定药物在特定人群,诸如老年人、儿童或患有糖尿病或其他病理病状的患者中的功效。
[0147]
在本说明书中提及的所有出版物和专利申请均以引用的方式并入本文,其程度如同每个单独的出版物或专利申请具体地和单独地表示为以引用的方式并入。
[0148]
从前述内容应当理解,虽然本文出于说明的目的已经描述了本发明的具体实施方案,但可以在不脱离本发明的精神和范围的情况下进行各种修改。因此,本发明仅受所附权利要求书的限制。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。