1.本发明属于医药技术领域,涉及一种贝利司他药物共晶及其制备方法和应用。
背景技术:
2.近年来,药物共晶技术由于其独特的物理化学性质而倍受关注,是一种可以快速有效的改善药物活性药物成分的物化性质的新手段。药物活性分子含有的官能团,能够利用氢键或者其它非共价键(如氢键、范德华力、π-π堆积、卤素键等)作用而与其它有机分子结合在同一晶格生成共晶,从而有效改善药物本身的结晶性能、稳定性、溶解度、溶出度和生物利用度及药效,成为药物固体制剂的一个新选择。药物共晶为剂型设计提供了更多的选择,可延长原有药物的市场周期,在药物学和生物医学领域显示出了潜在的应用前景。
3.贝利司他属于bcsⅱ类药物,受到低水溶性(0.14mg/ml)的限制,目前市面上以无菌冻干粉针形式进行销售,用于静脉注射。fda推荐剂量为1000mg/m2,在21天周期的第1-5天通过静脉输注给药,每天一次,给药30分钟,每21天重复周期。
4.为了提高贝利司他的水溶性,增加患者依从性,开发了多种贝利司他口服制剂。cn107698632a公开了一种基于乙酸的贝利司他衍生物,利用乙酸取代末端羟肟酸官能团,但使用了属于一类和二类溶剂的四氢呋喃和吡啶,具有一定的毒性和致癌性,大量吸入会引起中毒,出现头痛、呕吐、造血功能受损,带来一定的安全隐患;wo2019/002614en开发了一种贝利司他无定形固体分散体,贝利司他与pvpk30、pvpk12、pvpk25、pvpk90、pvpva64及其混合物通过喷雾干燥形式进行制备,但无定形固体分散体本身不稳定,易转化为晶体,且喷雾干燥设备复杂;wo2018/020406 en公开了一种贝利司他多晶型制备方法,在贝利司他与丙酮溶剂中加入极性质子溶剂,并加入反溶剂进行分离、干燥,制备得到贝利司他丙酮溶剂化物。发明人根据专利方法进行制备,并未得到贝利司他溶剂化物,重复性差,操作复杂,不利于工业放大。
5.为了提高药物的质量,解决贝利司他在水溶性低、依从性差等问题,以贝利司他为药物活性组分,寻找和合适的共晶配体,可以有效的提高其溶解度和溶解速率。目前还没有关于贝利司他药物共晶方面的报道。
技术实现要素:
6.为了解决现有技术的不足,本发明的目的是提供一种贝利司他药物共晶及其制备方法和应用,将药物贝利司他与共晶配体异烟肼或异烟酰胺形成共晶,在不改变活性药物组份贝利司他的共价结构的同时,对药物活性成分的理化性质产生巨大影响,从而提高药物的生物利用度,增强药物疗效。
7.本发明目的之一在于提供一种贝利司他药物共晶,所述贝利司他药物共晶是由贝利司他药物和共晶配体按照摩尔比1:1结合而成,所述共晶配体为异烟肼或异烟酰胺。
8.当所述共晶配体为异烟肼,贝利司他药物共晶的分子式为c
21h21
n5o5s;
9.优选地,使用cu-kα辐射,贝利司他异烟肼共晶以衍射角2θ表示的x-射线粉末衍射
图在18.1
±
0.2
°
、18.5
±
0.2
°
、18.9
±
0.2
°
、19.2
±
0.2
°
、19.9
±
0.2
°
、22.2
±
0.2
°
、22.9
±
0.2
°
、24.0
±
0.2
°
、24.2
±
0.2
°
、24.5
±
0.2
°
处有特征衍射峰。
10.优选地,贝利司他异烟肼共晶为三斜晶系,空间群为晶胞参数为晶胞参数为α=93.416(1)
°
,β=100.770(1)
°
,γ=101.188(1)
°
;晶胞体积为z=2。
11.在本发明中,贝利司他异烟肼共晶(记为共晶1)基本结构单元由一个贝利司他分子和的吡啶环上的氮原子形成氢键,在方向形成一维的链状结构。
12.当所述共晶配体为异烟酰胺,贝利司他药物共晶(记为共晶2)的分子式为c
21h20
n4o5s;
13.优选地,所述共晶配体为异烟酰胺,贝利司他药物共晶为三斜晶系,空间群为晶胞参数为α=96.427(1)
°
,β=101.025(1)
°
,γ=99.660(1)
°
;晶胞体积为z=2。
14.本发明的目的之二在于提供一种贝利司他药物共晶的制备方法,所述制备方法包括研磨法或溶剂介导法。
15.当共晶配体为异烟肼,制备方法为研磨法;
16.优选地,所述研磨法包括:将摩尔比为1:(0.8-1.3)的贝利司他与异烟肼加入到球磨机中,加入少量有机溶剂,球磨,得到贝利司他异烟肼药物共晶。
17.优选地,所述球磨的时间为10-60min,球磨的频率为15-30hz;
18.优选地,所述有机溶剂为乙醇、异丙醇、丙酮或乙酸乙酯中的任意一种或至少两种的组合;
19.优选地,以贝利司他的添加量为1g计,所述有机溶剂的添加量为0-800μl。
20.当所述共晶配体为异烟酰胺,制备方法为溶剂介导法;
21.优选地,所述溶剂介导法包括:将贝利司他和异烟酰胺按照摩尔比为1:(0.8-1.2)混合,加入有机溶剂,25-40℃反应12-48h,得到贝利司他异烟酰胺共晶。
22.优选地,所述有机溶剂包括异丙醇、乙腈或乙酸乙酯中的任意一种或至少两种的组合;
23.优选地,以贝利司他的添加量为1g计,所述有机溶剂的添加量为10-60ml。
24.优选地,所述制备方法还包括将反应后得到的混合物依次进行固液分离以及干燥。
25.本发明的目的之三在于提供一种如目的之一所述的贝利司他药物共晶在制备靶向药物中的应用。
26.相对于现有技术,本发明具有以下有益效果:
27.本发明制备出来的共晶本发明中制备共晶的方法为溶剂常温慢挥发法和液相辅助研磨法,这两种方法操作简便易行,便于在工业制药中大量推广,成本低廉。
28.1、本发明采用研磨或溶剂介导方法制备两种贝利司他药物共晶,工艺简单、条件温和、重复性好、绿色环保,适合大规模生产。
29.2、本发明中的贝利司他药物共晶能够有效地修饰药物的活性成分,除了保持了贝
利司他本身的治疗特性外,其物理化学性质上如溶解性、稳定性和生物利用度都有了一定程度的改善。
附图说明
30.图1药物活性组分贝利司他、共晶前驱体异烟肼及本发明提供的贝利司他药物共晶1的粉末x-射线衍射图。
31.图2药物活性组分贝利司他、共晶配体异烟酰胺及本发明提供的贝利司他药物共晶2的粉末x-射线衍射图。
32.图3本发明提供的贝利司他药物共晶1的晶体结构图。
33.图4本发明提供的贝利司他药物共晶2的晶体结构图。
34.图5本发明提供的贝利司他药物共晶1的热分析图谱,包括差式扫描量热图(dsc)及热失重分析图(tga)。
35.图6本发明提供的贝利司他药物共晶2的热分析图谱,包括差式扫描量热图(dsc)及热失重分析图(tga)。
36.图7本发明提供的贝利司他药物共晶1储存180天后的稳定性实验图谱。
37.图8本发明提供的贝利司他药物共晶2储存180天后的稳定性实验图谱。
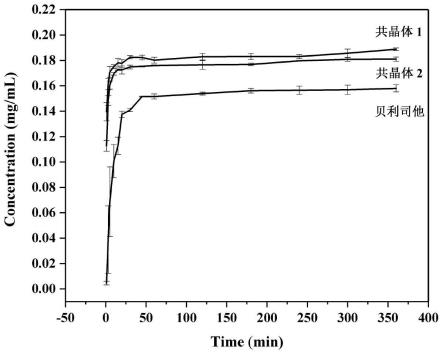
38.图9本发明提供的贝利司他药物共晶1、贝利司他药物共晶2、贝利司他在ph=6.8的磷酸盐缓冲溶液中的悬浮6h的粉末溶出曲线。
具体实施方式
39.应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅是帮助理解本发明,不应视为对本发明的具体限制。
40.检测仪器及方法
41.1.在环境温度下,pxrd图谱是使用粉末x-射线衍射仪rigaku d/max 2500(rigaku,日本)测量得到。测量条件如下:扫描范围为3.5-40
°
,扫描速率为8
°
/min,扫描步长为0.02
°
,发射靶为cu kα,波长为电压和电流分别为40kv和100ma。
42.2.将实施例中所得到的共晶单晶置于rigaku 007hf xtalab p200衍射仪中,选用mo kα为射线采集。晶体结构采用olex2直接法求解结构,采用shelxl-97程序包对f2进行全矩阵最小二乘优化,判别原子种类。
43.3.差式扫描量热(dsc)实验使用mettler toledo dsc 1/500进行测试,5-10mg药品放置在标准铝盘中以10℃/min的加热速率在氮气下加热。
44.4.tg分析采用mettler tga/dsc 1stare系统进行测试。称取5-10mg本发明所述方法制备得到的共晶,置于标准氧化铝坩埚中,在20ml/min氮气流量下,以10℃/min的升温速率扫描。
45.5.液相色谱柱:agilent extend c18色谱柱,4.6mm
×
250mm,5μm;柱温:30℃;流动相:0.1%磷酸水溶液和乙腈(60:40%v/v);流速:1ml/min;进样量:10μl;波长:272nm。
46.实施例1
47.称取31.8mg贝利司他固体与13.7mg异烟肼固体置于球磨罐中,加入20μl乙醇,以15hz的频率球磨10min,得到的产品室温干燥后为贝利司他药物共晶1。
48.实施例2
49.称取96.0mg贝利司他固体与41.1mg异烟肼固体置于球磨罐中,加入20μl乙酸乙酯,以25hz的频率球磨15min,得到贝利司他药物共晶1。
50.实施例3
51.称取0.199g贝利司他固体与0.0822g异烟肼固体放入球磨罐中,加入100μl异丙醇,以30hz的频率球磨20min,室温干燥后,得到贝利司他-药物共晶1的固体样品
52.实施例4
53.30℃,将31.8mg的贝利司他固体、12.2mg的异烟酰胺122.13固体、1ml乙腈加入到反应瓶中,搅拌48h,将所得悬浮液分离,在室温下干燥12h,所得固体就是贝利司他药物共晶2。
54.实施例5
55.40℃,将0.2786g的贝利司他固体、0.1024g的异烟酰胺固体、9ml乙酸乙酯加入到反应瓶中,搅拌48h,将所得悬浮液分离,在室温下干燥12h,所得固体就是贝利司他药物共晶2。
56.实施例6
57.30℃,将0.318g的贝利司他固体、0.122g的异烟酰胺固体、12ml异丙醇加入到反应瓶中,搅拌48h,将所得悬浮液分离,在室温下干燥24h,所得固体就是贝利司他药物共晶2。
58.以上所述实施例中,药物活性组分贝利司他,共晶前驱体异烟肼、贝利司他药物共晶1的粉末x射线衍射图谱分别如图1、图2所示。从图1中可以看到,贝利司他药物共晶1的x射线粉末衍射在衍射角度6.0
°
、11.0
°
、12.0
°
、12.5
°
、13.6
°
、15.0
°
、16.7
°
、18.1
°
、18.5
°
、18.9
°
、19.2
°
、19.9
°
、21.6
°
、22.2
°
、22.6
°
、22.9
°
、24.0
°
、24.2
°
、24.5
°
、25.6
°
、26.6
°
有特征峰,不同于原料药贝利司他及共晶前驱体异烟肼的特征峰的叠加,说明制备得到的物质可能是贝利司他药物共晶1。同样,贝利司他药物共晶2的x射线粉末衍射图谱如图2所示,在衍射角度12.3
°
、12.9
°
、13.59
°
、14.9
°
、16.1
°
、16.5
°
、18.1
°
、19.0
°
、19.3
°
、19.9
°
、22.1
°
、23.8
°
、24.5
°
、24.9
°
、25.3
°
、27.1
°
有特征衍射峰,不同于原料药贝利司他及共晶前驱体异烟酰胺的特征峰的叠加,说明制备得到的物质可能是贝利司他药物共晶2。
59.对贝利司他药物共晶1、贝利司他药物共晶2的晶体学结构(见图3、图4)进行分析,其晶体学参数如表1所示:
60.表1贝利司他药物共晶的结晶学参数
[0061][0062][0063]
本发明实施例中所制备的共晶通过差热扫描量热仪(dsc)进一步分析,如图5所示,贝利司他药物共晶1在161℃出现了尖锐的单峰,且不同于贝利司他172℃的熔点和异烟肼的170℃熔点,进一步证明了贝利司药物共晶1的形成;而图6,贝利司他药物共晶2在151℃出现了尖锐的单峰,同样证明了贝利司他药物共晶2的形成。
[0064]
对本发明实施例制备得到的共晶进行加速稳定性测试。共晶样品被称重并储存在40℃环境温度、75%相对湿度(rh),充满饱和氯化钠溶液的干燥器中,在180天后对样品进行称重并测定pxrd图谱。贝利司他药物共晶1的稳定性pxrd图谱如图7所示、贝利司他-药物共晶2的稳定性pxrd图谱如图8所示,二者的xrd图谱并没有明显变化,证明贝利司他药物共晶1、贝利司他药物共晶2均未发生转变,稳定性较好。
[0065]
采用浆式法测定贝利司他、贝利司他药物共晶1、贝利司他药物共晶2的溶出度。将含相同贝利司他摩尔量的原料药及共晶,研磨并过80和160目筛后分别加入到溶出仪中,以100rpm的桨速在37℃条件下进行。分别在1、3、5、10、15、20、30、45、60、120、180、240、300和360min时从容器中取出2ml溶液并通过0.45mm尼龙过滤器过滤,之后用高效液相色谱法测定溶液浓度并用标准曲线分析。
[0066]
共晶在体外缓冲溶液溶出度测试结果如图9所示,结果显示在ph=6.8的磷酸盐缓冲溶液中,两种共晶的溶解度相对于原料药提高了1.14-1.17倍。通过药物共晶技术提高原
料药水溶性,有望成为制备贝利司他固体口服制剂的突破口。
[0067]
申请人声明,以上所述仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,所属技术领域的技术人员应该明了,任何属于本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,均落在本发明的保护范围和公开范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。