1.本发明属于绿色化学制药领域,具体涉及一种制备他唑巴坦二苯甲酯的合成方法。
背景技术:
2.他唑巴坦(tazobactam),化学名为[2s-(2α,2β,5α)-3-甲基-7-氧代-3-(1h-1,2,3-三唑-1-甲基)-4-硫杂-1-氮杂双环-[3,2,0]-庚烷-2-羧酸-4,4-二氧化物。该药物是由日本大鹏制药公司开发的新型青霉烷砜类β-内酰胺酶抑制剂,于1992年以其复方药他唑巴坦/哌拉西林(1:8)在法国首次上市,用于治疗多种细菌感染。他唑巴坦是目前公认为抗菌谱最广、抗性最好、最有前途的β内酰胺酶抑制剂,具有抑酶活性强、稳定性高、活性低、毒性低等特点。化合物他唑巴坦二苯甲酯是合成他唑巴坦的上游产物,由于其合成收率很低,极大的限制了他唑巴坦生产工艺,所以他唑巴坦二苯甲酯的合成工艺改进空间很大。
[0003]
目前文献报道他唑巴坦二苯甲酯的合成方法有如下几种方法:
[0004]
文献报道有关他唑巴坦合成工艺按照三氮唑环引入方式的不同,主要有两种制备方法:叠氮化合物环加成合成三氮唑五元环和直接加侧链三氮唑环。
[0005]
曹琳等公开了一种制备他唑巴坦二苯甲酯的方法,其路线如下:
[0006][0007]
叠氮化合物环加成制备法,是以青霉烷酸二苯甲酯亚砜为原料,经过热裂解开环、氯化环合、取代上叠氮基、氧化、环合加成制得他唑巴坦二苯甲酯。虽然每个步骤都较简单,目前市场上大部分生产厂家也仍采用此路线,但是使用乙炔环合加成反应需要高温高压的条件,而且使用了高危险性的叠氮化钠容易造成安全事故,同时叠氮化反应中有大量六元环副产物生成难以分离,总收率较低,生产成本高。
[0008]
邓勇等人(邓勇,沈怡,钟裕国,唐维高.β-内酰胺酶抑制剂—他唑巴坦酸的合成[j].中国药物化学杂志,2001.)报道了一种制备他唑巴坦二苯甲酯的制备方法。
[0009][0010]
直接上三氮唑侧链是目前研究合成他唑巴坦的热点;但使用氯化硫酰做氯代试剂,后处理产生大量废酸废气,增加了后处理成本,对环境污染大;使用氯代物碱性条件直接与三氮唑反应,虽然也有少许产物生成,但得到的几乎全为六元环杂质。
[0011]
为了避免生成六元环这一副产物,cn102020663a公开了一他唑巴坦二苯甲酯亚砜不经热裂解开环,直接与2-三甲基硅基-1,2,3-三唑反应,上三氮唑,其路线如下:
[0012][0013]
这一方法反应步骤最短,但反应条件十分苛刻,要求反应体系严格无水,产物需要用到苯做洗脱剂来柱层析分离,使用苯对操作者造成了极大伤害;制备2-三甲基硅基-1,2,3-三唑时也需要严格无水,反应困难,需要高温加压,对设备要求高,且三甲基硅基-1,2,3-三唑贮存困难,故无法应用于工业化生产。
[0014]
此外,还有文献提出以3-甲基-[2-氧代-4-(2-苯并噻唑二硫基)-1-氮杂环丁烷基]-3-丁烯酸二苯甲酯与三氮唑银进行关键的c-n偶联反应时,由于反应过程中会生成不溶于反应溶剂的副产物噻唑二聚体,它会附着在三氮唑银固体颗粒表面,阻碍三氮唑银与其他原料接触,因此由于原料很难完全参与反应,使得反应难以顺利进行,导致收率低。而且该反应还需使用易升华的碘单质,当放大反应后,容易对操作者身体健康带来危害,存在非常严重的安全隐患。
技术实现要素:
[0015]
本发明目的在于提供一种生产安全操作,生产成本低,生产周期短,环境污染小的制备他唑巴坦二苯甲酯的方法。
[0016]
具体地,本发明提供了一种一步法制备他唑巴坦二苯甲酯的绿色生产工艺,其特征在于:以3-甲基-[2-氧代-4-(2-苯并噻唑二硫基)-1-氮杂环丁烷基]-3-丁烯酸二苯甲酯为原料,连续地经溴化关环反应、与三氮唑银发生亲核取代反应和氧化反应获得他唑巴坦二苯甲酯;
[0017]
其中,所述3-甲基-[2-氧代-4-(2-苯并噻唑二硫基)-1-氮杂环丁烷基]-3-丁烯酸二苯甲酯为如下结构所示的化合物:
[0018][0019]
具体反应方程式如下所示:
[0020][0021]
进一步地,本发明提供的一种一步法制备他唑巴坦二苯甲酯的绿色生产工艺,其特征还在于:
[0022]
所述溴化关环反应的反应产物为:2β-溴代甲基-2-甲基-6,6-二氢青霉烷酸二苯甲酯;
[0023]
所述2β-溴代甲基-2-甲基-6,6-二氢青霉烷酸二苯甲酯为如下结构所示的化合物:
[0024][0025]
进一步地,本发明提供的一种一步法制备他唑巴坦二苯甲酯的绿色生产工艺,其特征还在于:
[0026]
所述亲核取代反应的反应产物为:2α-甲基-2β-三唑代甲基-2-甲基-6,6-二氢青霉烷酸二苯甲酯:
[0027]
所述2α-甲基-2β-三唑代甲基-2-甲基-6,6-二氢青霉烷酸二苯甲酯为如下结构所示的化合物:
[0028][0029]
进一步地,本发明提供的一种一步法制备他唑巴坦二苯甲酯的绿色生产工艺,其特征还在于:
[0030]
所述溴化关环反应,以3-甲基-[2-氧代-4-(2-苯并噻唑二硫基)-1-氮杂环丁烷基]-3-丁烯酸二苯甲酯为原料,在氢溴酸,亚硝酸钠和相转移催化剂存在下进行,
[0031]
其中,3-甲基-[2-氧代-4-(2-苯并噻唑二硫基)-1-氮杂环丁烷基]-3-丁烯酸二苯
甲酯:氢溴酸:亚硝酸钠:相转移催化剂的摩尔比为1:5-10:1-2:0.02-0.1;
[0032]
所述溴化关环反应的反应温度设为-10
‑‑
5℃;
[0033]
所述溴化关环反应的反应时间为0.5-2小时。
[0034]
在该溴化关环反应过程中,实际还可采用溴化铜作为溴剂,但是,溴化铜相对来说成本较高,且会产生重金属残留。
[0035]
进一步地,本发明提供的一种一步法制备他唑巴坦二苯甲酯的绿色生产工艺,其特征还在于:
[0036]
所亲核取代反应中2β-溴代甲基-2-甲基-6,6-二氢青霉烷酸二苯甲酯为原料与三氮唑银的摩尔比设置为1:1-2;
[0037]
所述亲核取代反应的反应温度为20-25℃;
[0038]
所述亲核取代反应的反应时间为1-5小时。
[0039]
在本发明中,该亲核取代反应采用sn2机理,能够避免使用碘,从而提高了生产安全度,且由于机理上的差异相较于传统反应过程能够避免二聚体的产生,从而提高转化率,提高反应效率,降低原料成本。
[0040]
进一步地,本发明提供的一种一步法制备他唑巴坦二苯甲酯的绿色生产工艺,其特征还在于:
[0041]
所述氧化反应,以2α-甲基-2β-三唑代甲基-2-甲基-6,6-二氢青霉烷酸二苯甲酯为原料,经高锰酸钾,乙酸氧化得到他唑巴坦二苯甲酯;
[0042]
其中,2α-甲基-2β-三唑代甲基-2-甲基-6,6-二氢青霉烷酸二苯甲酯:高锰酸钾:乙酸的摩尔比为1:1.5-2:15-20;
[0043]
所述氧化反应的反应温度为20-25℃;
[0044]
所述氧化反应的反应时间为1-5小时。
[0045]
进一步地,本发明提供的一种一步法制备他唑巴坦二苯甲酯的绿色生产工艺,其特征还在于:
[0046]
所述他唑巴坦二苯甲酯的纯化方法如下所示:
[0047]
s1.反应完成后,于0-5℃降温搅拌5-20分钟;
[0048]
s2.滴加双氧水至反应液的紫黑色刚好褪去;
[0049]
s3.保温搅拌0.2-1小时后过滤;
[0050]
s4.滤饼采用醇与酮的混合溶剂于70-75℃搅拌打浆0.5-2小时后,再降温至室温;
[0051]
s5.经过滤,洗涤,干燥,得纯化产物。。
[0052]
此外,本发明还提供了一种一步法制备他唑巴坦二苯甲酯的绿色生产工艺,其特征在于:
[0053]
以青霉烷酸二苯甲酯亚砜为原料,连续地经开环反应,溴化关环反应、与三氮唑银发生亲核取代反应和氧化反应获得他唑巴坦二苯甲酯;
[0054]
其中,所述青霉烷酸二苯甲酯亚砜为如下结构所示的化合物:
[0055][0056]
反应路线如下:
[0057][0058]
上述一步法制备他唑巴坦二苯甲酯的绿色生产工艺,其特征还在于:
[0059]
所述溴化关环反应同上;
[0060]
所述三氮唑银发生亲核取代反应同上。
[0061]
上述一步法制备他唑巴坦二苯甲酯的绿色生产工艺,其特征还在于:
[0062]
所述开环反应为采用青霉烷酸二苯甲酯亚砜为原料与2-巯基苯并噻唑在回流分水的反应条件下制得3-甲基-[2-氧代-4-(2-苯并噻唑二硫基)-1-氮杂环丁烷基]-3-丁烯酸二苯甲酯;
[0063]
其中,所述青霉烷酸二苯甲酯亚砜与2-巯基苯并噻唑的摩尔比为1:0.95-1.5。
[0064]
在本发明中该四步反应是通过一步法工艺完成的,一步法工艺可以大大缩短生产周期,提高生产效率。而且操作简单,易于控制。
附图说明
[0065]
图1.产品质谱;
[0066]
图2.产品氢谱;
[0067]
图3.副产物质谱;
[0068]
图4.副产物氢谱。
具体实施方式
[0069]
下面通过具体实施案例进一步说明本发明,但本发明并不仅限于下面实例范围之中。下列实例中未具体注明条件的试验方法,按照常规方法和条件,或按照商品说明书选择。
[0070]
在本实施例中,具体采用如下反应过程进行:
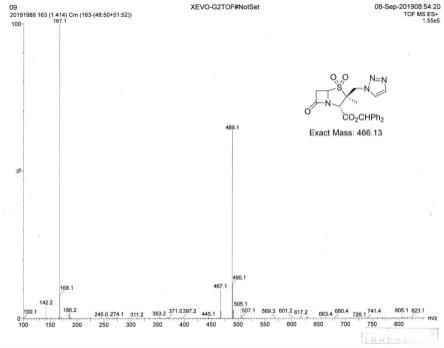
n,开始滴加30%双氧水,滴至反应液的紫黑色刚好褪去,保温搅拌0.5h,过滤,滤饼加300ml甲醇与丙酮混合(比例为1:1,1:2,1:3等)合溶剂于70-75℃搅拌打浆1h,再降温至室温,过滤,洗涤,干燥,得25.20g白色固体粉末即为他唑巴坦二苯甲酯,四步反应收率69.02%,hplc纯度95.03%(归一法),熔点202-204℃,es i-ms(m/z):489.1(m na)
。(见附录-图1)
[0083]1h nmr(400mhz,dmso-d6)δ(ppm):7.96(s,1h,-ch=ch),7.77(s,1h,-ch=ch),7.27
–
7.55(m,10h,-ph),6.99(s,1h,-chph2),5.22
–
5.36(m,3h,-chco2,-ch2n3,-chs),4.96(d,j=15.4hz,1h,-ch2n3),3.77(dd,j=16.6,4.5hz,1h,-ch2con),3.34
–
3.39(m,1h,-ch2con),1.12(s,3h,-ch3).(见附录-图2)
[0084]
后处理母液中分离出白色泡沫物2.56g,es i-ms(m/z):467.1(m h)
。(见附录-图3)
[0085]1h nmr(600mhz,cdc l 3
)δ(ppm):7.64(s,2h,-ch=ch),7.30
–
7.39(m,10h,-ph),6.93(s,1h,-chph2),5.12(d,j=14.6hz,1h,-ch2s),5.09(s,1h,-chco2),4.95(d,j=14.6hz,1h,-ch2s),4.62(dd,j=4.1,2.1hz,1h,-chs),3.46
–
3.59(m,2h,-ch2con),1.05(s,3h,-ch3).(见附录-图4)
[0086]
由他唑巴坦二苯甲酯(5)的质谱和氢谱,推测出该白色固体结构为六元环异构体杂质。
[0087]
实施例2
[0088]
(1)向1000ml三口瓶中加580ml甲苯,将反应瓶置于油浴锅中接分水器,升温回流,大约馏出80ml甲苯后,将青霉烷酸二苯甲酯亚砜(50g,91%)和2-巯基苯并噻唑(21.6g)加入到反应瓶中,继续回流分水反应1h,累积馏出液体100ml左右,降至室温,得到含热裂解开环物的甲苯溶液直接用于下步反应。
[0089]
(2)称取上述热裂解开环物的甲苯溶液投入2000ml三口瓶中加入600ml甲苯,-10℃下搅拌,开始滴加20%的氢溴酸水溶液(528g)滴毕,直接加入四丁基溴化铵(2.1g,),滴加2mo l/l nano2(33.5g)水溶液,滴毕,反应液棕黑色有泡沫,-10至-5℃保温反应1h左右,过滤,洗涤,干燥得到含溴化合的甲苯溶液直接用于下步反应。
[0090]
(3)将(2)中含溴化合物的甲苯料液置于1000ml三口瓶中置于20℃搅拌,一次性加入三氮银(24.6g),搅拌4h,原料基本反应完全,过滤,洗涤,减压蒸馏除溶剂得75g黄色油状物即为-甲基-2β-三唑代甲基-2-甲基-6,6-二氢青霉烷酸二苯甲酯直接用于下步反应。
[0091]
(4)将上步黄色油状物用750ml丙酮溶解,转入2000ml的反应瓶中,于0-5℃降温搅拌10mi n,加入600ml冰水,依次加入冰乙酸(152g),分批加入高锰酸钾(41.4g),用时20mi n(分4批),加毕。之后转移至20-25℃下反应。3h左右反应完毕。于0-5℃降温搅拌10mi n,开始滴加30%双氧水,滴至反应液的紫黑色刚好褪去,保温搅拌0.5h,过滤,滤饼加460ml甲醇与丙酮(1:2)混合溶剂于70-75℃搅拌打浆1h,再降温至室温,过滤,洗涤,干燥,得41.78g白色固体粉末即为他唑巴坦二苯甲酯,收率68.75%,hplc纯度95.15%(归一法),熔点202-204℃(文献值=202-205℃)。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。