growth reaction and continuous production process,journalofthe european ceramic society,1994,14:205-214)。
技术实现要素:
6.为了克服上述现有技术上的缺点与不足,本发明公开了一种超高纯度、均一的球形纳米氧化硅颗粒的制备方法,和传统的工艺的正加工艺不同,本发明利用有机硅源和溶剂水相体系相溶解的原理,提出了一种通过反加工艺的方法制备球形纳米氧化硅。本制备方法可以实现球形纳米氧化硅颗粒尺度为30~150nm之间可控制备,而且制备的纳米颗粒分布窄、均一性高、pdi《0.1,球形度高等特点。此外,本发明制备得到的纳米氧化硅的质量分数高于传统正加工艺制备方法,通过调节反加量的控制,可以实现制备的氧化硅浓度高的产品(此工艺的氧化硅浓度可以达到7~15wt.%,此数据远远的高于传统的方法得到的氧化硅浓度(小于6wt.%),且制备的超高纯度纳米氧化硅的金属杂质离子小于1ppm。
7.根据本技术的一个方面,提供了一种纳米氧化硅颗粒的制备方法,该方法为将水相溶液加入含有有机硅源的原料中,加热反应,得到所述的纳米氧化硅颗粒,所述水相溶液选自水、含有ph调节剂的水溶液中的一种。
8.可选地,所述有机硅源选自式i中的至少一种;
[0009][0010]
其中r1,r2,r3独立选自c
1-4
烷氧基、疏水基团、亲水基团、含有硅的基团;
[0011]
r4选自c
1-4
烷基中的至少一种;包括甲基、乙基、丙基、丁基;
[0012]c1-4
烷氧基包括甲氧基、乙氧基、丙氧基、丁氧基;
[0013]
所述疏水基团选自c
1-3
烷基、苯基、c
2-3
环氧基中的一种;包括甲基、乙基、丙基、苯基、c
2-3
环氧基;
[0014]
所述亲水基团选自氨丙基、巯丙基、氯丙基中的至少一种;
[0015]
所述含有硅的基团选自硅基三甲氧基、硅基三乙氧基、硅基三丙氧基中的一种。
[0016]
可选地,所述含有有机硅源的原料包括有机碱i、有机溶剂;
[0017]
所述有机硅源、有机碱i、有机溶剂的摩尔比:
[0018]
有机硅源:有机碱i:有机溶剂=1:0~5:0~5,其中有机硅源的摩尔数以氧化硅的摩尔数计;
[0019]
所述有机碱i选自单乙醇胺、二乙醇胺、三乙胺、二乙胺、吡啶、异丙醇胺、四乙基氢氧化胺、三乙烯二胺、吗啉中的至少一种;
[0020]
所述有机溶剂选自甲醇、乙醇、丙醇、异丙醇、苯、甲苯、乙苯、二甲苯、芳烃、环己烷中的至少一种。
[0021]
可选地,所述水相溶液ph值为7~12。
[0022]
可选地,所述反应温度为55~95℃。
[0023]
可选地,所述水相溶液中水的摩尔量与所述有机硅源的摩尔量之比为4~100:1;其中其中有机硅源的摩尔量以氧化硅的摩尔量计。
[0024]
可选地,水相溶液的加入需要控制一定的流量,所述水相溶液的加入速率的为0.2~50kg/(kg反应液体
×
hr);即为单位质量反应液体、每小时加入水相的质量为0.2~50kg;
[0025]
其中所述反应液体为水相溶液和含有有机硅源的原料的混合液体;
[0026]
可选地,所述ph调节剂选自无机碱、有机碱ii中的至少一种;
[0027]
所述无机碱选自氨水、氢氧化钠、氢氧化钾、氢氧化镁、氢氧化钙中的一种或者几种;
[0028]
所述有机碱ii选自单乙醇胺、二乙醇胺、三乙胺、二乙胺、异丙醇胺、四乙基氢氧化胺、三乙烯二胺、吗啉中的一种或者几种;
[0029]
可选地,所述方法中还包括陈化;
[0030]
所述陈化温度为55~95℃;陈化时间为0.5~4hr;
[0031]
优选地,所述陈化温度为65~95℃;陈化时间为0.5~2hr。
[0032]
根据本技术的又一个方面,提供一种纳米氧化硅颗粒,所述氧化硅颗粒由所述的方法制备获得。
[0033]
可选地,所述纳米氧化硅颗粒微观形貌为球形,颗粒粒径为30~150nm;单分散,颗粒的集中度pdi《0.1;
[0034]
优选地,所述纳米氧化硅颗粒的颗粒粒径可独立选自30nm、35nm、40nm、50nm、58nm、60nm、65nm、70nm、80nm、83nm、85nm、90nm、100nm。
[0035]
可选地,所述纳米氧化硅颗粒的固含量为7wt.%~20wt.%,
[0036]
优选地,固含量范围为7wt.%~15wt.%。
[0037]
可选地,所述纳米氧化硅颗粒中金属离子含量小于1ppm;
[0038]
优选地,所述纳米氧化硅颗粒中金属离子含量小于600ppb。
[0039]
本技术能产生的有益效果包括:
[0040]
1)本发明公开了一种超高纯度、均一的球形纳米氧化硅颗粒的制备方法,和传统的工艺的正加工艺(向溶液中加入硅源的方法)不同,本发明利用硅烷反应体系的相溶解的原理,提出了一种通过反加工艺,通过向硅源中加入水相溶液的方法进行制备的方法制备球形纳米氧化硅。本制备方法可以实现球形纳米氧化硅颗粒尺度为30~150nm之间可控制备,而且制备的纳米颗粒分布窄、均一性高、pdi《0.1,球形度高、纯度高等特点。
[0041]
2)由于采用了反加工艺,本发明制备得到的纳米氧化硅的质量分数高于传统正加工艺制备方法,通过调节水相溶液的反加量的控制,可以实现制备的氧化硅浓度高的产品(此工艺的氧化硅浓度可以达到7~15wt.%,此数据远远的高于传统的方法得到的氧化硅浓度(小于6wt.%)。
[0042]
3.本专利发明的制备方法工艺操作简单,易于工业化放大。
附图说明
[0043]
图1是实施例1制备的纳米二氧化硅的粒度分布检测图。
[0044]
图2是实施例2制备的纳米二氧化硅的粒度分布检测图。
[0045]
图3是实施例3制备的纳米二氧化硅的粒度分布检测图。
[0046]
图4是实施例4制备的纳米二氧化硅的粒度分布检测图。
[0047]
图5是实施例5制备的纳米二氧化硅的粒度分布检测图。
[0048]
图6是实施例6制备的纳米二氧化硅的粒度分布检测图。
[0049]
图7是对比例1制备的纳米二氧化硅的粒度分布检测图。
[0050]
图8是对比例2制备的纳米二氧化硅的粒度分布检测图。
[0051]
图9是实施例1制备的纳米二氧化硅的sem表征图。
[0052]
图10是实施例2制备的纳米二氧化硅的sem表征图。
[0053]
图11是实施例3制备的纳米二氧化硅的sem表征图。
[0054]
图12是实施例4制备的纳米二氧化硅的sem表征图。
[0055]
图13是实施例5制备的纳米二氧化硅的sem表征图。
[0056]
图14是实施例6制备的纳米二氧化硅的sem表征图。
[0057]
图15是对比例1制备的纳米二氧化硅的sem表征图。
[0058]
图16是对比例2制备的纳米二氧化硅的sem表征图。
具体实施方式
[0059]
下面结合附图和实施例子对本发明的具体实施方式做进一步详细的说明,但这些实施例并不对本发明构成限制。本领域技术人员在本发明原则范围内可对本发明技术方案的细节和形式进行修改,但这些修改均落入本发明的保护范围内。
[0060]
本发明实施例中的粒度数据是采用纳米马尔文激光粒度分析和电子扫描电镜检测得到的结果。
[0061]
如无特别说明,本技术的实施例中的原料均通过商业途径购买。
[0062]
其中,有机硅源试剂(正硅烷甲酯、正硅烷乙酯等)、醇类溶剂、四甲基氢氧化铵、乙醇胺等原料均购买于国药。
[0063]
其中:
[0064]
固含量的计算的方法是:
[0065]
固含量=干燥得到的固体颗粒质量m/(溶液的取样质量)。
[0066]
实施例1
[0067]
1、分别将22.5g正硅烷甲酯、5g的甲醇混合形成均一的溶液,然后加热到75℃时搅拌待用,形成的溶液a;
[0068]
2、将108g的去离子放入烧杯中,然后加入25%的四甲基氢氧化铵溶液,调节溶液的ph为10.5,搅拌均匀后待用,形成的溶液b;
[0069]
3、将上述的溶液b以10g/(kg反应液*hr)的速率加入到溶液a中,保持反应液的温度为75℃;
[0070]
4、将步骤3形成溶液在75℃的情况下,陈化1小时。
[0071]
所得的样品标记为1
#
,所得产品用马尔文激光粒度仪分析粒度(测试仪器为marlven nanozs90),其平均粒径为59.31nm,集中度pdi值为0.008,其结果见图1。
[0072]
实施例2
[0073]
1、分别将22.5g正硅烷甲酯、10g的甲苯混合形成均一的溶液,然后加热到75℃时搅拌待用,形成的溶液a;
[0074]
2、将108g的去离子放入烧杯中,然后加入25%的四甲基氢氧化铵溶液,调节溶液的ph为10.5;搅拌均匀后待用形成溶液b;
[0075]
3、将上述的溶液b以10g/(kg反应液*hr)的速率加入到溶液a中,保持反应液的温度为75℃;
[0076]
4、将步骤3形成溶液在75℃的情况下,陈化1小时。
[0077]
所得的样品标记为2
#
,所得产品用马尔文激光粒度仪分析粒度,其平均粒径为52.56nm,集中度pdi值为0.028,其结果见图2。
[0078]
实施例3
[0079]
1、分别将22.5g正硅烷甲酯、10g的甲苯混合形成均一的溶液,然后加热到75℃时搅拌待用,标记为溶液a;
[0080]
2、将56g的去离子放入烧杯中,然后加入25%的四甲基氢氧化铵溶液,调节溶液的ph为10.5;搅拌均匀后待用形成溶液b;
[0081]
3、将上述的溶液b以10g/(kg反应液*hr)的速率加入到溶液a中,保持反应液的温度为75℃;
[0082]
4、将步骤3形成溶液在75℃的情况下,陈化1小时。
[0083]
所得产品标记为编号3
#
,用马尔文激光粒度仪分析粒度,其平均粒径为87.08nm,pdi值为0.005,其结果见于图3。
[0084]
实施例4
[0085]
1:分别将22.5g正硅烷甲酯、10g的甲苯混合形成均一的溶液,然后加热到75℃时搅拌待用;形成的溶液a;
[0086]
2、将108g的去离子放入烧杯中,然后加入25%的四甲基氢氧化铵溶液,调节溶液的ph为10.5;搅拌均匀后待用形成溶液b;
[0087]
3、将上述的溶液b以5.0g/(kg反应液*hr)的速率加入到溶液a中,保持反应液的温度为75℃;
[0088]
4、将步骤3形成溶液在75℃的情况下,陈化1小时。
[0089]
所得的样品标记为4
#
,所得产品用马尔文激光粒度仪分析粒度,其平均粒径为83.12nm,集中度pdi值为0.03,其结果见图4。
[0090]
实施例5
[0091]
1:分别将22.5g正硅烷甲酯、10g的甲苯混合形成均一的溶液,然后加热到85℃时搅拌待用;形成的溶液a;
[0092]
2、将108g的去离子放入烧杯中,然后加入25%的四甲基氢氧化铵溶液,调节溶液的ph为10.5;搅拌均匀后待用形成溶液b;
[0093]
3、将上述的溶液b以10g/(kg反应液*hr)的速率加入到溶液a中,保持反应液的温度为85℃;
[0094]
4、将步骤3形成溶液在85℃的情况下,陈化1小时。
[0095]
所得的样品标记为5
#
,所得产品用马尔文激光粒度仪分析粒度,其平均粒径为83.32nm,集中度pdi值为0.016,其结果见图5。
[0096]
实施例6
[0097]
1:分别将22.5g正硅烷甲酯、6g的三乙胺和10g的甲苯混合形成均一的溶液,然后
加热到75℃时搅拌待用;形成溶液a;
[0098]
2、将108g的去离子放入烧杯中,形成溶液b;
[0099]
3、将上述的溶液b以10g/(kg反应液*hr)的速率加入到溶液a中,保持反应液的温度为75℃;
[0100]
4、将步骤3形成溶液在75℃的情况下,陈化1小时。
[0101]
所得的样品标记为6
#
,所得产品用马尔文激光粒度仪分析粒度,其平均粒径为122.9nm,集中度pdi值为0.028,其结果见图6。
[0102]
对比例1
[0103]
本对比例主要是采用的工艺进行合成纳米氧化硅,具体的步骤如下:
[0104]
1、将0.41g的浓氨水加入到71.6g的去离子水中搅拌均匀,然后加入23g乙醇进行搅拌得到混合溶液a;
[0105]
2、将17.9g的正硅酸乙酯和23g乙醇混合形成溶液b;
[0106]
3、将溶液a加热到60℃,然后将溶液b以1ml/min的速度加入到a溶液中;
[0107]
4、然后陈化,得到的纳米粒子标记为对比样品1
#
。
[0108]
对比例2
[0109]
本对比例主要是采用的工艺进行合成纳米氧化硅,具体的步骤如下:
[0110]
1、将0.41g的浓氨水加入到71.6g的去离子水中搅拌均匀,搅拌得到混合溶液a;
[0111]
2、将溶液a加热到60℃,然后将17.9g的正硅酸乙酯以1ml/min的速度加入到a溶液中;
[0112]
4、然后陈化,得到的纳米粒子标记为对比样品2
#
。
[0113]
测试例1
[0114]
分别对样品1
#
~6
#
和对比样品1
#
,2
#
进行形貌测试,测试仪器为jeol jsm-7800f scanning electron microscope(sem)。
[0115]
测试结果显示,样品1
#
~6
#
的粒径大小分别为60nm、52nm、85nm、83nm、85nm和125nm。
[0116]
测试例2
[0117]
对样品2
#
,3
#
,对比样品1
#
,2
#
分别进行干燥,干燥的方法是采用实施例2,3,的水溶液硅胶样品2
#
、3
#
,对比例1,2的水溶液硅胶样品对比样品1
#
,2
#
,取样品称量20g,然后在120℃下进行干燥处理12小时,然后称量得到的固体颗粒的质量m。
[0118]
通过上述的干燥方法,实施例2,3得到的氧化硅的固含量分别为7.2wt.%和12.4wt.%;对比例1,2得到的氧化硅的固含量分别为5.7wt.%和7.2wt%。
[0119]
测试例3
[0120]
分别对样品水溶液硅溶胶样品进行金属离子的检测,测试仪器为icp-ms(ns2000)。
[0121]
对样品3
#
的检测结果如下所示:
[0122]
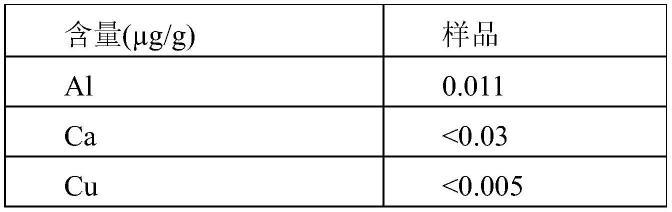
表1样品3#金属离子含量
[0123][0124][0125]
从上述的实施例1
#
~6
#
,可以发现采用本发明反加工艺可以很好的制备出球形的纳米氧化硅颗粒。
[0126]
从1
#
和2
#
的样品结果可以看出采用醇类和水相不相容的甲苯为溶剂,结果发现,尽管两种溶剂和水的相容性差别大,但是两种方法合成的颗粒尺度差别不是很大,分别为59.31nm和52.56nm,且颗粒的集中度都是比较高的。
[0127]
样品2
#
和3
#
的制备方法中,实施实例3中的溶剂水加入量是实施实例2的一半,结果发现3
#
样品的颗粒直径比2
#
样品的大,直径分别是87.08nm(3
#
样品)和52.56nm(2
#
样品),且颗粒的分布都是很好,pdi分别是0.005(3
#
样品)和0.028(2
#
样品)。
[0128]
样品4
#
的制备过程中,反加入溶剂水的速率相比于样品2
#
的制备过程降低了一倍。结果发现溶剂水相的加入速率越低,其造成合成的颗粒直径越大。由于加入的速率降低,样品4
#
的颗粒尺度为83.12nm,然而其制备的颗粒集中度pdi比较高,达到了0.03。
[0129]
和样品2
#
相比,5
#
样品的制备过程中主要提高了反应的温度,我们发现5
#
样品的颗粒尺度为83.32nm,集中度pdi为0.016,其制备的颗粒尺度大于2
#
样品,因此,增加了反应的温度,可以加快硅源之间的聚合反应,从而可以提高制备的颗粒尺度。
[0130]
相比于上述的制备过程,实施例6是将碱、有机硅源和溶剂一块混合,然后通过加入水相进行反应,结果发现制备的颗粒尺度达到了122.9nm,而且颗粒的分布比较集中,pdi为0.028。同样也验证了本发明提出的工艺是可以制备出均一的纳米氧化硅的颗粒。
[0131]
此次制备的上述样品1
#
~6
#
的电子扫描表征图9~14可以看出,本发明提出采用反加工艺制备纳米氧化硅的颗粒形貌均为球形颗粒,且颗粒的球形度比较高,从图中也可以看出颗粒的分布比较集中。
[0132]
针对于本发明提出的工艺的固含量,测试例2可以看出,其中的样品2
#
和3
#
的固含量分别为7.2wt.%和12.4wt.%,随着固含量的增加,纳米粒子的均一性和球形度都有不同
程度的上升,而且对样品3
#
的水溶液体系进行金属离子的检测,结果发现,其金属离子的含量小于151ppb,完全满足超高纯纳米氧化硅的要求。
[0133]
通过对比例1,2可以看出,采用正加工艺,如图15、16所示,在氧化硅固含量控制在6wt.%以下的情况下可以实现均一的球形纳米颗粒的制备,然而,当其中的固含量在7.2wt.%的时候,纳米粒子的均一性和球形度都有不同程度的下降。
[0134]
因此,本发明提出的一种超高纯度、均一的球形纳米氧化硅颗粒的制备方法,可以实现球形纳米氧化硅颗粒尺度为30~150nm之间可控制备,而且制备的纳米颗粒分布窄、均一性高、pdi《0.1,球形度高等特点,且制备的纳米氧化硅颗粒的固含量为7wt%~15wt%,金属离子的含量可以降低在1ppm以下。
[0135]
以上所述,仅是本技术的几个实施例,并非对本技术做任何形式的限制,虽然本技术以较佳实施例揭示如上,然而并非用以限制本技术,任何熟悉本专业的技术人员,在不脱离本技术技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
