1.本发明涉及有机化学领域,具体为一种苯磺酰氨基膦酸酯衍生物及其制备方法、应用。
背景技术:
2.抗菌药物的大量使用导致了细菌的出现耐药性,甚至导致超级细菌的产生。然而,新抗菌药物的开发远远落后于新耐药细菌的出现,如广泛使用喹诺酮类药物,在使用过程中所产生的对氟喹诺酮类耐药的大肠杆菌(frec),在临床治疗中非常棘手。因此,迫切需要开发高效低毒的抗菌药物,来应对新耐药菌的出现。
技术实现要素:
3.本发明意在针对现有技术的不足,提供一种苯磺酰氨基膦酸酯衍生物。
4.本方案中的一种苯磺酰氨基膦酸酯衍生物,其化学结构通式ⅲ所示:
[0005][0006]
其中,r1为氢、甲基、甲氧基、硝基、氟、氯、溴、碘中的一种;r2为氢、甲基、苯甲基、异丙基、异丁基中的一种;r3为氢、甲基、甲氧基、硝基、氟、氯、溴中的一种;r4为甲基、乙基、丙基中的一种。
[0007]
本技术还提供了所述苯磺酰氨基膦酸酯衍生物的制备方法,化学反应过程如下:
[0008][0009]
注:edci为1-乙基-(3-二甲基氨基丙基)碳化二亚胺盐酸盐,hobt为1-羟基苯并三唑,al(otf)3为三氟甲磺酸铝。
[0010]
步骤包括:
[0011]
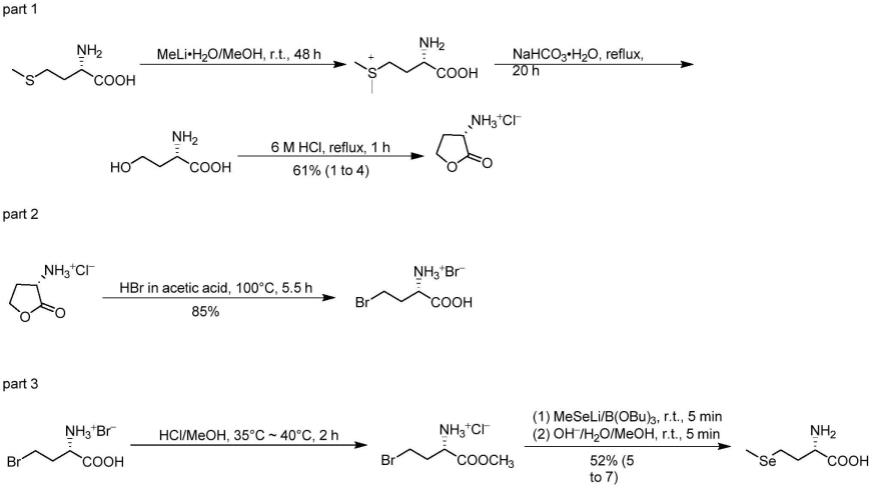
化合物ⅰ的制备:取一个干净的50ml反应瓶,将0.51ml(4mmol)苯磺酰氯加入到氨基酸(5mmol)的碳酸钠溶液(0.848g碳酸钠溶于10ml水)中,升温至70-80℃,搅拌反应0.5h,tlc监测反应,反应完毕,用浓盐酸调节ph至1,过滤,干燥,得到白色固体,即化合物ⅰ;
[0012]
化合物ⅱ的制备:依次称取亚磷酸二乙酯2.5405g(20mmol),芳香醛(25mmol),乙酸铵1.5401g(20mmol),加入50ml反应瓶中,室温搅拌10min,加入三氟甲磺酸铝,在90-100℃条件下搅拌反应,tlc监测,0.5h反应完毕,加入少量去离子水,用1mol/l的稀盐酸调节ph
至3-4,用环己烷萃取6次,取下层液,再用乙酸乙酯萃取6次,用1mol/l naoh溶液调节ph至8,再用乙酸乙酯萃取,取上层液,浓缩溶剂,得化合物ⅱ;
[0013]
化合物ⅲ的制备:依次称取化合物ⅰ(0.5mmol)、hobt 135.12mg(1mmol)于50ml反应瓶中,冰浴条件下,滴加三乙胺0.2ml(1.5mmol)及预先溶解的edci 191.7mg(1mmol),搅拌反应1h后,滴加化合物ⅱ(0.75mmol),滴加完毕,室温反应,tlc监测,待反应完毕,浓缩溶剂,经硅胶柱层析分离纯化,得化合物ⅲ。
[0014]
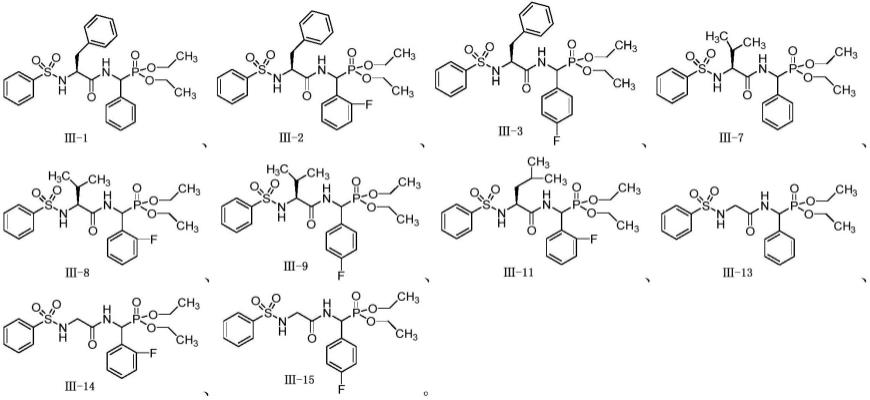
体外抗菌活性筛选结果表明,本发明的一种苯磺酰氨基膦酸酯衍生物具有潜在的抗菌活性,可与抗菌药物或抗菌活性成分组合使用,用于制备治疗和/或预防大肠杆菌(e.coli)药物,且部分目标物具有显著的抗革兰氏阴性[大肠杆菌(e.coli)]活性,尤其是其中的如下化合物对氟喹诺酮类耐药大肠杆菌(frec)的活性最为显著:
[0015][0016]
其中,化合物
ⅲ‑
4还对耐甲氧西林金黄色葡萄球菌具有显著的活性;化合物
ⅲ‑
2、
ⅲ‑
3、
ⅲ‑
8、
ⅲ‑
13、
ⅲ‑
14中任一项对大肠杆菌(e.coli)的活性与对照药诺氟沙星接近,可用于制备抗大肠杆菌药物。
具体实施方式
[0017]
下面结合实施例进一步介绍本发明,但本发明不仅限于下述实施例,可以预见本领域技术人员在结合现有技术的情况下,实施情况可能产生种种变化。
[0018]
本发明通式ⅲ化合物的制备过程如下:
[0019][0020]
部分化合物ⅲ的相关数据如下表1所示:
[0021]
表1
[0022]
[0023]
[0024][0025]
以下以部分化合物为例详述制备过程:
[0026]
实施例1:化合物
ⅲ‑
2的制备
[0027]
首先,取干净的50ml反应瓶,将0.51ml(4mmol)苯磺酰氯加入到5mmol的l-苯丙氨酸的碳酸钠溶液(0.848g碳酸钠溶于10ml水)中,加热至75℃,搅拌反应0.5h,反应完毕,用浓盐酸调节ph至1,过滤,干燥,得到白色固体,即化合物
ⅰ‑
2。
[0028]
然后,依次称取亚磷酸二乙酯2.5405g(20mmol),邻氟苯甲醛2.6437g(25mmol),乙酸铵1.5401g(20mmol),加入50ml反应瓶中,室温搅拌10min,加入三氟甲磺酸铝,在100℃条件下搅拌反应,0.5h反应完毕,加入少量去离子水,用1mol/l的稀盐酸调节ph至3,用环己烷
萃取6次,取下层液,再用乙酸乙酯萃取6次,用1mol/l naoh溶液调节ph至8,再用乙酸乙酯萃取,取上层液,浓缩溶剂,得化合物
ⅱ‑
2。
[0029]
最后,依次取化合物
ⅰ‑
2 159.69mg(0.5mmol)、hobt 135.12mg(1mmol)于50ml反应瓶中,冰浴条件下,滴加三乙胺0.2ml(1.5mmol)及预先溶解的edci 191.7mg(1mmol),搅拌反应1h后,滴加化合物
ⅱ‑
2(0.75mmol),完毕,室温反应,tlc监测,反应完毕,浓缩溶剂,经硅胶柱层析分离纯化,得白色固体,得化合物
ⅲ‑
2。
[0030]
实施例2:化合物
ⅲ‑
4的制备
[0031]
首先,取干净的50ml反应瓶,将0.51ml(4mmol)苯磺酰氯加入到5mmol的l-丙氨酸的碳酸钠溶液(0.848g碳酸钠溶于10ml水)中,加热至70℃,搅拌反应0.5h,反应完毕,用浓盐酸调节ph至1,过滤,干燥,得到白色固体,即化合物
ⅰ‑
4。
[0032]
接下来,依次称取亚磷酸二乙酯2.5405g(20mmol),苯甲醛2.6437g(25mmol),乙酸铵1.5401g(20mmol),加入50ml反应瓶中,室温搅拌10min,加入三氟甲磺酸铝,在95℃条件下搅拌反应,tlc监测,0.5h反应完毕,加入少量去离子水,用1mol/l的稀盐酸调节ph至3,用环己烷萃取6次,取下层液,再用乙酸乙酯萃取6次,用1mol/l naoh溶液调节ph至8,再用乙酸乙酯萃取,取上层液,浓缩溶剂,得化合物
ⅱ‑
1。
[0033]
最后,依次称取化合物
ⅰ‑
4 159.69mg(0.5mmol)、hobt 135.12mg(1mmol)于50ml反应瓶中,冰浴条件下,滴加三乙胺0.2ml(1.5mmol)及预先溶解的edci 191.7mg(1mmol),搅拌反应1h后,滴加化合物
ⅱ‑
1(0.75mmol),滴加完毕,室温反应,tlc监测,反应完毕,浓缩溶剂,经硅胶柱层析分离纯化,得白色固体,即化合物
ⅲ‑
4。
[0034]
实施例3:化合物
ⅲ‑
8的制备
[0035]
首先,取干净的50ml反应瓶,将0.51ml(4mmol)苯磺酰氯加入到5mmol的l-缬氨酸的碳酸钠溶液(0.848g碳酸钠溶于10ml水)中,加热至75℃,搅拌反应0.5h,反应完毕,用浓盐酸调节ph至1,过滤,干燥,得到白色固体,即化合物
ⅰ‑
8。
[0036]
接下来,依次称取亚磷酸二乙酯2.5405g(20mmol),邻氟苯甲醛25mmol,乙酸铵1.5401g(20mmol),加入50ml反应瓶中,室温搅拌10min,加入三氟甲磺酸铝,在100℃条件下搅拌反应,tlc监测,0.5h反应完毕,加入少量去离子水,用1mol/l的稀盐酸调节ph至3,用环己烷萃取6次,取下层液,再用乙酸乙酯萃取6次,用1mol/l naoh溶液调节ph至8,再用乙酸乙酯萃取,取上层液,浓缩溶剂,得化合物
ⅱ‑
8。
[0037]
最后,依次称取化合物
ⅰ‑
8(0.5mmol)、hobt 135.12mg(1mmol)于50ml反应瓶中,冰浴条件下,滴加三乙胺0.2ml(1.5mmol)及预先溶解的edci 191.7mg(1mmol),搅拌反应1h后,滴加化合物
ⅱ‑
8(0.75mmol),滴加完毕,室温反应,tlc监测,反应完毕,浓缩溶剂,经硅胶柱层析分离纯化,得白色固体,即化合物
ⅲ‑
8。
[0038]
实施例4:化合物
ⅲ‑
11的制备
[0039]
首先,取干净的50ml反应瓶,将0.51ml(4mmol)苯磺酰氯加入到5mmol的l-亮氨酸的碳酸钠溶液(0.848g碳酸钠溶于10ml水)中,加热至72℃,搅拌反应0.5h,反应完毕,用浓盐酸调节ph至1,过滤,干燥,得到白色固体,即化合物
ⅰ‑
11。
[0040]
接下来,依次称取亚磷酸二乙酯2.5405g(20mmol),邻氟苯甲醛25mmol,乙酸铵1.5401g(20mmol),加入50ml反应瓶中,室温搅拌10min,加入三氟甲磺酸铝,在96℃条件下搅拌反应,tlc监测,0.5h反应完毕,加入少量去离子水,用1mol/l的稀盐酸调节ph至3,用环
己烷萃取6次,取下层液,再用乙酸乙酯萃取6次,用1mol/l naoh溶液调节ph至8,再用乙酸乙酯萃取,取上层液,浓缩溶剂,得化合物
ⅱ‑
2。
[0041]
最后,依次称取化合物
ⅰ‑
11(0.5mmol)、hobt 135.12mg(1mmol)于50ml反应瓶中,冰浴条件下,滴加三乙胺0.2ml(1.5mmol)及预先溶解的edci 191.7mg(1mmol),搅拌反应1h后,滴加化合物
ⅱ‑
2(0.75mmol),滴加完毕,室温反应,tlc监测,待反应完毕,浓缩溶剂,经硅胶柱层析分离纯化,得白色固体,即化合物
ⅲ‑
11。
[0042]
实施例5:化合物
ⅲ‑
15的制备
[0043]
首先,取干净的50ml反应瓶,将0.51ml(4mmol)苯磺酰氯加入到5mmol的甘氨酸的碳酸钠溶液(0.848g碳酸钠溶于10ml水)中,加热至70℃,搅拌反应0.5h,反应完毕,用浓盐酸调节ph至1,过滤,干燥,得到白色固体,即化合物
ⅰ‑
15。
[0044]
接下来,依次称取亚磷酸二乙酯2.5405g(20mmol),对氟苯甲醛25mmol,乙酸铵1.5401g(20mmol),加入50ml反应瓶中,室温搅拌10min,加入三氟甲磺酸铝,在100℃条件下搅拌反应,tlc监测,0.5h反应完毕,加入少量去离子水,用1mol/l的稀盐酸调节ph至3,用环己烷萃取6次,取下层液,再用乙酸乙酯萃取6次,用1mol/l naoh溶液调节ph至8,再用乙酸乙酯萃取,取上层液,浓缩溶剂,得化合物
ⅱ‑
3。
[0045]
最后,依次称取化合物
ⅰ‑
15(0.5mmol)、hobt 135.12mg(1mmol)于50ml反应瓶中,冰浴条件下,滴加三乙胺0.2ml(1.5mmol)及预先溶解的edci 191.7mg(1mmol),搅拌反应1h后,滴加化合物
ⅱ‑
3(0.75mmol),滴加完毕,室温反应,tlc监测,待反应完毕,浓缩溶剂,经硅胶柱层析分离纯化,得白色固体,即化合物
ⅲ‑
15。
[0046]
本发明的抗菌活性测试:以诺氟沙星为对照药物,采用微量稀释法测定化合物
ⅲ‑
1~
ⅲ‑
15对金黄色葡萄球菌(s.aureus)、大肠杆菌(e.coli)、耐甲氧西林金黄色葡萄球菌(mrsa)和氟喹诺酮类耐药大肠杆菌(frec)的最小抑菌浓度(mic),数据如表2所示。
[0047]
表2
[0048][0049]
从上述实验结果可以清楚的看出,本发明所要保护的通式ⅲ的化合物具有潜在抗菌活性。部分化合物对氟喹诺酮类耐药大肠杆菌(frec)的抗菌活性远远优于对照药诺氟沙星,如化合物
ⅲ‑
1、
ⅲ‑
2、
ⅲ‑
3、
ⅲ‑
7、
ⅲ‑
8、
ⅲ‑
9、
ⅲ‑
11、
ⅲ‑
13、
ⅲ‑
14、
ⅲ‑
15对氟喹诺酮类耐药大肠杆菌(frec)的mic值远小于对照药诺氟沙星;部分化合物对大肠杆菌(e.coli)的活性与对照药诺氟沙星接近,如化合物
ⅲ‑
2、
ⅲ‑
3、
ⅲ‑
8、
ⅲ‑
13、
ⅲ‑
14。其潜在的抗菌活性可用于大肠杆菌(e.coli)和氟喹诺酮类耐药大肠杆菌(frec)引起的疾病的治疗。同时,也可与其它抗菌活性物质组合使用。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。