一种n,n-双氨丙基环己胺的制备方法
技术领域
1.本发明涉及一种氰基加氢的方法,具体涉及一种制备n,n-双氨丙基环己胺的方法。
背景技术:
2.n,n-双氨丙基环己胺是一种新型的脂环胺化合物,其分子结构兼顾脂环胺和脂肪胺的特点,主要用于环氧固化剂领域以及其它待开发的应用领域,可以用于饰品胶、美缝剂、环氧地坪等。目前在环氧固化剂领域可以应用的胺的类型很多,包括脂肪胺、脂环胺以及芳香胺等,各种胺类化合物具有各自的特点,其中脂环胺由于具有耐黄变性强、活性适中、挥发性低等优点备受市场的亲睐。当今社会,随着下游应用的迅猛发展,对具有特定结构的新分子胺有了一定的需求。为了满足下游客户产品多样化的需求,如今对脂环胺的改造方式也多种多样,包括环氧乙烷/环氧丙烷改性、丙烯腈改性等。
3.cn114835588a公开了一种双氰乙基脂环胺加氢制备双氨丙基脂环胺的方法。该方法以双氰乙基脂环胺为原料,在阴阳离子配体催化剂的作用下,通过加氢制得目标产物,但该发明的催化剂难以制备,将来产业化会存在一定的问题,此外该发明也没有进行催化剂的连续套用,没有说明催化剂稳定性的情况。
4.cn113372241a公开了一种脂肪族伯胺一步法合成双氰乙基叔胺的方法。该发明以乙醇酸水溶液为催化剂,将脂肪族伯胺加入至丙烯腈中,在加热回流条件下,一步法合成双氰乙基叔胺化合物,在反应结束后,减压蒸馏处理脱除低沸点的组分,以及在操作温度下将乙醇酸进行分解,就可获得收率高于95%的双氰乙基叔胺化合物。但该专利没有说明产品中酸含量的残留量以及如何利用该双氰来加氢得到相应的胺。
5.ep1229021a1也公开了一种脂环族连伯二胺氰乙基化的方法。该发明以水和乙酸作为催化剂(pka在-3.0到7.5之间),丙烯腈与脂环族连伯二胺反应生成相应的氰乙基化混合物。该发明也是在酸性催化剂的作用下制备氰乙基化合物,但没说明产品中酸含量的问题。
6.cn114591200a公开了一种双氰乙基叔胺的制备方法。该方法以单氰乙基仲胺和3-氰基丙酸为原料,在含fe催化剂的作用下得到双氰乙基叔胺,反应过程也利用了3-氰基丙酸的酸性催化作用。该发明没说明产品中酸残留量的问题以及如何利用该双氰来加氢得到相应的胺,同时3-氰基丙酸原料不易得且价格昂贵,会影响产品的经济性。
7.从以上文献报道可以看出,n,n-双氰乙基环己胺的合成是采用酸作为催化剂或者采用酸性原料来催化反应。因此综上所述,目前n,n-双氰乙基环己胺加氢过程中存在原料中含酸或者工艺条件不合适等情况导致催化剂易失活,寿命短,产品收率低等诸多问题,影响了其产业化进程。
技术实现要素:
8.基于现有技术中存在的上述不足,本发明提供了一种n,n-双氨丙基环己胺的制备
方法,所述方法是通过n,n-双氰乙基环己胺加氢反应制备n,n-双氨丙基环己胺,该方法可以利用现有商业化的雷尼催化剂,且对原料的酸含量要求不苛刻,催化剂寿命长,不易失活,方法简单,具有良好的产业化前景。
9.为了实现上述发明目的,本发明采用如下的技术方案:
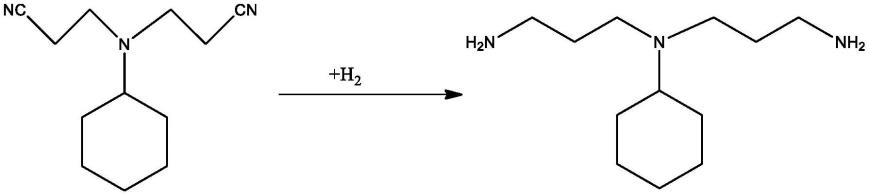
10.本发明提供一种n,n-双氨丙基环己胺的制备方法,所述方法是以n,n-双氰乙基环己胺为原料,在雷尼催化剂作用下通过加氢反应制备得到。
11.本发明的方法涉及的主要反应方程式如下:
[0012][0013]
在本发明实施方案中,一种n,n-双氰乙基环己胺的制备方法,步骤包括:
[0014]
1)将n,n-双氰乙基环己胺和溶剂a混合,配制成溶液;
[0015]
2)将雷尼催化剂、溶剂b和水混合,再加入碱性缓冲溶液和盐溶液,之后通入氢气调节体系至反应压力,并加热至反应温度,然后在氢气氛围下加入步骤1)配制的溶液,进行加氢反应,制得n,n-双氨丙基环己胺。
[0016]
本发明中,步骤1)所述n,n-双氰乙基环己胺,纯度为80-100wt%,优选90-100wt%;其中,控制酸含量≤1wt%,优选0-0.5wt%,其余为单氰乙基环己胺和微量杂质;其中所述的酸来源于合成过程中采用的酸催化剂或者采用的酸性原料的残留,例如盐酸、硫酸、磷酸、乙醇酸、三氟甲磺酸、对甲苯磺酸等中的一种或多种。
[0017]
所述n,n-双氰乙基环己胺为现有产品,可以采用n,n-双氰乙基环己胺纯品为原料,也可以采用精制前的粗品为原料;本发明对其来源没有特别要求,可以通过购买或自制得到,自制可以采用现有技术已公开的任意可实现方法,作为优选,在本发明一些具体示例中是通过酸催化单氰乙基环己胺与丙烯腈反应得到,例如可以参照专利cn113372241a、cn114890913a、cn114907216a等中公开的方法制备,其具体步骤本发明不再赘述。
[0018]
本发明中,步骤1)所述溶剂a选自甲醇、乙醇、四氢呋喃、二氧六环中的一种或多种,优选乙醇和/或四氢呋喃。
[0019]
本发明中,步骤1)配制的溶液中,n,n-双氰乙基环己胺的质量浓度为10-60wt%,优选40-50wt%。
[0020]
本发明中,步骤2)所述雷尼催化剂选自雷尼镍催化剂、雷尼钴催化剂中的一种或多种,优选grace2400、grace2800、grace 2724、迅凯1200、迅凯3300中的一种或多种;
[0021]
所述雷尼催化剂的用量,为步骤1)中n,n-双氰乙基环己胺质量的10-30wt%,优选15-25wt%。
[0022]
本发明中,步骤2)所述溶剂b选自甲醇、乙醇、四氢呋喃、二氧六环中的一种或多种,优选乙醇和/或四氢呋喃;
[0023]
所述溶剂b与步骤1)中n,n-双氰乙基环己胺的质量比为0.5-3:1,优选1-2:1;
[0024]
所述溶剂b可以与溶剂a相同或者不同,优选与溶剂a相同。
[0025]
本发明中,步骤2)所述水与步骤1)中n,n-双氰乙基环己胺的质量比为0.5-3:1,优选1-2:1。
[0026]
本发明中,步骤2)所述碱性缓冲溶液为碳酸钠-碳酸氢钠水溶液;
[0027]
所述碳酸钠-碳酸氢钠水溶液,其中碳酸钠和碳酸氢钠的总质量浓度为0.5-5wt%,优选1-3wt%;所述碳酸钠与碳酸氢钠的摩尔比为0.1-9:1,优选1-5:1;
[0028]
所述碱性缓冲溶液与步骤1)中n,n-双氰乙基环己胺的质量比为0.5-3:1,优选1-2:1。
[0029]
本发明中,步骤2)所述盐溶液为盐的水溶液,选自氯化钠、氯化钾、溴化钠、溴化钾的水溶液中的一种或多种,优选氯化钠的水溶液;
[0030]
所述盐溶液的浓度为10-30wt%,优选20-30wt%;
[0031]
所述盐溶液与步骤1)中n,n-双氰乙基环己胺的质量比为0.1-1:1,优选0.3-0.6:1。
[0032]
本发明中,步骤2)所述反应压力为3-8mpag,优选4-6mpag;所述反应温度为80-140℃,优选90-120℃。
[0033]
本发明中,步骤2),在氢气氛围下加入步骤1)配制的溶液,所述步骤1)配制的溶液采用连续加料方式,优选滴加加料,滴加时间为2-8h,优选4-6h;加料结束后继续保温进行加氢反应。
[0034]
本发明中,步骤2)所述加氢反应,在加料结束后的反应时间为0.2-2h,优选0.4-0.6h(不包括上述加料时间)。
[0035]
本发明中,步骤2)在加氢反应结束后,还包括降温、过滤等操作得到包含n,n-双氨丙基环己胺的母液,以及后续还可以通过精馏、薄膜蒸发等方法得到精制的纯品,为本领域常规操作,本发明不做具体限定。
[0036]
目前国内外对n,n-双氨基环己胺的合成鲜有报道,根据其结构特点,可以采用n,n-双氰乙基环己胺加氢的技术得到。但本发明人在对n,n-双氰乙基环己胺加氢的研究中发现,由于在制备原料n,n-双氰乙基环己胺的过程中需要加入酸性催化剂,而酸性催化剂在后处理的过程中难以完全除掉,这样导致n,n-双氰乙基环己胺产品中不可避免会残留酸,而在后续加氢制备n,n-双氰乙基环己胺的过程中,残留的酸会对催化剂的活性造成很大的影响,分析其原因主要是腈基官能团首先经历加氢生成亚胺中间体,而亚胺的反应活性很高,在催化剂表面如果亚胺不能迅速加氢生成伯胺,容易与反应中间体和产物发生缩合。并且酸的存在会促进缩合过程的发生,导致生成多种高沸点的大分子副产物,大分子副产物对催化剂表面孔道的堵塞、活性位点的覆盖会导致催化剂快速失活。为此,本发明研发人员通过进一步研究发现,采用雷尼催化剂,配合碱性缓冲溶液,并在反应体系中添加盐溶液,可以有效解决上述问题,消除残留酸的影响,降低对原料中酸含量的要求,使本发明方法具有更广泛的适用性,即使采用n,n-双氰乙基环己胺粗品为原料仍然可以得到优异的效果。
[0037]
与现有技术相比,本发明有益效果在于:
[0038]
(1)本发明采用常规的雷尼催化剂,同时引入碱性缓冲溶液一方面可以迅速被中和掉n,n-双氰乙基环己胺原料尤其是粗品中的残留的酸,从而避免酸促进缩合反应生成大分子副产的发生,延长催化剂的寿命;另一方面碱性缓冲溶液也能对雷尼催化剂进行改性,降低副产脱氨以及仲胺的生成,不仅提高主产品的收率,也能减少重组分对催化剂活性中
心的包裹,保持催化剂的高活性状态。
[0039]
(2)本发明在反应体系中添加盐溶液,可以促进溶剂与水的分相,由于n,n-双氰乙基环己胺难溶于水,n,n-双氨丙基环己胺易溶于水,从而使得整个体系呈现两相,n,n-双氰乙基环己胺主要在溶剂相,n,n-双氨丙基环己胺主要在水相,整个反应发生在两相界面。通过控制盐溶液的用量,保持n,n-双氰乙基环己胺在水相中一定的溶解度,降低原料n,n-双氰乙基环己胺对催化剂的毒害,从而提高催化剂的寿命。
[0040]
(3)采用本发明的工艺,反应条件温和,工艺简单,可以采用现有商业化的雷尼催化剂,对原料中的酸含量(≤1%)没有苛刻的要求,采用n,n-双氰乙基粗品也能保证催化剂性能稳定,催化剂可以套用30批次以上,n,n-双氨丙基环己胺的选择性≥95%,产品中单氨丙基环己胺和双氨丙基环己胺的总含量≥96wt%,缩合大分子副产含量≤3wt%,具有很好的产业化前景。
具体实施方式
[0041]
为了更好的理解本发明的技术方案,下面的实施例将对本发明所提供的方法予以进一步的说明,但本发明不限于所列出的实施例,还应包括在本发明的权利要求范围内其他任何公知的改变。
[0042]
以下实施例中进行气相色谱分析的条件为:安捷伦db-5色谱柱,进样口温度280℃,fid检测器温度300℃,柱流速1.3ml/min,氢气流速40ml/min,空气流速400ml/min,程序升温方式为:50℃保持2min,以5℃/min升温至80℃,然后以15℃/min升温至300℃,保持15min。
[0043]
以下实施例中进行原料n,n-双氰乙基环己胺的酸含量分析的条件如下:
[0044]
仪器:瑞士万通电位滴定仪;
[0045]
标准溶液:0.02mol/l氢氧化钾-甲醇标准溶液;
[0046]
溶剂:100ml甲醇;
[0047]
电极:非水相酸碱电极;
[0048]
操作步骤:称取10g样品,加入100ml甲醇,搅拌溶解后,在电位滴定仪上,以非水相酸碱电极为指示电极,以0.02mol/l氢氧化钾-甲醇标准溶液滴定。
[0049]
以下实施例或对比例中所用的主要原料来源如下,其它若未特别说明,均为普通市售原料:
[0050]
n,n-双氰乙基环己胺粗品:自制;以环己胺和丙烯腈为原料参照专利cn113372241a公开的方法制得反应液,之后将反应液进行分相得到n,n-双氰乙基环己胺粗品;
[0051]
雷尼镍催化剂:grace 2400/2800,购于grace催化剂公司;迅凯1200,购于上海迅凯新材料科技有限公司;
[0052]
雷尼钴催化剂:grace 2724,购于grace催化剂公司;迅凯3300,购于上海迅凯新材料科技有限公司。
[0053]
主要设备信息如下:
[0054]
高压反应釜:规格1l,制造商为烟台科立设备;
[0055]
平流泵:型号2pb00c,制造商为北京卫星。
[0056]
实施例1
[0057]
制备n,n-双氨丙基环己胺,步骤为:
[0058]
采用n,n-双氰乙基环己胺粗品为原料,组成包括95wt%n,n-双氰乙基环己胺,0.5wt%乙醇酸,4.4wt%单氰乙基环己胺,0.1wt%其它杂质;
[0059]
碱性缓冲溶液采用碳酸钠-碳酸氢钠水溶液,其中碳酸钠与碳酸氢钠总质量浓度为1wt%,二者的摩尔比为9:1。
[0060]
将n,n-双氰乙基环己胺粗品与乙醇配成50wt%的n,n-双氰乙基环己胺粗品乙醇溶液;
[0061]
在高压反应釜中加入10g雷尼钴催化剂(迅凯3300),50g乙醇,50g水,再加入50g碳酸钠-碳酸氢钠水溶液,15g氯化钠水溶液(盐浓度为20wt%),用氮气和氢气依次置换3遍后,充氢气至2mpag,开始升温,当反应温度升至100℃,将氢气压力充压至4mpag并且维持此压力,开始往反应体系中滴加50wt%的n,n-双氰乙基环己胺粗品乙醇溶液,当进料至100g时,总共滴加时间为4h,停止进料,继续反应0.2h后,降温、过滤得到n,n-双氨丙基环己胺反应液,取样进行气相色谱分析(用面积归一化法定量,各组分含量基于样品中扣除溶剂后的总质量计算得到),催化剂留在反应釜中继续套用。
[0062]
在相同的条件下催化剂套用30批次,得到的结果如下表1所示。
[0063]
表1
[0064][0065]
从上表数据可以看出,催化剂套用30批次,催化剂性能依然非常稳定。
[0066]
实施例2
[0067]
制备n,n-双氨丙基环己胺,步骤为:
[0068]
采用n,n-双氰乙基环己胺粗品为原料,组成包括90wt%n,n-双氰乙基环己胺,1.0wt%乙醇酸,8.9wt%单氰乙基环己胺,0.1wt%其它杂质;
[0069]
碱性缓冲溶液采用碳酸钠-碳酸氢钠水溶液,其中碳酸钠与碳酸氢钠总质量浓度为5wt%,二者的摩尔比为5:1。
[0070]
将n,n-双氰乙基环己胺粗品与甲醇配成60wt%的n,n-双氰乙基环己胺粗品甲醇溶液;
[0071]
在高压反应釜中加入15g雷尼镍催化剂(grace 2400),100g四氢呋喃,25g水,再加入100g碳酸钠-碳酸氢钠水溶液,5g氯化钾水溶液(盐浓度为30wt%),用氮气和氢气依次置换3遍后,充氢气至1mpag,开始升温,当反应温度升至80℃,将氢气压力充压至3mpag并且维持此压力,开始往反应体系中滴加60wt%的n,n-双氰乙基环己胺粗品甲醇溶液,当进料至83.3g时,总共滴加时间为6h,停止进料,继续反应0.4h后,降温、过滤得到n,n-双氨丙基环己胺反应液,取样进行气相色谱分析,催化剂留在反应釜中继续套用。
[0072]
在相同的条件下催化剂套用30批次,得到的结果如下表2所示。
[0073]
表2
[0074][0075][0076]
从上表数据可以看出,催化剂套用30批次,催化剂性能依然非常稳定。
[0077]
实施例3
[0078]
制备n,n-双氨丙基环己胺,步骤为:
[0079]
采用n,n-双氰乙基环己胺粗品为原料,组成包括80wt%n,n-双氰乙基环己胺,0.3wt%乙醇酸,19.6wt%单氰乙基环己胺,0.1wt%其它杂质;
[0080]
碱性缓冲溶液采用碳酸钠-碳酸氢钠水溶液,其中碳酸钠与碳酸氢钠总质量浓度为0.5wt%,二者的摩尔比为0.1:1。
[0081]
将n,n-双氰乙基环己胺粗品与四氢呋喃配成10wt%的n,n-双氰乙基环己胺粗品四氢呋喃溶液;
[0082]
在高压反应釜中加入5g雷尼钴催化剂(grace 2724),50g二氧六环,100g水,再加
入25g碳酸钠-碳酸氢钠水溶液,30g溴化钠水溶液(盐浓度为10wt%),用氮气和氢气依次置换3遍后,充氢气至3mpag,开始升温,当反应温度升至140℃,将氢气压力充压至6mpag并且维持此压力,开始往反应体系中滴加10wt%的n,n-双氰乙基环己胺粗品四氢呋喃溶液,当进料至500g时,总共滴加时间为2h,停止进料,继续反应2h后,降温、过滤得到n,n-双氨丙基环己胺反应液,取样进行气相色谱分析,催化剂留在反应釜中继续套用。
[0083]
在相同的条件下催化剂套用30批次,得到的结果如下表3所示。
[0084]
表3
[0085][0086]
从上表数据可以看出,催化剂套用30批次,催化剂性能依然非常稳定。
[0087]
实施例4
[0088]
制备n,n-双氨丙基环己胺,步骤为:
[0089]
采用n,n-双氰乙基环己胺纯品为原料,组成为100wt%n,n-双氰乙基环己胺;
[0090]
碱性缓冲溶液采用碳酸钠-碳酸氢钠水溶液,其中碳酸钠与碳酸氢钠总质量浓度为3wt%,二者的摩尔比为1:1。
[0091]
将n,n-双氰乙基环己胺纯品与二氧六环配成40wt%的n,n-双氰乙基环己胺二氧六环溶液;
[0092]
在高压反应釜中加入7.5g雷尼镍催化剂(迅凯1200),150g乙醇,50g水,再加入150g碳酸钠-碳酸氢钠水溶液,50g溴化钾水溶液(盐浓度为15wt%),用氮气和氢气依次置换3遍后,充氢气至3mpag,开始升温,当反应温度升至90℃,将氢气压力充压至8mpag并且维持此压力,开始往反应体系中滴加40wt%的n,n-双氰乙基环己胺二氧六环溶液,当进料至125g时,总共滴加时间为8h,停止进料,继续反应0.2h后,降温、过滤得到n,n-双氨丙基环己胺反应液,取样进行气相色谱分析,催化剂留在反应釜中继续套用。
[0093]
在相同的条件下催化剂套用30批次,得到的结果如下表4所示。
[0094]
表4
[0095][0096][0097]
从上表数据可以看出,催化剂套用30批次,催化剂性能依然非常稳定。
[0098]
对比例1
[0099]
参照实施例1中方法制备n,n-双氨丙基环己胺,不同之处仅在于:碳酸钠-碳酸氢钠缓冲液替换为等量的碳酸钠水溶液(浓度1wt%),其它参数和操作不变,制得n,n-双氨丙基环己胺,得到的结果如下表5所示。
[0100]
表5
[0101][0102]
对比例2
[0103]
参照实施例1中方法制备n,n-双氨丙基环己胺,不同之处仅在于:碳酸钠-碳酸氢钠缓冲液替换为等量的碳酸氢钠水溶液(浓度1wt%),其它参数和操作不变,制得n,n-双氨丙基环己胺,得到的结果如下表6所示。
[0104]
表6
[0105][0106][0107]
对比例3
[0108]
参照实施例1中方法制备n,n-双氨丙基环己胺,不同之处仅在于:不添加碳酸钠-碳酸氢钠缓冲液,其它参数和操作不变,制得n,n-双氨丙基环己胺,得到的结果如下表7所示。
[0109]
表7
[0110][0111]
对比例4
[0112]
参照实施例1中方法制备n,n-双氨丙基环己胺,不同之处仅在于:不添加盐溶液,其它参数和操作不变,制得n,n-双氨丙基环己胺,得到的结果如下表8所示。
[0113]
表8
[0114][0115][0116]
尽管本发明的内容已经通过上述优选实施例作了详细介绍,但应当认识到上述的描述不应被认为是对本发明的限制。本领域技术人员可以理解,在本说明书的教导之下,可对本发明做出一些修改或调整。这些修改或调整也应当在本发明权利要求所限定的范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
