用于筛查ivf胚胎的方法
1.相关申请
2.本技术要求于2019年10月22日提交的题为“用于筛查ivf胚胎的方法(method for screening ivf embryos)”的澳大利亚临时专利申请第2019903966号的优先权。所述较早申请的全部内容特此通过引用并入。
技术领域
3.本发明涉及筛查体外受精(ivf)胚胎的致病性基因变异,如单核苷酸多态性的方法。具体地,本发明涉及使用全基因组测序(wgs)数据筛查ivf胚胎的方法。
背景技术:
4.可以观察到遗传因果关系的已知综合征性疾病多达9,000种。约5,000个人类基因的一个或两个拷贝的单个突变可以引起这些病症之一。孕前基因筛查越来越多地用于确定女性或男性生殖伴侣的这些破坏性变异的携带状态,但通常仅限于由数百种高危疾病构成的子集。在体外对胚胎进行基因组筛查将限制导致儿童期发作性疾病的基因变异的传递。0.5%到5%之间的婴儿出生时患有基因病状或疾病;将全基因组测序(wgs)应用于ivf受孕胚胎提供了筛查遗传性综合征基因疾病的机会,此外还可以鉴定在技术上更具挑战性的新生突变。胚胎中引入了大约30个到100个新生突变,尽管所述新生突变仅带来引起与家族性疾病相关的疾病的绝对风险的一小部分,但当所述新生突变显性表达或作为复合杂合子引入时,所述新生突变往往会产生更严重的致病性表型。
5.二十多年来,临床体外受精(ivf)包含植入前基因检测(pgt),所述pgt应用了可获得的技术和方法。目的是通过筛查胚胎的染色体非整倍性和/或通过限制已知的遗传性单基因突变的传递来最大化健康婴儿的可能性。最新的主要pgt进展是应用低覆盖度下一代测序(ngs),以检测染色体非整倍性、结构变异(sv)和大拷贝数变异(cnv)。
6.迄今为止,wgs仅在有限数量的辅助生殖案例中被采用,主要是由于高昂的货币价格。同样,对来自胚胎的基因组dna进行wgs也是一个重大挑战,因为在从胚胎活检的少数细胞中,起始dna的数量很低(大约4个到12个拷贝)。在此方面,用于从ivf胚胎活检中扩增dna的量的方法会以每1,000个到10,000个碱基中约一个的比率引入突变。因此,来自ivf胚胎活检的wgs数据具有很高的错误率,因此难以准确确定胚胎是否存在具有致病性基因变异的风险。
7.因此,需要开发可以用于使用wgs数据筛查ivf胚胎的致病性基因变异的方法,所述方法可处理此类数据中存在的高错误率。
技术实现要素:
8.本发明人已经确定,通过将用于鉴定遗传变异的三重测试和用于wgs数据中新生变异的变异等位基因频率过滤器组合,可以成功地筛查ivf胚胎的致病性基因变异。因此,一方面,本发明提供了一种筛查体外受精(ivf)胚胎的致病性基因变异的方法,所述方法包
括:
9.a)从所述胚胎、所述胚胎的父本和所述胚胎的母本中获得全基因组测序数据,
10.b)将所述胚胎测序数据与参考基因组比对,并鉴定所述胚胎测序数据相对于所述参考基因组的变异,
11.c)将所述父本的测序数据和所述母本的测序数据与所述参考基因组比对,并鉴定所述亲本测序数据相对于所述参考基因组而存在的变异,
12.d)将步骤b)中鉴定的所述变异与步骤c)中鉴定的那些变异进行比较,以鉴定所述胚胎中遗传的基因变异,其中所述遗传的基因变异存在于所述胚胎测序数据和至少一个亲本的测序数据中,
13.e)通过变异等位基因频率(vaf)阈值过滤在步骤b)中鉴定的、在步骤d)中未被鉴定为遗传的基因变异的变异,其中具有高于所述阈值的vaf的经过滤的变异被鉴定为新生基因变异,以及
14.f)将所述遗传的基因变异和所述新生基因变异与已知致病性基因变异的数据库进行比较,以确定所述胚胎是否存在具有致病性基因变异的风险。
15.如技术人员将理解的,本发明的方法通过使用组合方法来解决易错胚胎源性wgs数据的问题,所述组合方法可以使用与所述亲本测序数据的比较(“三重测试”)来鉴定遗传的致病性变异并且通过使用合适的vaf阈值过滤器来鉴定新生致病性变异。
16.在一些实施例中,所述方法进一步包括使用一种或多种致病性预测算法来预测在步骤f)中未被鉴定为已知致病性基因变异的基因变异中的任何基因变异是否是致病性基因变异。因此,除了使用已知致病性基因变异的数据库来鉴定先前分类为致病性(或可能致病性)的变异外,本发明的方法还可以使用预测算法来评估与所鉴定的变异中的先前未被分类为所述数据库中的致病性变异的任何变异相关联的致病性风险。
17.在一些实施例中,所述一种或多种致病性预测算法包含sift、polyphen2 hvar、mutationtaster2、mutationassessor、fathmm、fathmm mkl。在一个实施例中,使用两种或三种或四种或五种或六种或更多种致病性预测算法。当使用多种致病性预测算法时,那些预测变异为致病性的算法的比例可以用作对所述变异的致病性的确定性的衡量。
18.在一些实施例中,将变异预测为致病性基因变异需要变异具有大于约2的mpc评分和/或大于约20的phred标度cadd评分。在一些实施例中,需要所述phred标度cadd评分大于25,或大于约30,或大于约35。
19.在一些实施例中,步骤e)中的所述vaf阈值介于0.25到0.45之间或介于0.3到0.4之间或为约0.35。在一些实施例中,所述vaf阈值是至少0.25、至少0.27、至少0.30、至少0.32或至少0.35。如本领域技术人员将理解的,可以根据胚胎的亲本愿意承担的相对风险量来选择所述vaf阈值。例如,更年轻(例如,更能生育)的夫妇可能倾向于选择较低的vaf阈值,以确保不会遗漏真阳性新生致病性基因变异,尽管可能会导致更多的假阳性新生致病性基因变异被鉴定。相反,年长夫妇,例如可供筛查的胚胎较少的年长夫妇可能倾向于选择较高的vaf阈值。
20.由于与来自经扩增的胚胎dna的wgs数据相关的高错误率,存在真阳性、低vaf、致病性新生变异可能被vaf阈值过滤掉的风险。因此,在一些实施例中,还检查具有低于所述阈值的vaf的变异的致病性潜力,无论它们是否是“真正的”基因变异。因此,在一些实施例
中,将具有低于所述阈值的vaf的变异与所述已知致病性基因变异的数据库进行比较以确定所述胚胎是否存在具有致病性基因变异的风险。在一些实施例中,通过一种或多种致病性预测算法来进一步评估vaf低于所述阈值且未被鉴定为已知致病性基因变异的变异以预测是否为致病性基因变异。
21.在一些实施例中,所述已知致病性基因变异的数据库是clinvar。其它数据库也是合适的。例如,在一些实施例中,所述已知致病性基因变异的数据库是hgmd、omim或acmg。在一些实施例中,将所述遗传的基因变异和所述新生基因变异与2个或3个或更多个已知致病性基因变异的数据库进行比较。
22.在一些实施例中,在步骤f)中确定基因变异是致病性基因变异需要所述基因变异在所述clinvar数据库中具有“致病性”或“可能致病性”的临床意义值。例如,此类变异被认为具有更高的风险,并且更有可能证明决定不移植胚胎是恰当的。其它数据库度量也可以用于评估致病性变异的临床意义的置信度。例如,数据库中与基因变异相关联的致病性的置信水平可以通过审查的程度或所述注释中的一致性水平来确定。例如,在一些实施例中,在步骤f)中确定基因变异是致病性基因变异需要所述变异在clinvar数据库中具有至少两星或至少三星或四星的审查状态。
23.在一些实施例中,所述基因变异包含单核苷酸多态性(snp)、插入或缺失(indel)、拷贝数变异(cnv)和/或结构变异。
24.在一些实施例中,所述遗传的基因变异包含常染色体显性、常染色体隐性、复合杂合和/或x连锁基因变异。
25.在一些实施例中,来自所述胚胎的所述全基因组测序数据通过以下来获得:
26.a)培养所述胚胎,
27.b)对所述胚胎进行活检,
28.c)从所述活检中扩增基因组dna,以及
29.d)对经扩增的基因组dna进行测序。
30.本领域技术人员将知道用于培养、进行活检、扩增dna和对dna进行测序的合适方法。
31.在一些实施例中,所述活检是滋养外胚层活检。在一些实施例中,所述滋养外胚层活检在培养的第5天或第6天进行。此类方法是有利的,因为它们使从胚胎获得的基因组dna的拷贝数最大化,而不会显著不利地影响胚胎的活力。
32.在一些实施例中,所述胚胎dna是从培养基而不是胚胎活检获得的,即从含“游离”dna的所述培养基或与囊胚腔液组合的囊胚培养条件培养基获得。
33.在一些实施例中,使用全基因组扩增(wga)方法从所述活检扩增所述基因组dna。在一些实施例中,使用多重置换扩增(mda)从所述活检扩增所述基因组dna。
34.任何核酸测序平台都适用于对所述基因组dna进行测序,包含高通量dna测序方法(通常也称为“下一代测序”或“ngs”)。因此,在一些实施例中,使用高通量测序方法对经扩增的基因组dna进行测序。在一些实施例中,使用dna纳米球测序对所述基因组dna进行测序。在一些实施例中,所述dna纳米球测序是用组合探针锚连接(cpal)进行的。
35.在一些实施例中,所述全基因组测序数据覆盖所述胚胎的基因组序列的至少60%、至少70%、至少80%、至少90%或至少95%。
36.将所述全基因组测序数据与参考基因组比对,使得可以将所述胚胎基因组序列(和亲本)与所述参考之间的任何差异鉴定为潜在的基因变异。在一些实施例中,所述参考基因组是人类参考基因组。在一些实施例中,所述参考基因组是某一基因组参考联盟人类版本(genome reference consortium human build)。在一些实施例中,所述参考基因组是基因组参考联盟人类版本37(genome reference consortium human build 37,grch37)或基因组参考联盟人类版本38(genome reference consortium human build 38,grch38)或任何未来的版本(即版本39或后续版本)。
37.在一些实施例中,筛查来自相同亲本的两个或更多个胚胎。在一些实施例中,筛查三个或四个或五个或六个或七个或八个或九个或十个或更多个胚胎。有利地,当筛查多个胚胎时,鉴定未被确定为存在具有致病性基因变异风险的胚胎的可能性更高,所述胚胎适合于移植。
38.在一些实施例中,所述方法进一步包括将所述胚胎移植到所述母本的子宫中。因此,如果对所述胚胎进行了致病性基因变异筛查并且所述胚胎被确定为存在具有致病性基因变异的风险或者具有可接受的低风险,则可以移植所述胚胎。
39.在一些实施例中,所述胚胎是人胚胎。在一些实施例中,所述胚胎是非人动物胚胎。因此,除了人ivf之外,本文所描述的方法还可以应用于其它动物胚胎,例如,筛查用于产生牲畜的胚胎。
40.在一些实施例中,所述亲本之一或两者具有致病性基因变异。因此,可以执行本文所描述的方法来筛查和选择用于移植的胚胎,所述胚胎被确定为不具有遗传的所述致病性基因变异。
41.另一方面,本发明提供了一种ivf方法,其包括:
42.a)用来自父本的精子使来自母本的卵子受精,
43.b)培养受精卵,从而产生胚胎,
44.c)使用本文所描述的方法筛查所述胚胎的致病性基因变异,以及
45.d)将所述胚胎移植到所述母本的子宫中。
46.另一方面,本发明提供一种筛查体外受精(ivf)胚胎的一种或多种表型性状的方法,所述方法包括:
47.a)从所述胚胎、所述胚胎的父本和所述胚胎的母本中获得全基因组测序数据,
48.b)将所述胚胎测序数据与参考基因组比对,并鉴定所述胚胎测序数据相对于所述参考基因组的变异,
49.c)将所述父本的测序数据和所述母本的测序数据与所述参考基因组比对,并鉴定所述亲本测序数据相对于所述参考基因组而存在的变异,
50.d)将步骤b)中鉴定的所述变异与步骤c)中鉴定的那些变异进行比较,以鉴定所述胚胎中遗传的基因变异,其中所述遗传的基因变异存在于所述胚胎测序数据和至少一个亲本的测序数据中,
51.e)通过变异等位基因频率(vaf)阈值过滤在步骤b)中鉴定的、在步骤d)中未被鉴定为遗传的基因变异的变异,其中具有高于所述阈值的vaf的经过滤的变异被鉴定为新生基因变异,以及
52.f)将所述遗传的基因变异和所述新生基因变异与具有已知表型性状的基因变异
harlow和david lane(编辑)《抗体:实验室手册(antibodies:a laboratory manual)》,冷泉港实验室,(1988);以及j.e.coligan等人(编辑),《当代免疫学指南(current protocols in immunology)》,约翰威利父子出版公司(包含迄今为止的所有更新)。
66.如本文所使用的,除非相反地说明,否则术语“约”是指指定值的 /-10%,更优选地 /-5%。
67.贯穿本说明书,词语“包括(comprise)”或如“包括(comprises)”或“包括(comprising)”等变体应当被理解为暗示包含所陈述要素、整数或步骤或要素组、整数组或步骤组,但不排除任何其它要素、整数或步骤或要素组、整数组或步骤组。
68.如本文所使用的,术语“或”旨在意指包含性的“或”,而不是排他性的“或”。也就是说,除非另外指明或根据上下文清楚的,“x采用a或b”旨在意指自然的包含性排列中的任何自然的包含性排列。也就是说,如果x采用a;x采用b;或x采用a和b两者,则“x采用a或b”在前述例子中的任何例子下都满足。进一步地,a和b中的至少一个和/或类似表达通常意指a或b或a和b两者。另外,本技术和所附权利要求中使用的冠词“一个(a)”和“一种(an)”总体上可以被解释为意指“一个或多个”,除非另有指定或根据上下文清楚的是针对单数形式。
69.如本文所使用的,术语“筛查”是指评估胚胎以确定其是否存在具有致病性基因变异的风险的过程。例如,此类过程可以用于选择合适的胚胎移植到女性的子宫中。
70.本文使用的术语“致病性”是指已知与或预测与疾病相关的基因变异。一种或多种基因变异与疾病的关联可以导致所述疾病或可以表示罹患所述疾病的遗传易感性,即风险。
71.如本文所使用的,术语“比对(aligned)”、“比对(alignment)”或“比对(aligning)”是指在核酸分子的顺序方面被鉴定为与来自参考基因组的已知序列匹配的一个或多个序列。此类比对可以手动或通过计算机算法完成,实例包含作为illumina基因组学分析流程的一部分分发的核苷酸数据有效局部比对(eland)计算机程序。比对中序列读段的匹配可以是100%的序列匹配或小于100%(非完美匹配)。
72.本文中的术语“等位基因”是指基因序列的序列变体。为了本技术的目的,等位基因可以但不必位于基因序列内。可以针对如snp等一个或多个多态性位置鉴定等位基因,而基因序列的其余部分可以保持未指定。例如,等位基因可以由存在于单个snp处的核苷酸定义,或由存在于多个snp处的核苷酸定义。
73.本文中的术语“测序”是指用于确定多核苷酸(例如基因组dna)的核苷酸序列的方法。优选地,测序方法包含作为非限制性实例的下一代测序(ngs)方法(即,高通量测序方法),ngs中克隆扩增的dna模板或单个dna分子以大规模平行方式测序(volkerding等人,2009;metzker等人,2010)。
74.术语“测序读段”或“读段”指具有足够长度(例如,至少约30bp)的dna序列,所述dna序列可以用于鉴定更大的序列或区域,例如可以比对并专门分配给染色体或基因组区域或基因的序列或区域。
75.本文中的术语“全基因组扩增”是指样品中存在的基因组dna序列被扩增以提供序列所表示的基因组的多个拷贝的过程。
76.术语“单倍型”是指包括一个或多个所关注基因变异的dna序列,所述变异包含在个体的单个染色体的子区域上。单倍型的基因变异可以是同一类型,例如所有snp,或者可
以是两种或更多种基因变异的组合,例如snp和str的组合。单倍型可以指单个基因、基因间序列或包含基因和基因间序列两者的更大序列,例如基因的集合或基因和基因间序列的集合中的一组基因变异。例如,单倍型可以指补体活化调控(rca)基因座中的一组基因变异,所述基因座包含补体因子h(cfh)、fhr3、fhr1、fhr4、fhr2、fhr5和f13b的基因序列和基因间序列(即,与基因区域中的基因变异处于连锁不平衡状态的插入基因间序列、上游序列和下游序列)。例如,单倍型可以是位于任何基因座处的一组母系遗传的等位基因或一组父系遗传的等位基因。
77.本文中的术语“单倍型分析”是指用于确定个体中一个或多个单倍型的过程,并且包含使用家族谱系、分子技术和/或统计推断。优选地,单倍型通过使用下一代测序技术进行测序来确定。
78.体外受精(ivf)
79.本文所描述的方法用于筛查ivf胚胎,使得可以鉴定存在具有致病性基因变异的风险的胚胎,从而允许选择合适的胚胎进行移植。ivf是其中卵子与精子在身体之外(即体外)组合的受精过程。所述过程涉及监测并在一些情况下刺激女性的排卵过程,从女性卵巢中取出一个或多个卵细胞(一个或多个卵子),并在实验室环境下让精子在合适的液体中使所述卵细胞受精。在受精卵(合子)经历胚胎培养约2天到6天后,将其植入同一或另一女性的子宫中,以期成功妊娠。通常,如果亲本难以自然受孕或者有将基因疾病传递给胚胎的风险,则他们将接受ivf。
80.本文所描述的方法适用于任何动物的ivf胚胎,只要可获得合适的参考基因组,使得可以比对测序数据以鉴定潜在的基因变异即可。例如,所述胚胎可以是人或其它非人动物胚胎。在一些实施例中,所述胚胎是人胚胎。在其它实施例中,所述胚胎是牛、绵羊、马、猪、狗、猫或其它非人动物胚胎。
81.如本文所描述的,获得“胚胎”的全基因组测序数据包含对来自以下细胞的基因组dna进行测序:来自在基因分型前不少于约40小时受精的胚胎的细胞;来自囊胚的细胞(通常是受精后第4天、第5天或第6天的胚胎);以及从胚胎活检的但起源于胚胎外(例如,滋养外胚层或极体)的细胞。因此,在一些实施例中,所述胚胎是两天、三天、四天、五天、六天或七天大的胚胎。包含此术语的复数形式,使得如本文所使用的术语“胚胎”考虑可以根据本发明的方法同时筛查或移植多于一个胚胎或囊胚。
82.在本文中进一步考虑,在条件允许的情况下,可以对胚胎的多于一个细胞进行活检。例如,可以对滋养外胚层的一个或多个细胞进行活检以获得基因组dna,用于根据本发明的方法进行测序。滋养外胚层(也称为“滋养层”)是囊胚的外层细胞,所述外层细胞为胚胎提供营养并发育成胎盘的很大一部分。它们是在妊娠的第一阶段期间形成的并且是首先从受精卵中分化出来的细胞。在一些实施例中,从胚胎活检至少2个、至少3个、至少4个、至少5个、至少6个、至少7个、至少8个、至少9个或至少10个细胞。
83.以这种方式测定多于一个细胞可以用于检测胚胎中的镶嵌性(胚胎中的细胞可能在基因上与胚胎中的其它细胞不同的情况),如果仅对单个细胞进行活检,则无法检测到所述镶嵌性。因此,如本文所考虑的,本发明的方法可以用于对第5天或第6天的胚胎进行活检,筛查胚胎的致病性基因变异,并且仍然允许在同一天新鲜移植所述胚胎。在一实施例中,基因样品是从胚胎活检的一个、两个、三个到五个、六个到十个或十一个到二十个细胞。
[0084]“移植”ivf胚胎是指将ivf胚胎放入女性受试者体内的过程,目的是胚胎将植入并引起可行的妊娠。女性受试者可以是胚胎的母本或任何其它适合移植胚胎的女性。
[0085]
如本文所考虑的,本发明的方法可以用于同时筛查一个或多个胚胎,使得可以鉴定和移植多于一个被认为不存在具有致病性基因变异的风险的ivf胚胎。可以适合移植的此类胚胎的数量可以由本领域技术人员根据常规方法确定。
[0086]
全基因组测序
[0087]
本文所描述的方法需要从ivf胚胎和胚胎的亲本获得全基因组测序数据,以便筛查胚胎的致病性基因变异。本文中的术语“全基因组测序(wgs)”是指可以确定生物体(例如人)的基因组的大部分的序列的过程。没有必要对整个基因组进行实际测序。可以使用本文所描述的任何测序技术进行全基因组测序。全基因组测序(wgs)也称为完全基因组测序、完整基因组测序或整个基因组测序。提及“全”基因组测序并不要求测序数据涵盖胚胎基因组中的每一个碱基。其只需要覆盖足够的基因组部分,以便可以预测胚胎是否存在具有致病性基因变异的风险。在一些实施例中,全基因组测序数据覆盖胚胎的基因组序列的至少60%、至少70%、至少80%或至少90%。在一些实施例中,全基因组测序数据覆盖父本的基因组序列的至少60%、至少70%、至少80%或至少90%。在一些实施例中,全基因组测序数据覆盖母本的基因组序列的至少60%、至少70%、至少80%或至少90%。具有高测序深度(或“覆盖深度”)以在数据中创建冗余也是有利的,这使得能够更准确地预测特定基因座处是否存在致病性基因变异。在一些实施例中,全基因组测序数据具有至少10x、至少20x、至少30x或至少40x的平均测序深度。
[0088]
可以通过任何方式获得全基因组测序,包含由另一方提供或通过制备合适的样品并对基因组dna进行测序。
[0089]
样品制备
[0090]
基因组测序数据可以从细胞dna中获得,所述细胞dna通过人工或机械方式从全细胞中提取基因组dna而来源于全细胞。用于从全细胞中提取基因组dna的方法是本领域已知的,并且根据来源的性质而不同。在一些情况下,将细胞基因组dna片段化可能是有利的。片段化可以是随机的,也可以是特异性的,例如,如使用限制性核酸内切酶消化所实现的。用于随机片段化的方法在本领域中是众所周知的,并且包含例如有限的dna酶消化、碱处理和物理剪切。在其它实施例中,样品核酸以细胞基因组dna的形式获得,使所述细胞基因组dna经受片段化成为大约500个或更多个碱基对的片段,并且可以容易地对其应用下一代测序(ngs)方法。
[0091]
为获得测序数据,所述方法可以包含例如通过扩增或纯化来进一步制备用于测序的基因组dna。可以使用任何合适的方法来制备用于测序的基因组dna。在一实施例中,通过扩增或通用扩增存在于基因样品中的dna来制备用于测序的基因组dna。此外,用于核酸分离和纯化的标准技术是已知的并且在以下文献中进行了描述:例如,miller(编辑)1972《分子遗传学实验(experiments in molecular genetics)》,纽约冷泉港的冷泉港实验室(cold spring harbor laboratory,cold spring harbor,n.y.);old和primrose,1994《基因操作原理(principles of gene manipulation)》,第5版,伯克利的加州大学出版社(university of california press,berkeley);schleif和wensink,1982《分子生物学实用方法(practical methods in molecular biology)》;glover(编辑)1985《dna克隆:第i
卷和第ii卷(dna cloning:vols.i and ii)》,英国牛津的irl出版社(irl press,oxford,uk);hames和higgins(编辑)1985《核酸杂交(nucleic acid hybridization)》,英国牛津的irl出版社;以及setlow和hollaender 1979《基因工程:原理和方法(genetic engineering:principles and methods)》,第1-4卷,纽约市的普莱南出版社(plenum press,new york city)。
[0092]
核酸扩增方法也是众所周知的,包含聚合酶链式反应(pcr)(《pcr方案:方法和应用指南(pcr protocols,a guide to methods and applications)》,innis编辑,纽约学术出版社(academic press,n.y.)1990;《pcr:一种实用方法(pcr:a practical approach)》,m.j.mcpherson等人,irl出版社(1991));连接酶链式反应(lcr)(landegren等人,1988);转录扩增(kwoh等人,1989);自持序列复制(guatelli等人,1990);qβ复制酶扩增(smith等人,1997)和其它rna聚合酶介导的技术,如基于核酸序列的扩增、nasba(us 4683195和us 4683202);3sr(自持序列反应);race-pcr(cdna端的快速扩增);plcr(聚合酶链式反应和连接酶链式反应的组合);sda(链置换扩增);以及soe-pcr(剪接重叠延伸pcr)。
[0093]
在一个实施例中,使用全基因组扩增(wga)方法从胚胎活检扩增基因组dna。在一个实施例中,使用多重置换扩增(mda)从胚胎活检扩增基因组dna。mda是一种非基于pcr的dna扩增技术。此方法可以将微量的dna样品快速扩增到合理的量,用于基因组分析。反应首先将随机六聚体引物退火到模板:dna合成由高保真酶,优选地φ29dna聚合酶在恒定温度下进行。与常规pcr扩增技术相比,mda产生的产物尺寸更大,错误频率更低。此方法已被积极用于全基因组扩增(wga)以获得全基因组测序数据。例如,合适的wga方法也在wo08051928中进行了描述。
[0094]
本文考虑用于分析胚胎和亲本核酸的常规方法包含允许分析来自少量细胞的核酸的方法。此类方法可以包含在实时pcr之前使用snp基因座特异性引物对dna进行“预扩增”。此类方法是本领域技术人员熟悉的方法的修改,并且用于进行此类预扩增的试剂盒可商购获得,例如来自应用生物系统公司(applied biosystems)的preamp cells-to-ct
tm
试剂盒。虽然这些试剂盒旨在预扩增源自rna的cdna,但这些试剂盒也可以成功地用于基因组dna。
[0095]
测序文库制备
[0096]
在一些实施例中,测序方法需要制备测序文库。测序文库制备涉及产生接头修饰的dna片段的随机集合,所述接头修饰的dna片段已准备好进行测序。多核苷酸的测序文库可以从dna或rna制备,包含dna或cdna的等效物、类似物,所述cdna即例如通过逆转录酶的作用从rna模板产生的互补或拷贝dna。多核苷酸可以来源于双链dna(dsdna)形式(例如,基因组dna片段、pcr和扩增产物)或可以来源于单链形式如dna或rna并被转化为dsdna形式的多核苷酸。例如,可以将mrna分子拷贝成适用于制备测序文库的双链cdna。初级多核苷酸分子的精确序列通常对文库制备方法不重要,并且可能是已知的或未知的。在一个实施例中,多核苷酸分子是dna分子。更具体地,多核苷酸分子表示生物体的整个基因互补,并且是基因组dna分子,例如cfdna分子,所述基因组dna分子包含内含子和外显子序列(编码序列)以及非编码调节序列,如启动子和增强子序列。仍更具体地,初级多核苷酸分子是存在于妊娠受试者的外周血中的人基因组dna分子,例如cfdna分子。为一些ngs测序平台制备测序文库要求多核苷酸具有特定的片段大小范围,例如0bp到1200bp。因此,可能需要多核苷酸(例
如,基因组dna)的片段化。cfdna以《300个碱基对的片段形式存在。因此,对于使用cfdna样品生成测序文库,cfdna的片段化不是必需的。通过机械手段,例如雾化、超声处理和水力剪切使多核苷酸分子片段化产生具有平末端以及3'和5'突出末端的异质混合物的片段。无论多核苷酸是被强制片段化还是以片段的形式自然存在,所述多核苷酸都被转化为具有5-磷酸和3'-羟基的平末端dna。
[0097]
通常,使用本领域已知的方法或试剂盒对片段末端进行末端修复,即平末端。平末端片段可以通过酶处理,例如使用多核苷酸激酶进行磷酸化。在一些实施例中,例如通过如taq聚合酶或klenow exo-聚合酶等某些类型的dna聚合酶的活性,将单个脱氧核苷酸,例如脱氧腺苷(a)添加到多核苷酸的3'末端。da加尾的产物与在后续步骤中连接到的接头的每个双链体区域的3'端上存在的“t”突出部相容。da加尾可防止两个平末端多核苷酸的自连接,使得有利于形成接头连接的序列。da加尾的多核苷酸连接到双链接头多核苷酸序列。相同的接头可以用于多核苷酸的两个末端,或者可以使用两组接头。连接方法是本领域已知的,并且利用如dna连接酶等连接酶将接头共价连接到d-a加尾的多核苷酸。接头可以含有5'-磷酸部分以促进与靶标3'-oh的连接。da加尾的多核苷酸含有从剪切过程中残留的或使用酶处理步骤添加的5'-磷酸部分,并且已经过末端修复,并且任选地通过一个或多个突出碱基延伸以产生3
′‑
oh用于连接。纯化连接反应的产物以去除未连接的接头、可能已经彼此连接的接头,并选择用于簇生成的模板的大小范围,在这之前可以进行扩增,例如pcr扩增。连接产物的纯化可以通过包含凝胶电泳和固相可逆固定化(spri)的方法获得。
[0098]
标准方案,例如用于使用例如illumina平台进行测序的方案指导用户在da加尾之前纯化经过末端修复的产物,并在文库制备的接头连接步骤之前纯化da加尾产物。经过末端修复的产物和da加尾的产物的纯化去除酶、缓冲液、盐等,以为后续酶促步骤提供有利的反应条件。在一个实施例中,末端修复、da加尾和接头连接的步骤不包含纯化步骤。因此,在一个实施例中,本发明的方法涵盖制备测序文库,其包括末端修复、da加尾和接头连接的连续步骤(us 20110201507)。在用于制备测序文库的不需要da加尾步骤的实施例中,例如用于使用roche 454和solid
tm
3平台进行测序的方案中,末端修复和接头连接的步骤不包含在接头连接前对经过末端修复的产物的纯化步骤。
[0099]
在所述方法的一个实施例的下一步骤中,制备扩增反应。扩增步骤将与流通池杂交所需的寡核苷酸序列引入接头连接的模板分子。扩增反应的内容是本领域技术人员已知的并且包含扩增反应所需的适当底物(如dntp)、酶(例如,dna聚合酶)和缓冲液组分。任选地,可以省略接头连接的多核苷酸的扩增。通常,扩增反应需要至少两个扩增引物,即引物寡核苷酸,所述至少两个扩增引物可以是相同的,并且包含“接头特异性部分”,所述接头特异性部分能够在退火步骤期间退火到要扩增的多核苷酸分子中的引物结合序列(或者如果模板被视为单链,则为其补体)。一旦形成,根据上述方法制备的模板文库就可以用于固相核酸扩增。如本文所使用的,术语“固相扩增”是指在固体支持物上进行或与固体支持物相关,使得所有或部分经扩增的产物在其形成时被固定在所述固体支持物上的任何核酸扩增反应。具体地,所述术语涵盖固相聚合酶链式反应(固相pcr)和固相等温扩增,除了正向和反向扩增引物之一或两者被固定在固体支持物上之外,它们是类似于标准溶液相扩增的反应。固相pcr涵盖:如乳液等系统,其中一种引物锚定在珠粒上,并且另一种引物在自由溶液中;以及在固相凝胶基质中的集落形成,其中一种引物锚定在表面上,并且一种引物在自由
溶液中。扩增后,可以通过微流体毛细管电泳对测序文库进行分析,以确保文库不含接头二聚体或单链dna。模板多核苷酸分子的文库特别适用于固相测序方法。除了为固相测序和固相pcr提供模板外,文库模板还为全基因组扩增提供模板。
[0100]
在一个实施例中,对接头连接的多核苷酸的文库进行大规模平行测序,所述大规模平行测序包含用于对数百万个核酸片段进行测序的技术,例如使用随机片段化的基因组dna与平坦、光学透明的表面的连接和固相扩增来创建具有数百万个簇的高密度测序流通池。可以使用如wo 9844151中所描述的热循环方法或其中温度保持恒定并使用试剂的变化进行延伸和变性循环的方法来制备聚簇阵列。本文提到的solexa/illumina方法依赖于将随机片段化的基因组dna连接到平坦、光学透明的表面。使连接的dna片段延伸并桥式扩增,以创建具有数百万个簇的超高密度测序流通池,每个簇含有同一模板的数千个拷贝(wo 0018957和wo 9844151)。簇模板使用稳健的四色dna边合成边测序技术来进行测序,所述技术采用具有可去除荧光染料的可逆终止子。可替代地,可以在珠粒上扩增文库,其中每个珠粒含有正向和反向扩增引物。序列读段的长度与特定的测序技术相关。ngs方法提供大小从数十个到数百个碱基对不等的序列读段。在本文所描述方法的一些实施例中,序列读段为约20bp、约25bp、约30bp、约35bp、约40bp、约45bp、约50bp、约55bp、约60bp、约65bp、约70bp、约75bp、约80bp、约85bp、约90bp、约95bp、约100bp、约110bp、约120bp、约130、约140bp、约150bp、约200bp、约250bp、约300bp、约350bp、约400bp、约450bp或约500bp。预计技术进步将实现大于500bp的单末端读段,当生成双末端读段时,能够实现大于约1000bp的读段。在一个实施例中,序列读段为36bp。本发明的方法可以采用的其它测序方法包含可以对》5000bp的核酸分子进行测序的单分子测序方法。大量序列输出由分析流水线传输,所述分析流水线将来自测序仪的主要成像输出转换为碱基串。集成算法软件包执行核心的主要数据转换步骤:图像分析、强度评分、碱基识别和比对。
[0101]
比对和变体识别
[0102]
一旦基因组dna的测序完成,就将所得的多个测序读段映射以确定其在基因组中的位置,这通过比对来实现。测序数据的比对通过将测序读段的序列与参考基因组的序列进行比较以确定经测序的dna分子的染色体来源来进行。
[0103]
在一些实施例中,所述参考基因组是人类参考基因组。在一些实施例中,所述参考基因组是某一基因组参考联盟人类版本。在一些实施例中,所述参考基因组是基因组参考联盟人类版本37(grch37)或基因组参考联盟人类版本38(grch38)或后续版本。
[0104]
许多计算机算法可用于比对序列,包含但不限于blast、blitz、fasta、bowtie、eland(美国加利福尼亚州圣地亚哥的illumina公司(illumina,inc.,san diego,calif.,usa))、burrows-wheeler aligner(li和durbin,2010)或gatk(depristo等人,2011;mckenna等人,2010)。用于鉴定多态性序列的测序信息的分析可以允许因参考基因组与胚胎或亲本基因组之间可能存在的微小多态性引起的小程度的错配(每个序列标签0-2个错配)。
[0105]
为了避免由于如等位基因或基因座丢失等扩增错误导致的误诊,在此应该理解,对所关注的等位基因进行测序可以包含对所述等位基因周围的核酸进行测序以确保扩增准确性。例如,致病等位基因可以与dna序列中附近的非致病等位基因物理连接(紧靠在一起)。dna中的这两个位点很可能一起遗传,除非所述位点之间有任何减数分裂重组。彼此靠
近的位点不太可能发生重组。因此,非致病等位基因可以用作致病等位基因的验证性标志物,以避免因疾病等位基因pcr丢失而误诊。此类技术是本领域技术人员熟悉的并且包含“单倍型分析”。合适的方法包含在wo 14145820和wo 15051006中描述的那些方法。
[0106]
一旦测序读段被映射到参考基因组,就分析比对的测序数据以定位序列数据相对于参考基因组的变异,这一过程称为“变体识别”。有大量软件包可以自动化此过程,包含freebayes、soapsnp、realsfs、samtools、gatk、beagle、impute2、mach、snvmix、varscan、deepvariant、somaticsniper、jointsnvmix、avocado、ngsep、vardict、reveel或haplotypecaller。在一些实施例中,使用haplotypecaller通过变异质量评分重新校准方法来鉴定测序数据中的变异。
[0107]
变体识别鉴定测序数据中存在的变异,所述变异可能是也可能不是胚胎或胚胎的亲本的基因组中存在的真正的基因变异。准确预测测序数据中的这些变异是否是真正的基因变异以及这些变异是否为致病性,对于从2到10个胚胎细胞中分离的dna的全基因组测序数据来说尤其成问题。为了获得足够量的dna用于高通量测序,通常对胚胎dna进行全基因组扩增(wga)。由于用于dna扩增的酶的固有错误率,此过程会将错误引入dna序列。在一些情况下,错误率是每1,000到10,000个碱基中1个。本发明分两个阶段解决此问题。首先,通过将三重测试(与父本和母本基因组测序数据比较)用于遗传的变异并将变体等位基因频率(vaf)阈值过滤器用于新生变异来评估测序数据中的变异以确定所述变异是否可能是真正的基因变异。其次,通过将变异与已知致病性基因变异的数据库进行比较来评估其引起/促成疾病的可能性。然后,在一些实施例中,可以使用一种或多种致病性预测算法进一步评估存在于测序数据中但未在数据库中分类为已知致病性(或可能致病性)变异的任何基因变异的潜在致病性。
[0108]
通过vaf阈值过滤器过滤未被鉴定为是从任一亲本遗传的基因变异,以鉴定潜在的新生基因变异。如本文所使用的,术语“变体等位基因频率”是指与支持所述变体的wgs数据中的基因组坐标重叠的测序读段部分。例如,如果有20个测序读段覆盖特定snp基因座,并且在这20个读段中,10个读段在所述位置具有“a”(其中a是参考基因组中的核苷酸)并且10个读段具有“g”(基因变异),那么在这种情况下的vaf将为50%。在实践中,来自胚胎活检的基因组dna的扩增和测序非常容易出错。因此,vaf过滤器用于去除低vaf变异,所述变异不太可能是胚胎基因组中存在的真正基因变异,但更有可能是由于扩增或测序错误造成的。因此,如本文所使用的,术语“过滤”是指根据某些条件(例如,vaf阈值)处理输入基因变异数据以产生输出。例如,输入数据可以包含所有未被鉴定为遗传的变异的变异,而输出可以只是具有高于阈值的vaf的变异。可以根据可以容忍的相对风险来选择精确的vaf阈值。较高的阈值更严格并且将使得假阳性更少,但可能会导致更多的假阴性。相反,较低的阈值不那么严格并且将使得假阳性更多,但假阴性更少。
[0109]
在一些实施例中,还将具有低于所述阈值的vaf的变异与已知致病性基因变异的数据库进行比较以确定胚胎是否存在具有致病性基因变异的风险。由于与来自经扩增的胚胎dna的wgs数据相关的高错误率,存在真阳性、低vaf、致病性新生变异可能被vaf阈值过滤掉的风险。因此,在一些情况下,(例如,通过使用“守门员”过滤器)针对任何已知的高风险致病性基因变异进一步评估低vaf(即,低于阈值)变异是有利的。在一些实施例中,通过一种或多种致病性预测算法来进一步评估vaf低于所述阈值且未被鉴定为已知致病性基因变
异的变异以预测是否为致病性基因变异。如果低vaf变异中的任何变异被鉴定为已知的致病性基因变异或被预测为致病性的,则技术人员可以人工评估与所述变异相关的风险是否足以证明不移植胚胎是恰当的,或是否需要对所述变异进行后续测试以确定其是否为真阳性。例如,可以使用测序质量度量、数据库注释源、基因/疾病严重性度量和其它可用的临床信息来执行人工评估,以确定是否要筛查胚胎。后续测试可以包含例如重新活检和直接pcr、重新测试全基因组扩增的dna,或测试胚胎培养基中由胚胎渗出的dna。
[0110]
候选变异也可以使用其它度量进行评估,如质量/深度(qd)、baseqranksum、strand bias-fisher's、映射质量、mqranksum、readposranksum、clippingranksum、gq_mean、gq_stddev或对称优势比(sor),所述度量可以使用基因组分析工具包(gatk;https://software.broadinstitute.org/gatk/)中免费提供的软件工具计算。在一些实施例中,将基因变异鉴定为新生变异包含计算其qd评分。在一些实施例中,将基因变异鉴定为新生变异需要的qd评分为至少5、至少6、至少7、至少8、至少9、至少10、至少11、至少12、至少13、至少14或至少15。在一些实施例中,将基因变异鉴定为新生变异需要的qd评分为至少12。
[0111]
wgs数据中存在的变异预测的置信水平也可以使用phred标度cadd评分来评估。phred标度cadd评分是对由自动dna测序生成的单个核碱基鉴定质量的衡量,并且与每个碱基的碱基识别错误概率呈对数相关。它最初是为phred碱基识别而开发的,以帮助人类基因组计划(human genome project)中的dna测序自动化。phred标度cadd评分分配给自动测序仪跟踪中的每个核苷酸碱基识别。phred标度cadd评分可以用于比较不同测序方法的功效。在一些实施例中,将变异预测为致病性基因变异需要变异具有大于约20的phred标度cadd评分。在一些实施例中,需要所述phred标度cadd评分大于25,或大于约30,或大于约35。
[0112]
在另一实施例中,dann(quang等人,2015)用于评估wgs数据中存在的变异的预测的置信水平。
[0113]
基因变异
[0114]
变异的类型
[0115]
术语“基因变异”、“多态性”和“变体”在本文中可互换使用,以指代胚胎的基因组(或任一亲本的基因组)的基因序列相对于参考基因组发生变异。每个趋异序列称为等位基因,并且可以是基因的一部分或位于基因间或非基因序列内。双等位基因变异具有两个等位基因,并且三等位基因变异具有三个等位基因,依此类推。如人类等二倍体生物体可以含有两个等位基因,并且对于等位基因形式可以是纯合的或杂合的。第一个鉴定的等位基因形式被任意指定为参考形式或等位基因;其它等位基因形式被指定为替代或变体等位基因。所选群体中最常出现的等位基因形式通常称为野生型形式。基因变异涵盖序列差异,所述序列差异包含单核苷酸多态性(snp)、串联snp、小规模多碱基缺失或插入(称为“indel”(也称为缺失插入多态性或dip))、多核苷酸多态性(mnp)、短串联重复(str)、限制性片段长度多态性(rflp)、缺失(包含微缺失)、插入(包含微插入)、重复、倒位、易位、倍增、复杂多位点变体、拷贝数变异(cnv)和其它结构变异,包括染色体中任何其它序列变化。基因组序列的差异包含不同类型变异的组合。例如,基因变异可以涵盖一种或多种snp和一种或多种str的组合。
[0116]
术语“单核苷酸多态性(snp)”是指群体中个体之间dna序列中的单碱基(核苷酸)
多态性。snp可以存在于基因的编码序列、基因的非编码区域内或基因之间的基因间区域中。由于基因密码的简并性,编码序列内的snp不一定会改变所产生蛋白质的氨基酸序列。两种形式产生同一多肽序列的snp被称为“同义的”(有时称为沉默突变),如果产生不同的多肽序列,则它们是“非同义的”。非同义变化可以是错义或“无义”,其中错义变化产生不同的氨基酸,而无义变化产生提前终止密码子。snp也可以由相对于参考等位基因的核苷酸缺失或核苷酸插入产生。单核苷酸多态性(snp)是人类群体中两个替代性碱基以明显的频率(》1%)出现的位置,并且是最常见的人类基因变异类型。
[0117]“indel”是生物体基因组中碱基的插入或缺失。其通常被分类为小的基因变异,长度为1个到10 000个碱基对。“微indel”通常定义为导致1个到50个核苷酸的净变化的indel。在基因组的编码区域中,除非indel的长度为3的倍数,否则会产生移码突变。例如,导致移码的常见微indel在犹太人或日本人群体中导致布卢姆综合征(bloom syndrome)。indel可以与点突变(snp)形成对比。indel将核苷酸插入序列中和从序列中删除核苷酸,而点突变(snp)是一种其在不改变dna中核苷酸总数的情况下替换核苷酸之一的取代形式。mrna编码部分中单个碱基对的indel变化导致mrna翻译期间的移码,这可能导致不同框中的不适当(提前)终止密码子。不是3的倍数的indel在编码区域中特别少见,但在非编码区域中相对常见。indel可能占人类所有序列多态性的16%到25%。
[0118]
如本文所使用的,术语“结构变异”用于指生物体的染色体结构的任何变异。其包括基因组中的多种变异,并且通常包含微观和亚微观类型、缺失、重复、拷贝数变体、插入、倒位和易位。通常,结构变异与snp相比影响更大的序列部分,但小于染色体异常(尽管定义具有一些重叠)。在一些实施例中,结构变体是影响超过50个碱基的区域的基因事件。结构变异的定义并不暗示关于频率或表型效应的任何内容。许多结构变体与基因疾病有关,然而也有许多与基因疾病无关。最近关于sv的研究表明,sv比snp更难检测。大约13%的人类基因组在正常人群中被定义为结构变体,并且在人类群体中至少有240个基因以纯合缺失多态性的形式存在,这表明这些基因在人类中是可有可无的。因此,结构变异可以在每个基因组中包括数百万个异质性核苷酸,并且可能是人类多样性和疾病易感性的重要原因。
[0119]
本文中的术语“拷贝数变异”是指一种结构变异,其是与合格样品中存在的通常约1kb或更大的核酸序列的拷贝数相比,测试样品中存在的所述核酸序列的拷贝数的变异。“拷贝数变体”是指约1kb或更大的核酸序列,其中通过比较测试样品中所关注的序列与合格样品中存在的序列发现拷贝数差异。拷贝数变体/变异包含缺失(包含微缺失)、插入(包含微插入)、重复、倍增、倒位、易位和复杂多位点变体。cnv涵盖染色体非整倍性和部分非整倍性。
[0120]
本文中的术语“短串联重复”或“str”是指当两个或更多个核苷酸的模式重复并且重复的序列彼此直接相邻时发生的一类多态性。所述模式的长度范围可以是2个到10个碱基对(bp)(例如,基因组区域中的(catg)n),并且通常位于非编码内含子区域中。通过检查若干个str基因座并对给定基因座处具体str序列的重复次数进行计数,可以创建个体的唯一基因谱。
[0121]
一旦在全基因组测序数据中鉴定出了候选基因变异,就评估所述候选基因变异导致疾病的可能性。因此,评估基因变异以确定其是否是致病性基因变异。如本文所提及的“致病性基因变异”是与特定疾病相关的基因变异。在此上下文中,受试者基因组中的致病
性变异不一定是其存在与否会决定所述受试者是健康还是患病的变异。“致病性”仅仅意指基因变异被认为以某种方式促成疾病。因此,隐性疾病的致病变体的单个拷贝被认为是致病性的,即使单独的所述单个拷贝不会引起疾病。类似地,致病性基因变异包含高外显率基因变异和低外显率基因变异。
[0122]
已知致病性基因变异的数据库
[0123]
例如,可以通过查询已知基因变异的数据库来鉴定致病性基因变异,所述已知基因变异被注释有其致病性水平。可替代地,对于不在此类数据库中或其致病性水平不确定的基因变异,可以使用致病性预测算法来确定所述变异是否可能是致病性的。
[0124]
已知致病性基因变异的合适数据库包含clinvar(https://www.ncbi.nlm.nih.gov/clinvar/)、clinvitae(http://clinvitae.invitae.com/)、leiden开放变体数据库(leiden open variant database,lovd;http://www.lovd.nl/)、人类基因变异数据库(human genetic variation database,hgvd;http://www.hgvd.genome.med.kyoto-u.ac.jp/)、在线人类孟德尔遗传(online mendelian inheritance in man,omim;https://www.omim.org/)、egl的变体分类目录(egl's variant classification catalog,emvclass;http://www.egl-eurofins.com/emvclass/emvclass.php)、arup突变数据库(http://www.arup.utah.edu/database/)或carver等位基因特异性变异数据库(https://www.carverlab.org/database)。
[0125]
在一个实施例中,clinvar数据库用于鉴定已知的致病性基因变异。clinvar是带有支持性证据的人类基因变异和表型之间关系的报告的可自由访问的公共档案。因此,clinvar记录了断言的人类变异与观察到的健康状况之间的关系,以及这种解释的历史。clinvar处理报告患者样品中发现的变异、关于其临床意义(即,致病性水平)的断言、关于提交者的信息以及其它支持性数据的提交文件。提交文件中描述的等位基因被映射到参考序列,并根据hgvs标准进行报告。
[0126]
根据美国医学遗传学和基因组学学院(acmg)指南,clinvar数据库条目通常具有五个临床意义(致病性)水平之一:良性、可能良性、意义不确定、可能致病性或致病性(richards等人,2015)。在一些实施例中,在步骤f)中确定基因变异是致病性基因变异需要所述基因变异在clinvar数据库中具有致病性或可能致病性的临床意义值。
[0127]
变异识别和临床意义断言的准确性的置信水平在很大程度上取决于支持性证据。由于支持性证据的可用性可能会有所不同,特别是在从已发表文献中汇总的回顾性数据方面,clinvar汇总了来自多个组的相关信息,以透明地反映一致和冲突的临床意义断言。审查状态也被分配给任何断言,以支持关于任何断言的可信度的交流。“审查状态”是对与断言的基因变异的临床意义相关的确定性水平的四星衡量。例如,具有四星审查状态的数据库条目被认为是“实践指南”,因为在变异的临床意义水平上存在高度一致性。相反,一星的审查状态反映了变体临床意义的一致性水平较低,例如在多个提交者提供了临床意义的证据,但存在冲突的解释的情况下。因此,在一些实施例中,在本发明的方法的步骤f)中确定基因变异是致病性基因变异需要所述变异在clinvar数据库中具有至少两星或至少三星或四星的审查状态。有关clinvar审查状态的另外的信息,可在https://www.ncbi.nlm.nih.gov/clinvar/docs/review_guidelines/中获得。
[0128]
可以用于权衡与已知致病性变体相关信息的背景的其它因素包含但不限于其外
显率、信息基础研究的统计效力、信息基础研究所涉及的对照的数量和类型、是否已知治疗剂基于信息可预测地起作用、是否已知通路中的多个突变会引起可预测的表型、知识库中是否存在矛盾的证据以及此类证据的数量/可信度、是否在健康个体中经常观察到破坏同一基因/通路的一个或多个变体、变体发生的位置或区域是否在进化上高度保守,和/或与变体相关的拟表型是否存在并可预测地起作用。
[0129]
致病性预测算法
[0130]
对于未知致病性的基因变异(即,不存在于已知致病性变异的数据库中或存在但审查状态较低),可以通过使用一种或多种致病性预测算法来计算所述基因变异为致病性的可能性。可以使用被配置成评估基因变异导致疾病的可能性的任何合适的算法。
[0131]
例如,这些算法可以预测基因变体是否被预测为无害(例如,使用功能预测算法,如sift或polyphen)。以下算法可以单独使用或组合使用作为致病性预测算法的一部分:sift、polyphen、polyphen2、panther、snps3d、fastsnp、snap、ls-snp、pmut、pupasuite、snpeffect、snpeffectv2.0、f-snp、mapp、phd-snp、mutdb、snp功能门户(snp function portal)、polydoms、snp@promoter、auto-mute、mutpred、snp@ethnos、nssnpanalyzer、snp@domain、stsnp、mtsnpscore、mutationtaster2、mutationassessor、fathmm或基因组变异服务器(genome variation server)。可以利用这些算法和本领域已知的试图预测变异对蛋白质功能、活性或调节的影响的其它合适算法。例如,预测的转录因子结合位点、ncrna、mirna靶标、增强子和utr可以并入到算法中以进行数据分析。可以不同地处理与编码区域相关的变体和与非编码区域相关的变体。类似地,可以不同地处理与外显子相关的变体和与内含子相关的变体。进一步地,可以不同地处理编码区域中的同义变体与非同义变体。在一些情况下,在分析密码子变化时可以考虑受试者的翻译机制。
[0132]
在一些实施例中,致病性预测算法确定与变体相关的序列是否在进化上是保守的。可以预期在进化上高度保守的那些序列中出现的变体会更有害,并且因此在一些实施例中,致病性预测算法可以根据应用保留(或去除)这些变体。可以用于量化核苷酸水平进化保守程度的一种度量是基因组进化速率分析(gerp)。
[0133]
在一些实施例中,致病性预测算法评估与变体相关的氨基酸置换的性质。例如,可以计算格兰瑟姆矩阵评分(grantham matrix score)。类似地,在一些实施例中,根据多态性表型分析(例如,polyphen2)和/或从容忍中区分不容忍(sorting intolerant from tolerant,sift)算法来评估变异。
[0134]
在一些实施例中,所述一种或多种致病性预测算法包含sift、polyphen2 hvar、mutationtaster2、mutationassessor、fathmm或fathmm mkl。在一些实施例中,使用两种或三种或四种或五种或所有六种算法。因此,报告变异被预测为致病性的算法的数量可以用作预测中的置信水平。
[0135]
用于预测基因变异的致病性的合适方法和算法包含例如在wo 2015061422、us 20120310863、us 20140359422和us 20160371431中描述的那些。
[0136]
在一些实施例中,为了预测基因变异是否是致病性的,生成mpc评分。如本文所使用的,“mpc评分”是指如samocha等人,2017(biorxiv,http://dx.doi.org/10.1101/148353)中所描述的“错义不良、polyphen-2和约束评分”。mpc评分衡量氨基酸取代发生在错义约束区域时的有害性增加。其可以用于将来自正交有害性度量的信息组合成一个评
分。在一些实施例中,确定基因变异是致病性的需要所述基因变异具有大于或等于2的mpc评分。
[0137]
由致病性基因变异引起的疾病
[0138]
与可以通过本方法鉴定的致病性基因变异相关的疾病是基因疾病,所述基因疾病是至少部分由基因或染色体异常引起的疾病。此类疾病包含单基因疾病(monogenic disease),即单基因疾病(single gene disease),和多基因疾病,即复杂疾病。单基因疾病包含常染色体显性、常染色体隐性、x连锁显性、x连锁隐性和y连锁。本发明的方法可以用于筛查携带此类疾病的等位基因的亲本的胚胎,从而选择合适的胚胎进行移植。
[0139]
本方法可以用于筛查存在罹患常染色体显性疾病的风险的胚胎。在常染色体显性疾病中,一个人受所述疾病感染只需要基因的一个突变拷贝。通常,受感染的受试者具有一个受感染的亲本,并且后代有50%的机会遗传突变基因。常染色体显性病状有时具有降低的外显率,这意味着虽然只需要一个突变拷贝,但并非所有遗传所述突变的个体都会继续罹患所述疾病。常染色体显性疾病的实例包含但不限于家族性高胆固醇血症、遗传性球形红细胞增多症、马凡综合征(marfan syndrome)、1型神经纤维瘤病、遗传性非息肉病性结直肠癌和遗传性多发性外生骨疣和亨廷顿氏病(huntington's disease)。
[0140]
本方法也可以用于筛查存在罹患常染色体隐性疾病的风险的胚胎。在常染色体隐性疾病中,基因的两个拷贝必须发生突变,受试者才会受到常染色体隐性疾病的感染。受感染的受试者通常具有未受感染的亲本,所述亲本各自携带突变基因的单个拷贝(并且被称为携带者)。各自携带突变基因的一个拷贝的两个未受感染的人每次妊娠都有25%的机会生下受疾病感染的孩子。这类疾病的实例包含囊性纤维化、镰状细胞病、泰-萨二氏病(tay-sachs disease)、尼曼-皮克病(niemann-pick disease)、脊髓性肌萎缩、罗伯茨综合征(roberts syndrome)、粘多糖贮积症、糖原贮积病和半乳糖血症。如湿耳垢与干耳垢等某些其它表型也以常染色体隐性方式确定。
[0141]
本方法也可以用于筛查存在罹患x连锁显性疾病的风险的胚胎。x连锁显性疾病是由x染色体上的基因突变引起的。只有少数疾病具有此遗传模式,典型的实例是x连锁低磷性佝偻病。男性和女性都受到这些疾病的感染,其中男性通常比女性受到更严重的感染。一些x连锁显性病状,如雷特氏综合征(rett syndrome)、2型色素失调症和爱卡迪综合征(aicardi syndrome)通常在男性中是致命的,并且因此主要见于女性。此发现的例外是极其罕见的情况,其中患有克兰费尔特综合征(klinefelter syndrome)(47、xxy)的男孩也遗传了x连锁显性病状,并且在疾病严重程度方面表现出与女性更相似的症状。男性和女性传递x连锁显性疾病的机会不同。患有x连锁显性疾病的男性的儿子都不会受到感染(因为他们接受了其父亲的y染色体),而他的女儿们都将遗传所述病状。患有x连锁显性疾病的女性每次妊娠都有50%的机会生下受感染的胎儿,但应该注意的是,在如色素失调症等情况下,通常只有女性后代可以存活。另外,尽管这些病状本身不会改变生育能力,但患有雷特氏综合征或爱卡迪综合征的个体很少生育。
[0142]
本方法也可以用于筛查存在罹患x连锁隐性疾病的风险的胚胎。x连锁隐性病状也是由x染色体上的基因突变引起的。男性比女性受感染的频率更高,并且男性和女性传递所述疾病的机会也不同。患有x连锁隐性疾病的男性的儿子不会受到感染,而他的女儿将携带一个突变基因拷贝。作为x连锁隐性疾病(xrxr)携带者的女性有50%的机会生下受感染的
儿子,并有50%的机会生下携带一个突变基因拷贝的女儿,并且因此是携带者。x连锁隐性病状包含但不限于严重疾病a型血友病、杜氏肌营养不良(duchenne muscular dystrophy)和莱施-奈恩综合征(lesch-nyhan syndrome)以及常见和不太严重的病状,如男性型脱发和红绿色盲。由于偏移的x失活或单体x(特纳综合症(turner syndrome)),x连锁隐性病状有时可能会出现在女性身上。
[0143]
本方法也可以用于筛查存在罹患y连锁疾病的风险的胚胎。y连锁疾病是由y染色体上的突变引起的。因为男性从他们的父亲那里遗传了y染色体,所以受感染的父亲的每个儿子都会受到感染。因为女性从她们的父亲那里遗传了x染色体,所以受感染的父亲的女性后代永远不会受到感染。由于y染色体相对较小并且含有的基因很少,因此y连锁疾病相对较少。通常症状包含不育,这可以在一些生育治疗的帮助下避免。实例是男性不育和耳廓多毛症。
[0144]
本方法还可以用于筛查存在罹患基因疾病的风险的胚胎,所述基因疾病是复杂、多因素或多基因的,这意味着所述疾病可能与多个基因与生活方式和环境因素组合的影响有关。多因素疾病包含例如心脏病和糖尿病。虽然复杂疾病经常在家族中聚集,但它们没有明确的遗传模式。在谱系上,多基因疾病确实倾向于“家族遗传”,但遗传并不像孟德尔疾病(mendelian disease)那样简单。强烈的环境组分与许多复杂疾病相关,例如,血压。本方法可以用于鉴定与多基因疾病相关的致病性基因变异,所述多基因疾病包含但不限于哮喘、如多发性硬化症等自身免疫性疾病、癌症、纤毛类疾病、腭裂、糖尿病、心脏病、高血压、炎症性肠病、精神发育迟缓、情绪疾病、肥胖症、屈光不正和不育。
[0145]
计算机实施的方法
[0146]
设想本公开的方法至少部分地可以由如计算机等系统来实施。例如,所述系统可以是包括一个或多个连接到存储器的可以一起操作的处理器(为方便起见称为“处理器”)的计算机系统。存储器可以是非暂时性计算机可读介质,如硬盘驱动器、固态盘或其它介质。软件,即可执行指令或程序代码(如分组为代码模块的程序代码)可以存储在存储器上,并且当由处理器执行时,可以使计算机系统执行功能,如确定要执行任务以帮助用户:接收全基因组测序数据;处理数据,例如,将数据与参考基因组比对以鉴定变异;比较胚胎与亲本之间的变异,以鉴定遗传的基因变异;向所鉴定的变异应用各种过滤器,例如,vaf阈值过滤器;将变异与已知致病性变异的数据库进行比较;和/或输出已在胚胎基因组中鉴定的任何致病性基因变异。
[0147]
在另一实施例中,所述系统可以耦接到用户界面以使所述系统能够从用户接收信息和/或输出或显示信息。例如,用户界面可以包括图形用户界面、语音用户界面或触摸屏。
[0148]
在一实施例中,所述系统可以被配置成通过如无线通信网络等通信网络与至少一个远程装置或服务器进行通信。例如,所述系统可以被配置成通过通信网络从装置或服务器接收信息,并且通过通信网络将信息传输到相同或不同的装置或服务器。在其它实施例中,所述系统可以与直接用户交互隔离。
[0149]
实例
[0150]
材料和方法
[0151]
研究参与者
[0152]
因单基因疾病而接受pgt的夫妇为对他们自己和他们的为研究而选择的活检的胚
胎进行wgs签署了进一步的知情同意书(handyside等人,2010)。每个参与的男性和女性伴侣都有权选择报告或保留其dna和活检的胚胎样品的结果。所述研究和方案根据接受的建议获得了蒙纳士健康人类研究伦理委员会(monash health human research ethics committee)的批准。所有受试者均根据《赫尔辛基宣言(declaration of helsinki)》被给予书面知情同意书。
[0153]
文库制备和测序
[0154]
来自五对夫妇的储存的基因组dna(gdna)样品被选择用于在华大基因(bgi genomics)(中国香港大埔(tai po,hong kong))进行wgs,所述样品曾被用作核映射(karyomapping)pgt病例的参考模板。使用reliaprep
tm blood gdna miniprep system(美国普洛麦格公司(promega,usa))从全血中提取dna。为了分离胚胎dna,icsi创建的属于五个pgt病例的胚胎在培养的第 5天或第 6天接受了使用激光或机械技术的滋养外胚层(te)活检,以取出估计的4个到10个te细胞。按照制造商的说明,将活检细胞在1x pbs缓冲液(美国细胞信号传导技术公司(cell signalling technologies,usa))和1x聚乙烯吡咯烷酮(澳大利亚库克医疗公司(cook medical,australia))的溶液中洗涤三次,然后使用suremda系统(美国illumina公司(illumina,usa))通过多重置换扩增进行全基因组扩增(wga)。用于wgs的样品是基于核映射质量控制度量来选择的,所述度量指示humancytosnp-12beadarray上的snp检出率》96%并且等位基因丢失率和误检率《1%。将1ug到2ug的基因组dna和wga产物发送到华大基因(中国香港大埔)以利用bgi-seq500进行测序。简而言之,用e220 covaris(covaris公司(covaris inc.))将dna样品片段化到大约350bp,然后进行3'末端修复、接头连接和通过连接介导的pcr进行的扩增、单链分离和环化。dna纳米球是通过滚环扩增产生的,放置在图案化的纳米阵列中,所述纳米阵列由bgi-seq500进行双末端读取(patch等人,2018)。
[0155]
读段处理
[0156]
通过bgi online(van der auwera等人,2013),根据基因组分析工具包(gatk)执行读段处理直到变异识别格式(vcf)。使用burrows-wheeler比对器(burrows-wheeler aligner)(li和durbin,2010)将样品映射到人类参考基因组(grch37/hg19),使用picard工具(https://broadinstitute.github.io/picard/)去除pcr重复,并与gatk(depristo等人,2011;mckenna等人,2010)和通过变异质量评分重新校准方法用haplotypecaller识别的变异进行局部重新比对。
[0157]
snp和indel分析
[0158]
分析遵循用于解释序列变异的《acmg标准和指南(acmg standards and guidelines)》(rehm等人,2013;richards等人,2015)。将每个伴侣和胚胎bam和原始vcf文件导入varseq(美国goldenhelix公司(goldenhelix,usa))。针对以下遗传模式布置变异过滤工作流:i.显性杂合,ii.隐性纯合,iii.复合杂合,iv.x连锁,v.新生和vi.“守门员”过滤器组,以用于捕获潜在等位基因丢失或低变体等位基因频率区域内的变异(图1)。对于未分类的变异(即,未在已知致病性基因变异数据库中发现的变异),针对以下六种预测算法启用了严格的致病性功能预测过滤器:sift、polyphen2 hvar、mutationtaster2、mutationassessor、fathmm和fathmm mkl(ng和henikoff,2003;adzhubei等人,2013;schwarz等人,2014;reva等人,2011;shihab等人,2014)。如果六种算法中的任何一种算法
预测变异为“破坏性”,则保留所述变异以在过滤器链中继续评估。》2的mpc评分和/或》35的最终phred评分终止了突变预测过滤器组(图2)(samocha等人,2017;kircher等人,2014)。使用expansionhunter工具计算串联重复,以确定胚胎和男性和女性生殖伴侣的串联重复数(dolzhenko等人,2017)。在华大基因对以下基因座进行了计算:所有成人和胚胎样品上的cbl原癌基因(cbl)、atrophin 1(atn1)、ataxin 2(atxn2)、ataxin 3(atxn3)、亲联蛋白3(junctophilin 3,jph3)、钙通道、电压依赖性、p/q型、α1a亚基(cacna1a)、肌营养不良性肌强直症蛋白激酶(dmpk)、胱抑素b(cstb)、ataxin 10(atxn10)、ataxin 7(atxn7)、亨廷顿蛋白(huntingtin,htt)、蛋白磷酸酶2、调节亚基bβ(ppp2r2b)、ataxin 10(atxn1)、染色体9开放阅读框72(c9orf72)、frataxin(fxn)、雄激素受体(ar)和脆性x智力迟钝1(fmr1)。
[0159]
拷贝数和结构变异
[0160]
cnv识别是使用cnvnator(v.0.2.7)(abyzov等人,2011)进行的,并且结构变异是使用breakdancer(fan等人,2014)和crest(wang等人,2011)进行的。第二次分析是通过分箱成10kb窗口并使用clingen基因剂量敏感性(clingen gene dosage sensitivity)(27-09-2017)进行注释来进行的,所述clingen基因剂量敏感性通过》95%的识别loh进行过滤(kearney等人,2011;riggs等人,2018)。将cnvnator和breakdancer识别导入varseq中,与来自每个亲本的遗传的cnv进行比较,并归类为对单倍剂量不足或三倍剂量敏感性具有剂量致病性。三重识别杂合性丢失(loh)区域(》100%和95%的变异),与夫妇的loh区域进行比较。将过滤应用于“剂量致病性的充分证据”或“与常染色体隐性表型相关的基因”的单倍剂量不足和三倍剂量敏感性类别,并使用每个样品先证者的目标拷贝数状态,通过对重复应用》2.0的比率和》0的z评分,以及《0.5的比率与《0的z评分,平均目标平均深度》5且缺乏qc标志(高对照变异、低对照深度、低z评分,在区域iqr内)来识别致病性。针对常染色体隐性snp和indel变异对具有隐性遗传的cnv进行交叉检查(图3)。
[0161]
实例1-pgt变异验证
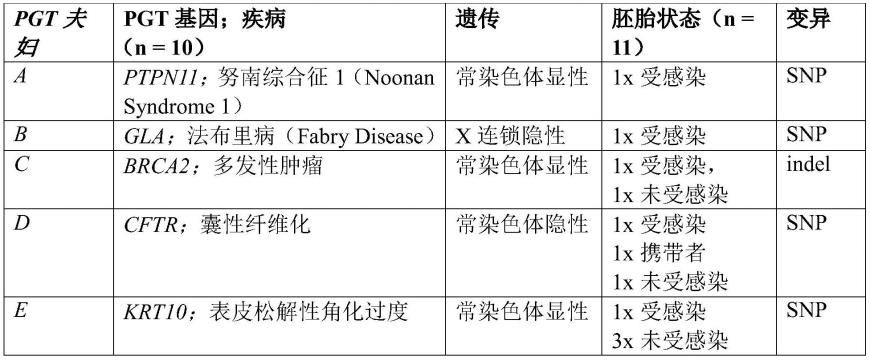
[0162]
所有参与的夫妇都同意wgs并选择接收他们自己和pgt胚胎的结果。三个家庭因常染色体显性病状接受了pgt,一个为常染色体隐性并且一个为x连锁(表1)。对于五个家庭中的三个,可能的携带者状态情形(即受感染/携带者/未受感染)中的每种情形中至少有一个整倍体胚胎可用。将从vcf文件识别的杂合子变异与先前通过核映射获得的变异进行比较,显示出与wgs识别的一致性》99%。
[0163]
表1
–
来自pgt的亲本中的先前已知的变异和受感染的胚胎数量
[0164]
[0165]
实例2
–
经扩增的胚胎dna的测序和映射到伴侣基因组dna
[0166]
来自胚胎的经扩增的滋养外胚层活检dna与夫妇基因组的测序深度相当(平均48.2x对46.1x)。对于原始和干净读段、比对的碱基以及转换突变与颠换突变的比率,胚胎与亲本基因组dna样品等效(tv/ti;2.071对2.081,表2)。
[0167]
表2
–
测序和变异统计
[0168][0169][0170]
胚胎和夫妇的基因组覆盖率在4x和10x下相当。然而,在20x下,与亲本dna的覆盖率96.4%相比,活检的胚胎的覆盖率下降到87.5%。snp/indel识别的组装和映射度量在胚胎与夫妇之间高度一致,但新型snp除外,对于胚胎,平均识别85,527个新型snp(s.d.29,576.6),而夫妇平均具有21,663个新型snp(s.d.1102.4)。因此,变异过滤器被布置成去除假阳性新生变异,同时保留任何被认为临床上可执行的变异。对于父本和母本测序数据,在0.5处观察到变异等位基因频率(vaf)峰值,这与针对真杂合变异的预期一致。另一个假阳性峰值出现在约0.25处(范围为0.08到0.34,图4)。因此,利用vaf《0.35在胚胎测序数据中识别的新生变异被过滤掉以降低假阳性的风险,但被过滤掉的高风险新生变异被保留用于手动检查和潜在管理(使用“守门员”过滤器)。有趣的是,观察到与涉及碱基改变的snp相
比,涉及》1bp缺失的snp的vaf更高,但是并未基于此改变过滤,因为上限大致一致。
[0171]
实例3-变异三重识别
[0172]
先前在pgt中鉴定的变异在男性/女性伴侣和胚胎中得到证实,其中在所有11个胚胎中完全一致(表1)。尽管通过显示出与亲本中的突变一致证实了突变的存在,但一个胚胎的pgt变异具有明显较低的变异等位基因频率(vaf=0.143;3/21读段),所述突变由遗传病理学实验室通过测序为夫妇分型以进行pgt。对于胚胎,与常见snp的连锁不平衡用于已知的突变携带者、受感染和未受感染的亲本和/或同胞。与clinvar审查状态介于1星与2星之间(共4星)的1.27个分类的和0.45个未分类的新生显性变异相比,在消除作为pgt指征的变异后,每个胚胎平均有0.82个“分类的”和1.27个“未分类的”隐性新生变异被识别。胚胎中识别为杂合性丢失(loh)的区域的平均数量显著高于夫妇(3733个区域,s.d.87对5460个,s.d.1609)。通过杂合性丢失(loh;》95%和100个变异)手动检查位于等位基因丢失和/或低覆盖的胚胎区域内的任一成体携带的致病性变异,以确定胚胎覆盖》10个读段未覆盖的变异。由于覆盖阈值或loh低,胚胎测序中平均有2.3个被分类为可能致病性/致病性(根据clinvar)的变异(sd
±
1.2)缺失,而所有非pgt变异在证据水平上被列为clinvar审查状态“2星”(0星表示无断言标准,即证据最少;4星最高,表示实践指南)。低覆盖率的缺失致病性变异通过缺失区域最近的侧接snp分期以确定携带者状态。平均4.5个各自在胚胎中发现的来自可能致病性或致病性变异的变异(s.d.3.7)和5.5个未分类的变异(s.d.3.4)被认为可能为致病性的,需要单倍型管理以考虑到潜在遗传的但缺失的致病性变异的丢失。
[0173]
为了避免过滤掉真阳性新生突变,使用“守门员”过滤器容器来捕获临床相关的变异以进行手动管理。消除pgt变异后,在11个胚胎中共检测到17个具有3星审查状态的变异,其中没有一个是临床上可执行的必需/发育迟缓基因。审查状态分类揭示,只有“守门员”过滤器容器有缺失识别,其中平均值为2.36(s.d.3.86);没有一个守门员变异导致复合杂合子具有遗传的和传递的变异。在胚胎样品中的任何胚胎样品中都没有发现clinvar审查状态1星——“冲突的解释”。类似地,在女性中没有复合杂合子、纯合常染色体隐性(即alt/alt)或x连锁,或在胚胎或男性或女性伴侣基因组中的acmg偶然发现变异中没有可能致病性/致病性。在11个胚胎中有109个候选致病性新生突变,其中9个变异在多个胚胎中重复出现,除了两个之外,所有变异都在多于一个家庭中出现。鉴定了14个候选新生常染色体显性致病性变异(表3)。其中,10个变异的vaf《0.4,表明其中大部分为假阳性识别的概率很高(表3)。
[0174]
表3-在活检的胚胎中鉴定的常染色体显性新生突变。
[0175][0176][0177]
如图6和图7所示,还根据质量/深度(qd)评分评估候选新生突变。由于测序伪影或
早期扩增阶段碱基错误掺入,观察到胚胎新生识别中的qd值表现出低vaf(00.3),这与qd值《12一致。虽然可以使用较低的值,但这是基于qd值的用于鉴定胚胎中的新生突变的适当假阳性过滤器的一个实例。
[0178]
实例4-拷贝数和结构变异
[0179]
除了染色体间结构变异和结构缺失外,经扩增的胚胎和亲本样品的cnv识别增加,表明假阳性率很高(表1)。通过将读段分箱在10kb窗口clingen基因剂量敏感性评分中来评估cnv的致病性和遗传性。如根据核映射结果所预期的,没有检测到预测为致病性的cnv。夫妇和胚胎平均有2.0个有害常染色体隐性结构变异(sv),相比之下,三倍剂量敏感性在夫妇和胚胎中作为常染色体隐性起作用分别平均有5.21个和8.05个sv。
[0180]
实例5-串联重复疾病基因座分析
[0181]
对于expansion hunter评估已知致病基因座处的串联重复数的17个基因座,没有亲本样品表明致病性重复数。在胚胎样品中,大多数测试的基因座在每个胚胎中提供了至少一个在传递准确性方面的一致识别。在三个基因座处,两个等位基因都不一致;fmr1、atxn1和atxn3。
[0182]
讨论
[0183]
成功完成了对人胚胎整个基因组的致病性突变筛查,证明了全基因组测序筛查活检的ivf胚胎的致病性基因变异的可行性。包含使用孕前测试引入的新生突变和前突变三核苷酸重复疾病在内,如果任何一对夫妇如此选择,则儿童疾病的风险、真正全组已知病因的疾病都是可以预防的。
[0184]
总而言之,经过多重置换扩增(mda)的胚胎活检样品和相应的亲本基因组dna样品用作用于生成dna文库的模板,随后对所述dna文库进行测序以获得全基因组测序数据。多个三重测试用于通过疾病/变异分类和变异过滤将变异分类为“可能致病性”或“致病性”。对胚胎和相应的亲本的基因组进行测序,并且使用已知致病性变异的数据库和功能预测算法来解释以检测致病性突变的传递或引入。新生变异识别提出了独特的挑战;定制的过滤器组最小化了由来自单碱基插入的mda扩增掺入错误引起的假阳性,所述单碱基插入以vaf《0.35进行识别。确定新生变异识别的有效性需要克服任何早期扩增循环聚合酶碱基掺入错误、等位基因丢失和错误比对的读段,它们会产生假阳性变异。4个到10个细胞的滋养外胚层活检技术赋予胚胎发育和植入率的优势是通过最大化胚胎基因组测序覆盖率而带来另外的益处。通过等位基因比率和单倍型评分最小化扩增和测序伪影有效地将候选新生突变的数量最小化为可以在必要时手动管理的数量。
[0185]
通过vaf阈值对新生变异进行的初始严格过滤被“守门员”过滤器组抵消,所述过滤器组旨在执行逆向低敏感性功能,但可检测临床上可执行的(高风险)变异。最终,0.35个过滤器的vaf阈值充当新生致病性变异的置信度的指南。在胚胎结果之一中在非常低的vaf的情况下出现的先前鉴定的pgt突变的存在例证了每种疾病/变异亚型需要特定的过滤器组。为了规避在低覆盖或缺失覆盖区域中传递的非孟德尔致病性变异未被检测到的情形,“未传递的”变异过滤器手动检查单倍型侧接的未识别变异,以确认每个位点的结果。来自所研究胚胎的mda扩增的dna表现出的均匀覆盖表明,在覆盖率低的区域中出现致病性新生突变的可能性很小。这些ii类错误率通过“守门员”低覆盖率评估过滤器得到减轻,尽管loh和vaf过滤器可以在一定程度上指导决策。针对loh区域避免填补,以专注于可以直接从数
据中确定的内容。
[0186]
已知“可能致病性”和“致病性”变异的变异检测是根据已证明具有高到完全外显率致病性的变异的公开可用数据库(即,clinvar)进行的。此外,使用被描述为与发育迟缓基因相组合的必需基因的非详尽基因列表作为另外信息,以为临床指导提供高风险变异的实例。
[0187]
总之,本公开证明了在ivf诊所中利用wgs来筛查胚胎的遗传的和新生致病性基因变异的有效性。
[0188]
本领域技术人员将理解的是,在不脱离广泛描述的本发明的精神或范围的情况下,可以对如在特定实施例中示出的本发明进行多种变化和/或修改。因此,本实施例应当在所有方面都被视为是说明性的而非限制性的。
[0189]
本文所引用的所有出版物都以全文引用的方式并入本文中。当提及url或其它此类标识符或地址时,应理解此类标识符可以变化,并且互联网上的特定信息可能会有所增删,但可以通过搜索互联网找到等效信息。对此的引用证明了此类信息的可用性和公开传播。
[0190]
已包含在本说明书中的文件、法令、材料、装置、物品等的任何讨论仅仅是出于为本发明提供上下文的目的。这不应视为承认任何或所有这些事项形成现有技术基础的一部分,或者是与本发明相关的领域中的公知常识,因为其在本技术的每个权利要求的优先权日期之前就已经存在。
[0191]
参考文献
[0192]
abyzov等人(2011)《基因组研究(genome research)》21:974-984
[0193]
adzhubei等人(2013)《当代人类遗传学实验指南(current protocols in human genetics)》7:7.20单元到7.20单元
[0194]
bentley等人(2009)《自然(nature)》6:53-59
[0195]
depristo等人(2011)《自然遗传学(nature genetics)》43:491-498
[0196]
dolzhenko等人(2017)《基因组研究(genome research)》
[0197]
fan等人(2014)《生物信息学实验指南(curr.prot.bioinformatics)》2014:10.1002/0471250953.bi1506s45
[0198]
guatelli(1990)《美国国家科学院院刊(proc.natl.acad.sci.usa)》87:1874
[0199]
handyside等人(2010)《医学遗传学杂志(journal of medical genetics)》47:651-658
[0200]
harris等人(2008)《科学(science)》320:106-109
[0201]
kearney等人(2011)《医学遗传学(genetics in medicine)》13:680
[0202]
kircher等人(2014)《自然遗传学》46:310-315
[0203]
kozarewa等人,(2009)《自然方法(nature methods)》6:291-295
[0204]
kwoh(1989)《美国国家科学院院刊》86:1173
[0205]
landegren(1988)《科学》241:1077
[0206]
li等人,(2004)《临床化学(clin chem)》50:1002-1011
[0207]
li和durbin(2010)《生物信息学(bioinformatics)》26
[0208]
margulies等人(2005)《自然》437:376-380
[0209]
mckenna等人(2010)《基因组研究》20
[0210]
metzker(2010)《自然综述(nature rev)》11:31-46
[0211]
ng和henikoff(2003)《核酸研究(nucleic acids research)》31:3812-3814
[0212]
patch等人(2018)《公共科学图书馆
·
综合(plos one)》13:e0190264
[0213]
quang等人(2015)《生物信息学》31;761-763
[0214]
rehm等人(2013)《医学遗传学:美国医学遗传学学院官方期刊(genetics in medicine:official journal of the american college of medical genetics)》15:733-747
[0215]
reva等人(2011)《核酸研究》39:e118
[0216]
richards等人(2015)《遗传学与医学(genet med)》17:405-24
[0217]
riggs等人(2018)《人类突变(human mutation)》39:1650-1659
[0218]
samocha等人(2017)《生物预印本资料库(biorxiv)》,2017
[0219]
schwarz等人(2014)《自然方法》11:361
[0220]
shihab等人(2014)《人类基因组学(human genomics)》8:11
[0221]
smith(1997)《临床微生物学期刊(j.clin.microbial.)》35:1477-1491
[0222]
soni和meller(2007)《临床化学》53:1996-2001
[0223]
van der auwera等人(2013)《生物信息学实验指南》11:11.10.1-11.10.33
[0224]
volkerding等人(2009)《临床化学》55:641-658
[0225]
wang等人(2011)《自然方法》8:652-654
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。