一种泛癌种超早期筛查方法
1.相关申请的交叉参考
2.本技术引用申请人于同一申请日提交的、发明名称为“一种突变的 人端粒酶rna基因及其在预防和治疗泛癌种中的应用”,其公开内容全部 并入本文作为参考。
技术领域
3.本发明涉及一种用于对受试者的泛癌种超早期筛查方法。更具体地, 本发明涉及对受试者的外周血或其它生物样品中的基因组dna进行遗传 缺陷端粒的测定而在早期确定受试者是否患有癌症和/或是否存在患癌 高风险的方法。本发明还涉及用于这种筛查的试剂盒。
背景技术:
4.癌症早期发现,特别是早期筛查是有效提高癌症预防和治愈最重要 的手段之一。癌症早期筛查的概念和技术要求不同于临床早期辅助诊断 和辅助治疗。首先,早期筛查技术的应用对象是没有任何患癌临床症状 的正常人群,所以癌症早期筛查技术必须对泛癌种有效,至少一次性筛 查对前十位高发癌种有效,且要求技术简单、方便、易行、安全、无创, 有效筛查时间节点要显著早于临床技术的诊断,筛查费用是普通人群可 接受。目前,国内外的癌症早期筛查技术几乎都是基于癌细胞生长到一 定阶段脱落进入外周血中的残留物展开的技术研发,包括cfdna,ctdna, ctc,dna甲基化以及基因突变谱(panel)等各种超微量检测技术正逐 步走向临床应用。然而,这些技术都面临一些类似的严重瓶颈问题,即 筛查的时间节点与现有临床诊断技术并没有显著提早。由于残留物半衰 期短、含量甚微,加上造血干细胞遗传性变异干扰等,导致这些早期筛 查技术很难进一步提升,严格地说这些技术都不是真正意义上的癌症早 期筛查技术,只能在临床辅助诊断和辅助治疗中实现市场应用。据全球 领先企业报道的相关技术测试数据,这些技术在“回顾性”测试中,灵 敏度和特异性均表现出优异,但在“前瞻性”测试中,阳性预测值(ppv) 和阴性预测值(npv)往往不理想,特别是癌症早期筛查的核心指标ppv 远达不到要求。因此,这些技术对特定单一癌种或有限癌种的较早期辅 助诊断和辅助治疗比较有效,但它们都几乎不能用于癌症的早期筛查。 学术界也在质疑这些基于癌细胞残留的超微量液体活检技术的可行性, 认为它们存在很多不确定性(razavi,p.,et al.,2019,nat med 25: 1928-1937)。另一种途径是针对基因组dna的ngs深入解析,包括wes、 wgs、dna甲基化、癌症相关基因谱等,似乎显示出更加可靠和稳定, 但它们仍然面临一个短期难以逾越的障碍。虽然测序技术和费用已接近 市场应用的要求,可是要寻找到某(些)基因改变与癌症病症的关系却 非常困难,还需要做大量医学与表观遗传的基础研究,诸如生物体内基 因与基因之间、基因与性状之间、性状与性状之间的相互作用堪比互联 网,单个基因仅仅是基因网络中的某个节点而已。除极少数的基因以外, 几乎现有的测序结果就是一部“天书”,对癌症早期筛查帮助甚微。
5.因此,本领域中迫切需要有能够实现泛癌种早期筛查的技术和方法。
技术实现要素:
6.发明人为探索大多数癌症的细胞端粒酶异常升高却端粒超短,甚至 不及无端粒酶活性的正常细胞这一共性问题,对六种癌症的正常组织、 癌旁组织和癌组织的端粒3’末尾重复序列进行了检测,发现全部正常组 织都是以agggtt为主,但癌变组织则很低,发生了严重紊乱。
7.发明人又对癌细胞端粒酶特性进行了深入解析,发现它与正常胚性 细胞无差别,需要与端粒3’末尾两个碱基配对结合才能有效催化、合成、 延伸端粒。
8.发明人基于癌细胞端粒3’末尾紊乱和端粒酶催化特性,再结合伴随 细胞分裂端粒逐渐缩短,对ncbi提供的人类全基因组数据库中已定位 的27个端粒序列进行了详细分析,出乎意料地发现,在近染色体大约100~1000bp的端粒区域内,有18个端粒存在着(5’agggtc3’)n、 (5’gggttc3’)n、(5’agggtg3’)n和(5’gggttg3’)n等n次重复序列, 这些重复序列与正常的端粒3’末尾重复序列(5’agggtt3’)n、 (5’gggtta3’)n存在重大差异。由于细胞分裂过程中dna的半保留复 制,细胞经过数十次分裂之后,端粒就会缩短至这些存在重大差异的区 域,某(些)端粒3’末尾的2个碱基就会出现tc或tg结尾。发明人 推测这种tc或tg结尾的端粒影响了端粒酶的结合,可能导致端粒酶 异常升高又限制了端粒的延伸。因此在本发明中,发明人将具有这种以 tc或tg结尾的端粒重复序列的端粒称为“遗传缺陷端粒”。
9.为了端粒酶能与遗传缺陷端粒结合,发明人对端粒酶rna进行了单 碱基定点突变重组,修复出现严重缺陷的端粒,其结果令发明人振奋, hela癌细胞生长明显减缓,bcap37端粒酶活性大幅下降,癌变细胞开 启了程序性凋亡。这些结果表明,端粒的tc或tg遗传缺陷端粒与细 胞早期癌变直接相关,也是导致细胞癌变的关键驱动因子之一,它的出 现预示着细胞处于癌变阶段或者发生癌变的风险之中。无疑,通过对细 胞端粒3’末端遗传缺陷端粒进行测定,可以评估细胞是否癌变或者是否 处于发生癌变的风险之中。
10.发明人还对来自不同癌症的受试者的生物学样品和多种癌细胞系的 基因组进行了端粒3’末端重复序列的测定,发现了它们的部分端粒变成 了tc或tg遗传缺陷端粒,即端粒3’末端重复序列为5’agggtc3’、 5’gggttc3’、5’agggtg3’和/或5’gggttg3’。
11.发明人进一步发现,来自受试者的生物学样品基因组的遗传缺陷端 粒存在的总量与其患癌状况或者患癌风险高度相关,同时发现不同的染 色体臂出现的这种遗传缺陷端粒与细胞癌变或癌变风险的器官或组织相 关。所述生物学样品不一定是患癌组织或有患癌风险的组织的样品,可 以是外周血样品。发明人所获得的相关论据还表明外周血也是最佳的样 品。因此,通过对来自受试者的生物学样品内细胞的端粒末端重复序列 5’agggtc3’、5’gggttc3’、5’agggtg3’和/或5’gggttg3’的含 量进行测定,或进一步定位它们所在的染色体臂,可以评估该受试者是 否患有癌症和/或处于发生癌症的高风险之中,以及可能发生的组织。
12.因此,在一方面,本发明提供了一种鉴定受试者是否患有癌症和/或 确定受试者是否有患癌症风险的方法,其中包括:a)对来自受试者生物 样品中的基因组dna,测定是否存在遗传缺陷端粒,存在遗传缺陷端粒 的数量,包括扩增的产物总量、扩增dna条带数量、各扩增dna条带的 分子量和产物量,以及遗传缺陷端粒在染色体臂上的定位分布;和b)已 知的患癌受试者和非患癌受试者的“回顾性”测试大数据形成的参考值 对比。对具体未知
患癌与否的受试者可以做出在某一时间范围内可能或 不可能患癌的风险判断,对已患癌和/或患癌风险高的受试者还可以提供 最可能发生的具体组织,帮助受试者警惕和预防。
13.在另一方面,本发明提供了作为细胞癌变早期驱动因子的生物标志 物,其是遗传缺陷端粒,它的3’末端重复序列具有5’agggtc3’、 5’gggttc3’、5’agggtg3’或5’gggttg3’的特征。本发明还涉及 利用这种tc和/或tg遗传缺陷端粒用于泛癌种的辅助诊断与治疗,包括 治疗前、治疗中和/或治疗后,以及复发监测。还可以用于对治疗药物、 治疗的方案等实施疗效的监控和评价。
14.在又一方面,本发明涉及一种用于泛癌种超早期筛查的试剂盒,其 中包含有用于测定来自受试者生物样品的基因组dna内端粒3’末端重复 序列5’agggtc3’、5’gggttc3’、5’agggtg3’和/或5’gggttg3
’ꢀ
的存在情况所需的成分,以及任选地使用说明书。
15.本发明还涉及所述试剂盒用于泛癌种超早期筛查的用途,包括医疗 机构对泛癌种的辅助诊断、辅助治疗、癌症复发监测、抗癌药物评价以 及具体治疗方案的疗效监控和评价等等。
16.发明详述
17.大多数癌症是一种衰老性疾病,因少数衰老细胞发生遗传变异,逃 避了程序性凋亡,持续分裂,形成组织异常增生性疾病,它也是人体细 胞为了逃避衰老或死亡,在基因组上的进化与适应。研究也发现衰老细 胞更具有这种异常增殖的能力(nishiguchi,m.a.等,2018,cell reports, 24,3383-3392)。研究证明随着人年龄的增加而机体逐渐衰老,体现在基 因组上除了dna甲基化明显改变外,更重要的是染色体末端的端粒长度 不断地缩短。大量研究表明这种缩短的速率除了遗传因素以外,还受到 外部环境多因素综合影响,包括生活、工作环境,饮食习惯,性格修养 等,所以端粒的缩短具有“生命时钟”的作用,与癌症关联更密切。进 一步研究表明是一部分的端粒长度逐渐变短,另一部分保持相对恒定 (muezzinler,a.等,2013,ageing res rev 12:509-519),只有体内正常的 胚性细胞,包括生殖细胞、干细胞等能通过端粒酶的催化作用将端粒延 伸,维系全部端粒的正常长度和细胞可持续分裂。然而,90%以上的癌 细胞都具有端粒酶活性异常增高的特征,但端粒却比没有端粒酶活性的 体细胞还更短,端粒酶还能使这部分变短的端粒保持恒定,逃避细胞程 序性凋亡,从而实现癌细胞的“永生性”。端粒的缩短和端粒酶的异常激 活与细胞分裂、衰老、癌变直接关联(greider,c.w.等,1996,annu revbiochem 65,337-365;blasco,m.a.,2005,nat.rev.genet.6:611
–
622; turner,k.j.,et al.,2019,cells,8:73-92),2009年诺贝尔医学或生 理学奖授予了三位端粒和端粒酶的发现者。
18.端粒位于染色体的末端,是一种基因组dna末端与多种蛋白结合形 成的特殊结构。除了相关蛋白以外,人端粒dna均由简单重复序列 (ttaggg)n组成,且3’端凸出形成大约24-96bp长度的单链结构。在 体内,3’末端单链回插进入双链区域,形成“t-loop”和“d-loop”结构,为 端粒蛋白tin2,tpp1,trf1,trf2,pot1,pot1a等提供了结合基础,形 成末端“帽子”结构,以有效阻止核酸外切酶对基因组的破坏(tian,x. 等,2010,appl biochem biotechnol,160:1460-1472),这个loop结构 对实现端粒及染色体自身保护和稳定十分重要。一旦失去loop结构,细 胞将启动程序性凋亡。理论上,人端粒3’末端存在着六种可能的结尾重 复序列,即1)5’ttaggg3’;2)5’gttagg3’;3)5’ggttag3’;4) 5’gggtta3’;5)5’agggtt3’;6)5’tagggt3’。发明人利用一种端粒 3’末尾重复序列检测方法对17例不同年龄
的正常人全血基因组中的15 个端粒进行了检测,结果显示,它们的3’末尾重复序列存在显著偏好, 以5’agggtt3’结尾占比在26
±
7.7%,且随年龄的增加而逐渐减少。 核酸动力学分析也显示5’agggtt3’结尾的重复序列明比其余的5种结 尾方式更加稳定,这也佐证了体内端粒3’末尾重复序列偏好的存在。
19.发明人进一步对乳腺癌、直肠癌、结肠癌、肝癌、肺癌和胃癌等六 种不同癌症患者的癌组织、癌旁组织和正常组织进行相同遗传背景下的 同步检测研究,更重要的发现癌组织的5’agggtt3’所占比例均比它的 癌旁组织、正常组织显著下降,病理早期小于19.0%,晚期小于15.4%, 不及六种结尾的理论平均值16.7%。这种癌组织下降的程度与病理进程 呈现明显的线性关系,且与癌症种类无关,指示端粒3’末尾重复序列 5’agggtt3’的下降在癌症中是一个普遍现象。这提示端粒3’末尾重复 序列与细胞癌变可能有直接关联。
20.端粒3’端重复序列的非随机性结尾主要是由于端粒酶rna模板对 端粒3’末端碱基的选择性偏好所致,它即是端粒酶的结合位点,即“底 物”,又是端粒酶催化合成的“产物”。发明人基于trap方法(imam,s. a.,et al.,1997,anticancer res 17:4435-4441)和端粒3’末端重复序列 检测方法相结合,利用六种3’末尾2个碱基差异的寡核苷酸为前导序列 对hela细胞端粒酶的体外催化合成端粒的效率和特性进行了深入研究, 发现端粒酶与端粒3’末尾的2个碱基配对形成有效结合位点,每次转位 合成6个碱基,且不同的结合位点直接决定了端粒酶的转位效率和合成 效率,从而影响端粒的延伸,其作用效率从高到低依次是tt》gt》gg, 分别达到177、110和89.7个相对单位。
21.结合癌细胞基因组端粒3’末尾重复序列的紊乱以及端粒酶的催化特 性,发明人根据ncbi提供的人类全基因组数据库信息,对已定位的27 个端粒序列进行了详细分析,令人惊奇地发现在距染色体大约 100~1000bp端粒区域内,有18个端粒存在着(5’agggtc3’)n、 (5’gggttc3’)n,(5’agggtg3’)n和(5’gggttg3’)n等n次异常重 复序列。发明人推测,由于细胞分裂过程中dna半保留复制的缺陷,导 致端粒dna重复序列缩短,每分裂一次将缩短约60-180bp。细胞经过 数十次分裂之后,端粒就会缩短至端粒遗传缺陷区域(即含有 5’agggtc3’,5’gggttc3’,5’agggtg3’或5’gggttg3’的端粒 区域),某(些)端粒3’末尾的2个碱基就会出现tc或tg结尾。由于 它们伴随人类基因组的遗传,故在本发明中,发明人分别将这种结尾的 端粒定义为tc或tg遗传缺陷端粒。
22.不局限于具体理论,发明人推测由于tc或tg遗传缺陷端粒的出现, 完全改变了端粒酶催化合成端粒时的最佳结合位点,严重影响端粒酶正 常催化作用和催化效率,一方面扰乱了以5’agggtt3’为主要方式的转 位控制,导致延伸的端粒3’结尾紊乱,干扰loop的形成。另一方面,由于 催化效率低,合成端粒长度受限,端粒末端的结合蛋白专递出“端粒酶 缺乏”的错误信号,细胞重启端粒酶表达,最终导致本应启动程序性凋 亡的细胞,相反地误导了端粒酶基因的重启,规避了细胞的程序性凋亡, 恢复分裂能力,实现“永生”。
23.为了进一步论证tc和tg遗传缺陷端粒与细胞癌变的直接关系,发 明人采用了重组人端粒酶rna基因定点突变的方法,利用端粒酶rna基 因的单碱基突变体与遗传缺陷端粒的tc或tg的有效配对结合,以修复 hela细胞内的遗传缺陷端粒,结果显示绝大部分hela细胞出现了程序 性凋亡。借助于腺病毒ad5基因载体,将该突变体的腺病毒颗粒注入至 bcap37细胞株的移植瘤中,结果也显示瘤体生长速率减小三分之一,瘤 体石蜡切片显示细胞凋亡明显。经瘤体基因组dna分析,呈现非常典型 的程序性凋亡特征,还令发明人十分惊
奇的是端粒酶活性下降了75.1%。 这些发现阐明tc和tg遗传缺陷端粒的出现是人体细胞衰老或癌变的重 要特征,也证实它们与细胞癌变直接相关,是极其重要的细胞早期癌变 驱动因子之一,可作为创新药物的研发崭新靶点。tc和tg遗传缺陷端 粒也是极其重要的细胞早期癌变的生物标志物,可用于泛癌种超早期筛 查。因此,针对细胞tc和tg遗传缺陷端粒的定量检测能达到对泛癌种 超早期筛查的目的。同时,开发出本发明的泛癌种超早期筛查的伴随干 预、预防和治疗的相关药物。
24.发明人还发现,人体外周血基因组端粒状态的检测不仅能反映其它 人体组织的端粒状态,还是遗传缺陷端粒检测的最佳组织。它是“液相
”ꢀ
组织,发生遗传缺陷端粒的少数细胞与其它正常细胞处于一个相对均匀 体中,使得非常微量的样本也具有很好的代表性,而其它“固相”组织 则不可能。在时间节点上,外周血也是遗传缺陷端粒更早发生的组织, 这个结论也得到《科学》杂志的一篇论文中大数据的支持(demanelis etal.,2020,science 369:1333,https://doi.org/10.1126/science.aaz6876)。 另外,从基础细胞遗传学可知,真核细胞的有丝分裂与减数分裂完全不 同。有丝分裂不存在细胞的完全脱分化以及姐妹染色体的联会过程,分 裂后产生的细胞与分裂前完全相同。发明人通过对模式生物—水稻的细 胞内端粒行为的研究,发现只有通过减数分裂才可能改变姐妹染色体之 间的端粒长度差异,而有丝分裂则不能。水稻中有籼稻和粳稻两个生物 亚种,前者的平均端粒长度比后者长3倍多(ohmido,n.,et al.,2000, mol gen genet 263:388-394)。利用这个特点,将籼稻(广陆矮4号)为 父本与粳稻(日本晴)为母本,通过人工授粉杂交,获得杂种f1,并对 f1和父本和母本同时对其中第四条染色体长臂的端粒(chr.04q)长度进 行分析,结果显示f1中来自父本的染色体端粒长度与父本相似,f1来自 母本的染色体的端粒长度与母本相似,即f1的姐妹染色体之间端粒长度 相差较大,说明有丝分裂不能改变姐妹染色体之间端粒的长度。在哺乳 动物中,世界著名的体细胞克隆羊—“多利”,它的寿命比正常相同品种 的羊缩短了约6.7年,这与它的细胞核来自6龄羊的乳腺细胞密切相关。这 种提前衰老和死亡或许也佐证了尽管通过不同体细胞的核、质融合完全 改变了动物细胞的整个脱分化和再分化过程,但结果也不能改变或恢复 端粒的长度,回拨“端粒时钟”(kishigami,s.,et al.,2008,exp cell res 314:1945-1950),显然有丝分裂不能改变哺乳动物细胞端粒长度的遗传 行为。人体最初来源于一个受精卵细胞,是经若干次有丝分裂之后形成 的个体,所以人体各个组织包括外周血基因组中端粒行为的一致性,随 年龄的增加只是它们的绝对长度发生了同步的缩短改变,也就是说外周 血基因组端粒状态能直接反映人体及其各个组织的端粒状态。这个结论 得到世界权威杂志《科学》中发表的一篇大数据论文的充分论证和支持。 该论文利用近1000名遗体捐赠者,25种不同人体组织,6391个样本对端 粒长度变化进行了深入细致的研究(demanelis et al.,2020,science 369: 1333,https://doi.org/10.1126/science.aaz6876),结果显示只有睾丸组织 的端粒长度最长,且显著长于其它组织,说明只有减数分裂才能恢复端 粒长度,而有丝分裂则不能。来自这篇报道中更重要的发现是在25种组 织中外周血端粒长度最短,充分说明外周血可以作为其他组织中端粒长 度的替代物。不仅如此,发明人还认为外周血中端粒最短说明它在各个 组织中也是最先可能出现tc和tg遗传缺陷端粒的组织。所以,利用外 周血对端粒的筛查在时间节点上无疑也是最佳选择,只要对外周血基因 组中tc和tg遗传缺陷端粒的定量检测就能达到对泛癌种超早期筛查的 目的。
25.因此,本发明公开了一种基于外周血dna的泛癌种超早期筛查方法。 它涉及对tc和tg遗传缺陷端粒的定量检测,可将癌症筛查提早至细胞 发生癌变前后,实现泛癌种超早期筛查。
26.如本文中所使用的,术语“泛癌种”是指细胞端粒酶异常升高的癌 症种类,这类癌症大约占人类癌症种类的90%以上,大多数来源于上皮 组织中细胞的癌变,也包括肉瘤,母细胞瘤等。
27.如本文中所使用的,术语“超早期筛查”是指对组织或细胞癌变前 后不久,癌细胞或癌组织尚未脱落、坏死、凋亡或分泌进入外周循环系 统之前的有效筛查。它的有效筛查时间节点显著早于现有基于ctdna、 cfdna、ctc、dna甲基化和/或癌基因突变谱(panel)等的各种早期 筛查技术。
28.如本文中所使用的,术语“受试者”是指本发明技术所涉及的测试 对象和筛查对象。为了便于本文准确描述可以将受试者分成三种类型:a 类受试者为经现有临床诊断技术已确诊的癌症患者,可以是已知或未知 进展期肿瘤,可以是已采用医疗干预或未干预的癌症患者;b类受试者 为无任何患癌病症的正常受试者,且经过2~5年跟踪调查期内也没有出 现任何患癌病症的受试者;c类受试者为无任何患癌病症的正常受试者, 可能在未来的一段时间内是或不是癌症患者。
29.如本文中所使用的,术语"核酸"、"核酸序列"、"寡核苷酸"和"引 物"表示任何基于核糖核苷酸或脱氧核糖核苷酸的分子,包括dna、 ctdna、cfdna、或其任何组合。因此,核酸分子、寡核苷酸或引物可 包括天然存在的核苷酸、非天然核苷酸或其组合。
30.如本文所使用的,术语“寡核苷酸”是指脱氧核糖核苷酸或核糖核 苷酸碱基的单链序列、天然核苷酸的已知类似物或其混合物。寡核苷酸 通常长度较短(例如在长度少于100个核苷酸),其可通过任何适当的方 法、包括例如直接化学合成或适当序列的克隆和限制性酶切来制备。通 常在本发明的情境中,寡核苷酸被设计为识别和结合位于另一个核酸分 子内的特定的互补核苷酸序列。寡核苷酸还可包括除与所述互补序列杂 交所需的那些碱基之外的碱基。寡核苷酸可能能够或可能不能作用于聚 合酶介导的延伸的引物。并非要求寡核苷酸中的所有碱基与所述寡核苷 酸被设计用以结合的序列互补;寡核苷酸只需要包含足够的互补碱基来 使所述寡核苷酸识别并且结合该序列。寡核苷酸序列可以是未修饰的核 苷酸序列或可以通过多种本领域技术人员已知的方法进行化学修饰或缀 合。
31.如本文所使用的,术语"引物"是指结合单链模板核酸分子的特定 区域并且通过酶促反应起始核酸合成(从引物的3'末端延伸并且与模 板分子的序列互补)的寡核苷酸。根据常规,引物通常称为'正向'和' 反向'引物,所述引物之一与核酸链互补,并且所述引物的另一个引物 与该链的互补链互补。
32.如本文所使用的,术语"dna接头"是指短的双链寡脱氧核糖核苷 酸,分别由连接链和选择链组成的粘性端接头。它可以通过dna连 接酶而连接到与选择链3’端凸出的碱基互补的基因组遗传缺陷端粒 dna的末端。
33.如本文所使用的,术语"生物样品"是指包含人基因组dna的任 何生物样品。生物样品可以是人体组织、细胞、分泌物或其预处理样 品。
34.如本文所使用的,术语“检测值”是通过本发明的方法对来自待筛 查的受试者的生物样品的基因组dna进行分析而获得的数值,可被 描述为基因组dna中出现的tc和/或tg
遗传缺陷端粒的扩增dna 条带的总量(ng)和各dna条带的分量(ng)以及对应的dna分子量(bp), 以及遗传缺陷端粒的染色体臂定位
35.如本文所使用的,术语“参考值”指通过检测大量的a类受试者和 b类受试者获得的检测值,可以包含遗传缺陷端粒在不同染色体臂上 的分布与癌症种类的关联性参数,经数学建模运算和人工智能技术处 理,所获得的一组泛癌种风险阈值,可以包含或不包含年龄、性别以 及外部环境因素等。可被用作对受试者的患癌风险和患某类癌的风险 评判比对标准或参数。
36.如本文所使用的,术语“端粒遗传缺陷”指在人类染色体端粒的 dna重复序列中,部分近染色体的端粒重复序列存在多次串联式异常, 且这种异常是伴随基因组遗传,与正常端粒重复序列序列相比存在缺 陷,包括(agggtc)n,(gggttc)n,(agggtg)n和/或 (gggttg)n。
37.如本文所使用的,术语“遗传缺陷端粒”指随人体细胞的逐次分裂, 端粒将逐渐缩短,当端粒缩短至端粒遗传缺陷区域时,端粒3’末尾的 2个碱基出现了tc或tg缺陷结构的端粒。例如5
’……
agggtc 3’、 5
’……
gggttc 3’、5
’……
agggtg 3’和/或5
’……
gggttg 3’。
38.如本文所使用的,术语“内参对照”指为了尽可能减少不同受试者 样本在检测过程中的各种不确定性误差,内参对照可从人基因组中选 择一条两端具有相同平末端内切酶切口,大小在0.5~2.0kb的单拷贝 保守基因组dna片段或看家基因的dna片段,设计该片段的特异性 扩增引物,与同一受试者的待检测样本同步pcr扩增,获得的扩增产 物量,可以用于矫正该样本的扩增产物量。
39.在一个方面,本发明提供了一种对受试者进行泛癌种的超早期筛查 的方法,其中包括对来自受试者的生物样品的基因组dna中的遗传缺陷 端粒,即端粒的3’末尾序列5’agggtc3’、5’gggttc3’、5’agggtg3
’ꢀ
和5’gggttg3’中的一种或多种进行定量检测,和/或将遗传缺陷端粒 定位在不同染色体臂上,以及任选地将所获得的检测水平与参考值进行 比较的步骤。
40.用于本发明的基因组dna可以通过常规方法制备,参考《分子克隆 实验手册》,确保制备的基因组dna完整,无降解,可以通过pcr测定 其端粒3’末端序列。在一个实施方案中,首先利用限制性核酸内切酶将 制备的基因组dna进行限制性酶切,然后与合适的dna接头连接,通 过pcr扩增待检测的端粒,并对其产物进行定量分析。
41.本发明的方法可以包括:基因组dna样品制备;限制性核酸内切酶 处理基因组dna;酶切样品纯化;纯化的酶切dna片段与特异于待检 测端粒的dna接头连接;将连接样品纯化;pcr扩增待检测的端粒序 列,并对其产物进行定量分析和/或对其产物进行dna测序,并与人类 全基因组信息进行比对,明确其对应的染色体臂。
42.在一个实施方案中,本发明的方法包括以下步骤:
43.1)对来自所述受试者生物样品分离的基因组dna进行内切酶处理, 并进行纯化;
44.2)将步骤1)中纯化的酶切基因组dna与接头连接,所述接头特异针 对待检测的本发明所述的遗传缺陷端粒3’末尾重复序列,其由两条寡核 苷酸链组成。其中一条寡核苷酸链是5’端磷酸化修饰、3’端c3 spacer修 饰的18个碱基的连接链(ll),其序列可以是例如 (seq id no.5),另一条寡核苷酸链是选自3’端c3 spacer修
饰的17个碱 基序列(seq id no.1)、 (seq id no.2)、(seq id no.3)或 (seq id no.4)的选择链(sl,分别为sl1、sl2、 sl3和sl4),它们分别适用于不同的遗传缺陷端粒序列;
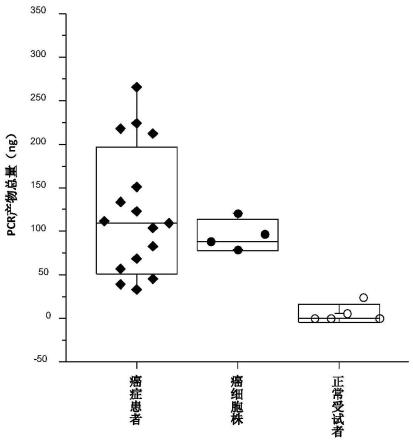
45.3)对步骤2)中获得的连接样品进行纯化;
46.4)对步骤3)中获得的纯化样品进行pcr扩增和定量分析。
47.在本发明中,受试者可以是哺乳动物,例如灵长类,特别是人。受 试者可以是无任何患癌病症的正常个体,或者是处于患癌风险中的个体, 或者是疑似肿瘤患者,或者是有不良嗜好,或肿瘤相关遗传家族史的个 体,或在特殊的生活和工作环境中的个体等等。所述生物样品是拥有基 因组dna的样品,可包括例如血液、尿、便、痰、胸膜液、腹膜液、 支气管和支气管肺泡灌洗液、组织切片、表皮、粘膜等。可以例如穿 刺技术抽吸活组织检查获得样品。可以是经过处理的样品,例如石蜡 包埋的组织,固定液固定的组织,防腐处理的组织,冰冻组织等。血 液可以是全血、白细胞,优选地是所述生物样品是外周血。
48.所述恶性肿瘤可以是端粒酶异常升高的肿瘤或癌,约占恶性肿瘤类 型的90%以上,例如肺腺癌、肝癌、乳腺癌、胃癌、肾上腺皮质癌、 宫颈癌、卵巢癌等等源于上皮组织的癌,以及肉瘤,母细胞瘤。
49.在一些实施方案中,可在分析之前从细胞或组织中提取基因组 dna。本领域技术人员清楚如何从来自受试者的生物样品中提取基因组 dna,或采用“全自动基因组dna提取仪”制备基因组dna,确保基因 组末端的完整,无降解,参考例如《分子克隆操作手册》(sambrook和 manitis等人主编)等的教导。可选择地,可另外处理细胞以使dna可用 于分析,例如可以在或可以不在对细胞蛋白质进行变性或降解的情况 下使用细胞裂解物,可使用或不使用防止dna降解的保护剂,dna清 洁剂等。优选地在血液收集管中使用edta抗凝剂,并对基因组dna做 进一步纯化,确保基因组的完整性和纯度,以用于步骤1)。
50.在本发明的方法中,可以采用任何合适的核酸内切酶对分离自受试 者样品的基因组dna实施片段化处理,条件是在设计的正向引物与端粒3
’ꢀ
末尾重复序列之间不存在该内切酶的酶切位点。优选地所述内切酶是平 端内切酶,例如ecorv酶、alui、haeiii、sma i、snab i等,但不局限 于此,本领域技术人员可以通过常规实验,结合上述条件确定合适的内 切酶。
51.在本发明的方法中,对通过核酸内切酶酶切过的受试者基因组dna 进行纯化和富集目标片段。这样的dna纯化和富集方法是现有技术中已 知的,包括例如苯酚/氯仿纯化法、磁珠纯化和富集方法等。可以选择性 回收合适长度的酶切后的片段,例如小于2.5kb、2.0kb、1.5kb、1.0kb或 0.9kb、0.8kb、0.7kb、0.6kb、0.5kb的目标片段。优选地回收小于1.5kb 的片段。经过纯化的受试者基因组dna可以用于后面的连接反应中。
52.用于本发明的dna接头可以寡核苷酸形式提供,然后在使用时制作成 接头。或者,用于本发明的dna接头可以是在合成寡核苷酸链制成的 dna接头之后提供。所述接头是由两条人工合成的寡核苷酸链组成,一 条是5’端磷酸化修饰、3’端c3 spacer或c6 spacer修饰的连接链(ll), 以5
’‑
x
’‑y’‑3’
的方式设计序列;另一条是3’端c3 spacer或c6 spacer修饰 的选择链(sl),针对不同遗传缺陷端粒序列,分别以sl1,5
’‑
x-gaccct-3’; sl2,5
’‑
x-gaaccc-3’;sl3,5
’‑
x-caccct-3’;和sl4,5
’‑
x-caaccc-3’的方 式设计序列。x’和x反向互补,长度可以是8-25个、10-20个或10-15个核 苷酸;y’和y反向互补,长度可以是2-20个、4-15个或6-12个核苷酸(y 是反向引物的一部分,详见反向引物设计)。优选地连接链为18个核苷酸, 其中x’为11个核苷酸,y’为7个核苷酸,即(seqid no.5);优选地选择链为17个核苷酸,其中x为11个核苷酸,即sl1, (seq id no.1);sl2, (seq id no.2);sl3,(seq id no.3);和sl4, (seq id no.4)。ll连接链可以与sl1、sl2、 sl3和sl4选择链分别制成lk1、lk2、lk3和lk4的粘性接头,适用 于不同的遗传缺陷端粒。
53.将寡核苷酸制成dna接头的方法是现有技术中已知的,本领域技术 人员可以很容易完成。例如,可以将1-100mm连接链(ll)溶液和 1-100mm的相应选择链溶液的等摩尔当量在缓冲液中混匀、离心,然后 进行pcr降温循环。在一种实施方式中,可以通过在98℃/1min、97℃ /1min,然后每循环降低1℃,直到12℃停止,获得相应的dna接头溶液。
54.dna接头溶液可以直接与前面步骤中制备的纯化的内切酶酶切过的 受试者基因组dna片段,通过dna连接酶进行连接。dna连接反应的条 件是本领域技术人员已知的,并且可以通过常规实验进行。优选地,所 述连接酶是高特异粘性端连接酶,例如t7 dna连接酶,但不局限于此, 本领域技术人员可以通过常规实验确定合适的连接酶。
55.连接反应获得的产物可以直接用于进行pcr扩增测试,或者可以在 经dna纯化方法进行纯化后用于pcr扩增测试。合适的dna纯化方法是 本领域中已知的,包括例如苯酚/氯仿提取法、磁珠提取法等。在一个实 施方案中,pcr扩增中采用的引物包括:正向引物(fprim),它的设计可 以是特别针对单个端粒或一组端粒进行,具体地是在染色体的近端粒区 域设计出特异的正向引物,或从多个已定位的染色体近端粒区域的同源 保守序列中设计一个能特别代表多个端粒的正向引物,该正向引物也可 能涵盖了其它拥有相同同源保守序列的未知或未定位的端粒。优选地采 用能一次扩增已定位的15个端粒(chr01q,chr2q,chr04q,chr7q,chr8p, chr9p,chr10q,ch12q,chr13q,chr15q,chr16p,chr21q,chr22q, chryq和chrxq)的同源正向引物,所述的同源正向引物是: 5
’‑
gaatcctgcgcaccgagattctc-3’(seq id no.6;fprim);反向引物以 5
’‑
y-x-z-3’的方式设计序列,x的长度可以是8-25个、10-20个或10-15个 核苷酸,y的长度可以是2-20个、4-15个或6-12个核苷酸,且x和y分别与 连接链中的x’和y’反向互补(x’和y’详见接头设计)。z的长度可以是 1~6个核苷酸,并与遗传缺陷端粒3’末端重复序列反向互补。优选地y是6 个核苷酸,x是11个核苷酸,z是3个核苷酸,即反向引物:核苷酸,x是11个核苷酸,z是3个核苷酸,即反向引物:(seq id no.7;rprim1,适用于lk1);适用于lk1);(seq id no.8;rprim2,适用于lk2);rprim2,适用于lk2);(seq id no.9;rprim3,适用于lk3);和和(seq id no.10;rprim4,适用于lk4)。
56.pcr扩增条件的优化是本领域技术人员熟知的,例如改变pcr扩增 的温度循环条件;在反应液中添加增效剂提高pcr扩增的特异性和/或扩 增效率,例如peg、甘油、二甲基
亚砜等;也可以使用或不使用热启动 dna聚合酶等。优选地选用甘油以及热启动聚合酶,使用降温pcr (touchdown pcr)技术。
57.然后对pcr扩增的产物进行定量测定。pcr扩增产物的定量测定 可以通过常规方法完成,例如电泳法、色谱法等,通过对电泳条带或洗 脱峰值进行定量来定量测定pcr扩增结果,也可以用荧光定量pcr、 数字pcr等,通过对引物荧光标记或一系列不同模板浓度梯度的pcr 达到pcr定量测定。可以通过对pcr扩增产物进行电泳分离、染色、 成像。还可以进一步对具体目标dna条带进行dna序列测定,并与人 类全基因组序列进行比对,明确目标dna条带对应的染色体(臂)上, 获得遗传缺陷端粒在染色体上的分布图谱与不同组织的细胞癌变的相关 性,为癌症溯源建立重要数据库。
58.通过合适的软件对图像中扩增的每一条dna条带进行pcr扩增量 和分子量的同步定量测定,可用的软件包括例如syngene tools, quantityone,image j等软件。在一个实施方式中,以标准量化的dnamarker为标量,通过质量控制体系完成对pcr产物进行琼脂糖电泳分 离,染色,成像记录。采用syngene tools软件对图像中的dna条带进 行定量分析,包括pcr扩增产物总量(ng)和每一条带的分子量(bp) 以及对应的扩增产物量(ng)获得pcr扩增产物的带谱检测数据。基于 获得这些数据,对a类受试者的前后纵向检测参数综合对比可以监控、 评估该受试者癌症的进展、治疗效果或癌症复发的评估;与同类癌种患 者的横向对比,可以辅助优化治疗方案;可以进一步对a类受试者的扩 增带普的dna序列分析信息,定位遗传缺陷端粒,得到对应的染色体遗 传缺陷端粒与癌种的关联性,为下一步的治疗方案和药物选用提供重要 参考。通过对a类和b类受试者的大数据积累,建立数学模型,结合人 工智能算法分析判断细胞癌变最可能出现的组织和器官,获得检测的综 合参考阈值。每位c类受试者的检测值,结合人工智能学习技术与参考 值进行计算对比,可以做出受试者在未来一定时间范围内可能或不可能 患癌的风险判断。例如,c类受试者,若出现清晰的pcr扩增条带指示 出受试者血液中已存在遗传缺陷端粒,再依据pcr扩增总量、具体条带 的分子量和扩增量与参考值对比获得进一步的信息和评估,可以进一步 对扩增的dna条带进行dna测序分析,获得目标dna条带所在的染 色体(臂)的信息,达到癌症超早期筛查和预警的目的。
59.本发明的泛癌种超早期筛查方法基于的是细胞tc和tg遗传缺陷端 粒的筛查,是一项全新的独创筛查技术,可以将筛查的时间节点提前至 细胞癌变前后,真正实现了癌症的超早筛,显著优于现有筛查技术。它 利用了人体各个组织细胞基因组端粒之间的一致性。特别是,外周血与 其它组织、器官(睾丸中处于减数分裂的组织除外)基因组端粒变化的高度 一致性特点,以微量全血例如0.2ml为筛查样本,取样方便、无创伤。
60.在另一方面,本发明提供了一种用于泛癌种超早期筛查的试剂盒, 其中包含用于测试受试者中tc和tg遗传缺陷端粒的寡核苷酸链和pcr 检测引物及相关配套试剂。
61.在一个实施方式中,所述试剂盒中包含至少五种寡核苷酸,即5’x
‑ꢀ
gaccct3’、5’x-gaaccc3’、5’x-caccct3’和5’x-caaccc3’,它们分别是 5’agggtc3’、gggttc3、5’agggtg3’和5’gggttg3’遗传缺陷 端粒的选择链,分别被称为选择链(sl)sl1、sl2、sl3和sl4,其中 各个x独立地具有8~25个、10-20个或10-15个核苷酸,以及序列为5
’ꢀ‑
x
’‑y’3’
的相应连接链,其中x和x’反向互补;y’和y反向互补(y是pcr 反向引物的一部分)。所述至少五种寡核苷酸可以以连接链与选择链之间 形成的接头(lk)lk1、lk2、lk3和lk4形式存
在。优选地,所述连 接链的序列为5’tcgtagactgcgtaccga 3’(seq id no.5),并且所述选择链 的序列分别为5’gcagtctacgagaccct3’(seq id no.1)、 5’gcagtctacgagaaccc3’(seq id no.2)、5’gcagtctacgacaccct3’(seq idno.3)、和5’gcagtctacgacaaccc3’(seq id no.4)。
62.在另一个实施方案中,一个或多个所述连接链的5’端具有磷酸化修 饰,以及连接链和选择链的3’端具有c3 spacer或c6 spacer修饰。
63.在进一步的实施方案中,所述试剂盒中还包含扩增引物,包括例如 正向引物:5’gaatcctgcgcaccgagattctc3’(seq id no.6);以及选自下述 的一种或多种反向引物:5’cggtacgcagtctacgagac3’(prim1,seq id no. 7)、5’cggtacgcagtctacgagaa3’(prim2,seq id no.8)、5
’ꢀ
cggtacgcagtctacgacac3’(prim3,seq id no.9)和5
’ꢀ
cggtacgcagtctacgacaa3’(prim4,seq id no.10),它们分别可用于连 接链ll与选择链sl1、sl2、sl3和sl4形成的接头形式lk1、lk2、lk3 和lk4的检测。
64.任选地,本发明的试剂盒中还可以包含选自下述的一种或多种:阳 性对照、阴性对照、内参对照、dna marker标样、缓冲液、限制性内 切酶、连接酶、pcr扩增酶或其任何组合。本发明的试剂盒中还可以任 选地包含使用说明书。
65.对本发明的筛查方法和试剂盒的测试结果显示,在用于多种癌症患 者血液样品的回顾性测试中灵敏度达到100%(16/16),特异性可达到97.6% (41/42),其中一位受试者虽然检测出一个低分子量条带,但pcr扩增 总量很低,经过56个月跟踪回访,身体健康,其实际特异性达到100%。 可覆盖端粒酶异常升高的癌种,大约占癌种的90%,满足泛癌种要求。 同时,针对tc缺陷端粒的检测还可用于多种癌症治疗过程中的伴随诊断 和监测。该技术基于受试者组织、分泌物,特别是外周血的基因组dna, 取样方便、微量、无创,所需设备少,操作便捷,数据稳定性,重复性 更好,成本低廉,拥有更加广阔的市场应用潜力,显著优于现有技术。
66.在又一方面,可以针对tc和/或tg遗传缺陷端粒的末端单个碱基, 直接实施特异性荧光探针标记,或通过基因芯片检测方法,都能实现对 tc和/或tg遗传缺陷端粒的定量检测,达到类似上述的泛癌种的超早期 筛查的目的。
附图说明
67.下面结合附图对本发明的具体实施方式作进一步详细说明。
68.图1和图2a、图2b、图2c、采用本发明的ll链和sl1链制备dna 接头,对不同种类的癌症样本与非癌症样本全血基因组dna检测结果。 dna marker,分子量从大到小依次是2000bp、1000bp、750bp、500bp、 250bp和100bp,其中750bp的标准量为60.0ng。并采用syngene genetool 软件对pcr扩增产物进行的定量分析结果,包括dna条带的分子量 (bp)和产物量(ng)。图中,虚线所示dnamarker标准样品,实线所 示受试者样品。图1-1~1-4所示4个癌细胞系,分别是肺癌、结肠癌、肝 癌和宫颈癌;图1-5所示正常肝细胞系(lo2);图2a中:图2-1~2-7所 示肺癌例;图2-8~图2-9所示结肠癌;图2b中:图2-10所示结肠癌; 图2-11所示直肠癌;图2-12~2-13所示宫颈癌;图2-14所示卵巢癌;
69.图2-15所示贲门癌;图2-16所示牙龈癌;图2-17~图2-18所示正常受 试者(b类受试者);图2c中:图2-19~图2-21所示正常受试者(b类受 试者)。图中所示全部癌症患者中
可能接受过或正在接受各种治疗,除了 结肠癌患者3癌细胞已转移以外,其它患者都未发生转移。
70.表1、将图1中的受试例的扩增的dna条带分子量和pcr产物量 汇总表,包括计算出的受试例的端粒数量。在7例肺癌中,的“粗体 下 划线”dna片段来源于一个相同tc遗传缺陷端粒;在3例结肠癌中, 的“粗体 斜体”dna片段来源于另一个相同的tc遗传缺陷端粒。
71.图3、将表1中,各个测试例的pcr产物总量进行统计分析的结果。 其中,癌症患者16例(
◆
),癌细胞系4个(
●
),以及正常受试者5例 (
○
)。
72.图4显示了hterc真核表达载体构建的示意图。
73.图5显示了携带htert启动子的hterc腺病毒穿梭载体构建示 意图。
74.图6显示了hterc野生型真核表达的重组体在hela细胞中的表 达。
75.图7显示了突变的hterc基因对癌细胞系
‑‑
hela细胞株的生长作 用。
76.图8显示了hoechst 33342染色观察hela细胞的凋亡特征。
77.图9显示了突变的hterc基因对人乳腺癌实体瘤生长的抑制作用。
78.图10显示了突变的hterc基因对人乳腺癌细胞内端粒酶的抑制作 用。
79.图11显示了切片观察hterc基因单碱基突变对人乳腺癌实体瘤细 胞的凋亡作用。图12显示了突变的hterc基因对人乳腺癌细胞的程序性凋亡作 用。
具体实施方式
80.本发明中涉及如下序列:
81.sl1人工合成序列,3’端c3 spacer修饰
[0082]5’
gcagtctacgagaccct 3’(seq id no.1)
[0083]
sl2人工合成序列,3’端c3 spacer修饰
[0084]5’
gcagtctacgagaaccc 3’(seq id no.2)
[0085]
sl3人工合成序列,3’端c3 spacer修饰
[0086]5’
gcagtctacgacaccct 3’(seq id no.3)
[0087]
sl4人工合成序列,3’端c3 spacer修饰
[0088]5’
gcagtctacgacaaccc 3’(seq id no.4)
[0089]
ll人工合成序列,5’端磷酸化,3’端c3 spacer修饰
[0090]5’
tcgtagactgcgtaccga 3’(seq id no.5)
[0091]
引物fprim人工合成序列,
[0092]5’
gaatcctgcgcaccgagattctc 3’(seq id no.6)
[0093]
引物rprim1人工合成序列,
[0094]5’
cggtacgcagtctacgagac 3’(seq id no.7)
[0095]
引物rprim2人工合成序列,
[0096]5’
cggtacgcagtctacgagaa 3’(seq id no.8)
[0097]
引物rprim3人工合成序列,
[0098]5’
cggtacgcagtctacgacac 3’(seq id no.9)
[0099]
引物rprim4人工合成序列,
[0100]5’
cggtacgcagtctacgacaa 3’(seq id no.10)
[0101]
实施方案
[0102]
下面提供实施本发明的泛癌种超早期筛查方法的一种方案:
[0103]
1)基因组dna样品制备:
[0104]
用宝生物工程(大连)有限公司(takara)提供的minibestwhole blood genomic dna extraction kit试剂盒,根据试剂盒说明书分 别提取、制备人血液基因组dna样品。最后,用1xte,ph7.6缓冲液替 代试剂盒提供的“elution”洗脱液。
[0105]
2)寡核苷酸合成及接头制作:
[0106]
a)上海生物工程公司合成序列表中所列的寡核苷酸,并按要求进行 化学修饰。
[0107]
b)采用双蒸水(ddh2o)分别将合成的寡核苷酸溶解成20mm溶液, 待用。将20ul灭菌双蒸水(ddh2o)和5.0ul接头缓冲液[200mm tris-hclph7.6,600mm kac,20mm mg(ac)2]加入到pcr薄壁管中,加入12.5ul 20mm连接链(ll)溶液和12.5ul 20mm选择链(sl1)溶液,混匀、离心。 放入pcr仪器中,执行下列程序:98℃/1min,97℃/1min(-1℃/cycle,83 cycles),12℃停止。并标记为:lk1。
[0108]
通过相同的方法制备dna接头lk2、lk3和lk4。
[0109]
3)内切酶处理基因组dna:
[0110]
用1xte ph7.6缓冲液将制备的基因组dna稀释成20ng/ul,取25ul (500ng dna)与25ul酶切混合液[5ul 10x ecor v buffer,17ul ddh2o 和3.0ul 15u/ul ecor v(takara)]混合后,37℃保温2小时,并标记为:m1。
[0111]
4)酶切样品纯化:
[0112]
每份酶切样品补入50ul ddh2o,再加入40ul磁珠混合液(hieffngstm smarter dna clean beads试剂盒),并按试剂盒要求进行样品 纯化。用28.0ul 0.1xte,ph7.6洗脱,吸取26.5uldna纯化样品,并标记 为:mc1。
[0113]
5)纯化的酶切dna片段与接头连接:
[0114]
取12.0ul mc1 dna片段,加入15ul 2x t7 dna ligase buffer,1.5ul5.0um lk1,1.5ul t7 dna ligase(biolab neb)混合、离心,25℃保 温2小时,并标记为:mcl1。
[0115]
6)连接样品纯化:
[0116]
每份mcl样品补入70ul ddh2o,再按第4步方法进行样品纯化,并 用34ul 0.1xte ph7.6洗脱,吸取32.5ul dna纯化样品,并依次标记为: mclc1。
[0117]
7)pcr扩增纯化的样品:
[0118]
取出8ul mclc1,加入2ul ddh2o,再加入10ul pcr混合液[4.5ulddh2o,2.0ul 10x pcr buffer,1.6ul 2.5mm dntp,0.5ul 25um fprim, 0.5ul 25um rprim1,0.6ul 100%甘油和0.3ul hs taq(takara)混匀、离 心。执行touchdown pcr反应程序(bio-rad s1000tm thermal cycler): 94℃/4min;{94℃/45sec,67℃(-1.0℃/循环)/60sec,72℃/60sec}共6个循环, (94℃/35sec,60℃/60sec,72℃/60sec)共28个循环,72℃/15min,4℃ /forever。
[0119]
8)pcr产物电泳分离、染色、成像:
[0120]
将pcr产物样品和60ng(4ul)dna标样(dl2000 dna marker, takara),在1.5%琼脂糖凝胶上分离;然后,用0.1%溴化乙锭进行标 准化染色,并用genegenius bio-imaging system(syngene,synopticsltd,cambridge,uk),记录成像。
[0121]
9)定量分析,取得结果:
[0122]
利用genetool(syngene,synoptics ltd,cambridge,uk)图像定 量分析工具。根据dna标样说明书,对泳道m的dna条带分子量2000bp、 1000bp、750bp、500bp、250bp和100bp进行定义,其中750bp条带定义 60ng dsdna。然后,分别对pcr产物各目标条带的分子量和扩增产物量 进行测定。
[0123]
具体实施例
[0124]
实施例1各癌细胞系中5’agggtc3’遗传缺陷端粒的测定
[0125]
实施例1.1
[0126]
选取肺癌细胞系,经常规培养,收集足够量的细胞,提取基因组dna。 其余所有步骤完全等同前述测试方案,定量测定结果记录为图1-1。
[0127]
实施例1.2
[0128]
选取结肠癌细胞系,经常规培养,收集足够量的细胞,提取基因组 dna。其余所有步骤完全等同前述测试方案,定量测定结果记录为图1-2。
[0129]
实施例1.3
[0130]
选取肝癌细胞系,经常规培养,收集足够量的细胞,提取基因组dna。 其余所有步骤完全等同前述测试方案,定量测定结果记录为图1-3。
[0131]
实施例1.4
[0132]
选取宫颈癌细胞系,经常规培养,收集足够量的细胞,提取基因组 dna。其余所有步骤完全等同前述测试方案,定量测定结果记录为图1-4。
[0133]
实施例1.5
[0134]
选取正常肝细胞系(lo2),经常规培养,收集足够量的细胞,提取 基因组dna。其余所有步骤完全等同前述测试方案,定量测定结果记录 为图1-5。
[0135]
以上癌细胞系和正常肝细胞系的定量测试结果总结于表1中。从中可 以看出,与正常肝细胞系中的测定结果相比,各癌细胞系中5’agggtc3
’ꢀ
遗传缺陷端粒存在的量显著更高,表明5’agggtc3’遗传缺陷端粒的更 高含量预示着细胞的癌变性质和/或趋势。
[0136]
实施例2一些癌症受试者中5’agggtc3’遗传缺陷端粒的测定
[0137]
实施例2.1
[0138]
选取肺癌受试者1(腺癌伴间质浸润,未转移)的edta抗凝静脉血 样,通过前述测试方案进行测试,定量测定结果记录为图2-1。
[0139]
实施例2.2
[0140]
选取肺癌受试者2(肺结节型腺癌,22只淋巴结慢性炎伴炭末沉着, 未转移)的edta抗凝静脉血样,通过前述测试方案进行测试,定量测定 结果记录为图2-2。
[0141]
实施例2.3
[0142]
选取肺癌受试者3(肺结节型,4只淋巴结慢性炎伴炭末沉着,未转 移)的edta抗凝静脉血样,通过前述测试方案进行测试,定量测定结果 记录为图2-3。
[0143]
实施例2.4
[0144]
选取肺癌受试者4(肺结节型,未转移)的edta抗凝静脉血样,通 过前述测试方案进行测试,定量测定结果记录为图2-4。
[0145]
实施例2.5
[0146]
选取肺癌受试者(未转移)的edta抗凝静脉血样,通过前述测试方 案进行测试,定量测定结果记录为图2-5。
[0147]
实施例2.6
[0148]
选取肺鳞癌受试者1(肺鳞癌,未转移)的edta抗凝静脉血样,通 过前述测试方案进行测试,定量测定结果记录为图2-6。
[0149]
实施例2.7
[0150]
选取肺鳞癌受试者2(肺鳞癌,未转移)的edta抗凝静脉血样, 通过前述测试方案进行测试,定量测定结果记录为图2-7。
[0151]
实施例2.8
[0152]
选取结肠癌受试者1(中分化腺癌,35只淋巴结慢性炎,未转移) 的edta抗凝静脉血样,通过前述测试方案进行测试,定量测定结果记 录为图2-8。
[0153]
实施例2.9
[0154]
选取结肠癌受试者2(晚期,已转移)的edta抗凝静脉血样,通过 前述测试方案进行测试,定量测定结果记录为图2-9。
[0155]
实施例2.10
[0156]
选取结肠癌受试者3(中分化腺癌,31只淋巴结慢性炎,未转移)的 edta抗凝静脉血样,通过前述测试方案进行测试,定量测定结果记录为 图2-10。
[0157]
实施例2.11
[0158]
选取直肠癌受试者(肠溃疡型,14只淋巴结慢性炎,未转移)的edta 抗凝静脉血样,通过前述测试方案进行测试,定量测定结果记录为图2-11。
[0159]
实施例2.12
[0160]
选取宫颈癌受试者1(未转移)的edta抗凝静脉血样,通过前述测 试方案进行测试,定量测定结果记录为图2-12。
[0161]
实施例2.13
[0162]
选取宫颈癌受试者2(未转移)的edta抗凝静脉血样,通过前述测 试方案进行测试,定量测定结果记录为图2-13。
[0163]
实施例2.14
[0164]
选取卵巢癌受试者(未转移)的edta抗凝静脉血样,通过前述测试 方案进行测试,定量测定结果记录为图2-14。
[0165]
实施例2.15
[0166]
选取贲门癌受试者(贲门隆起型中-低分化腺癌,1只淋巴结慢性炎, 未转移)的edta抗凝静脉血样,通过前述测试方案进行测试,定量测定 结果记录为图2-15。
[0167]
实施例2.16
[0168]
选取牙龈癌受试者(未转移)的edta抗凝静脉血样,通过前述测试 方案进行测试,定量测定结果记录为图2-16。
[0169]
实施例2.17
[0170]
选取正常受试者1(无任何患癌临床症状,经4.5年跟踪回访为健康受 试者)的edta抗凝静脉血样,通过前述测试方案进行测试,定量测定结 果记录为图2-17。
[0171]
实施例2.18
[0172]
选取正常受试者2(无任何患癌临床症状)的edta抗凝静脉血样, 通过前述测试方案进行测试,定量测定结果记录为图2-18。
[0173]
实施例2.19
[0174]
选取正常受试者3(无任何患癌临床症状,经4.5年跟踪回访为健康受 试者)的edta抗凝静脉血样,通过前述测试方案进行测试,定量测定结 果记录为图2-19。
[0175]
实施例2.20
[0176]
选取正常受试者4(无任何患癌临床症状)的edta抗凝静脉血样, 通过前述测试方案进行测试,定量测定结果记录为图2-20。
[0177]
实施例2.21
[0178]
选取正常受试者2(无任何患癌临床症状)的edta抗凝静脉血样, 通过前述测试方案进行测试,定量测定结果记录为图2-21。
[0179]
结果分析
[0180]
表1汇总了来自实施例2.1至2.21的全部数据。
[0181]
表1不同癌种5’agggtc3’结尾端粒检测结果
[0182][0183]
结果清晰地显示,无论是肺癌、结肠癌、直肠癌、卵巢癌、宫颈癌 还是不常见牙龈癌,其受试者(a类受试者)的全血基因组dna中都能 扩增出不同数量的清晰dna目标条带,pcr产物总量平均达到123.8ng, 而对照非癌症受试者(b类受试者)平均仅有6.0ng,而且多数非癌症受 试者没有检测到5’agggtc3’遗传缺陷端粒的存在。同时,对癌细胞株 的测试也得到类似结果,癌细胞系平均96.1ng,而对照只有5.6ng。无论 是癌症受试者还是癌细胞
株的dna条带的含量均极显著增高。
[0184]
基于每个实施例内的pcr扩增的dna条带,把每一条带的分子量除以一个完整重复序列的碱基数6bp之后得到的不同的非整倍数数量,即tc端粒数量(个)。例如结肠癌2,扩增出981bp、576bp、443bp、375bp和275bp共5个条带,对计算得到的非整倍数是0.500、0.000、0.8333、0.500和0.8333。其中,相同非整倍数对应的981bp和375bp,443bp和275bp分别来源于同一端粒,只是发生5’agggtc3’的位点差异。所以,含有5’agggtc3’遗传缺陷端粒数是3个。当然,这仅仅是一种简单初略的判断,可以通过dna测序分析,与人类基因组进行比对,得到准确信息。粗略分析结果显示在同一患癌病例的基因组中可检测到2~5个来源于不同染色体臂的5’agggtc3’遗传缺陷端粒。在7例肺癌中就出现了2~4个遗传缺陷端粒,但它们中至少有1个遗传缺陷端粒是源于相同的染色体臂。在3例结肠癌中也至少有1个遗传缺陷端粒也来源于相同的染色体臂。然而,在结肠癌中这个遗传缺陷端粒所在染色体臂却不同于在肺癌中的那个遗传缺陷端粒。这指示出不同癌种类型以及进展期不同与5’agggtc3’遗传缺陷端粒存在数量以及所在染色体臂有密切关联,为超早期筛查中的阳性受试者进一步提供癌症风险的类型,以及通过dna测序或结合其它方法可能实现癌症组织溯源,包括家族遗传、外部环境多因素等,达到更加精准的预防目的。
[0185]
将患癌受试者、癌细胞系和正常受试者的pcr扩增产物量作一个统计分析图(图3),结果清晰显示患癌受试者和癌细胞系与正常受试者相比存在极显著差异,显示出100%的灵敏度和100%的特异性,表明泛癌种超早期筛查技术的有效性和可靠性。
[0186]
可替代地或者额外地,通过将实施例1中sl1分别替换成sl2、sl3或sl4,同时将prim1替换成对应的prim2、prim3或prim4,其余步骤保持不变,能够分别对5’gggttc3’、5’agggtg3’和5’gggttg3’等遗传缺陷端粒实施测试,获得更多的泛癌种超早期筛查参数。
[0187]
实施例3靶向人染色体端粒3’末尾两个碱基为tc和/或tg的遗传缺陷端粒的hterc基因单碱基突变体对癌细胞的凋亡作用
[0188]
不局限于具体理论,推测靶向遗传缺陷端粒tc或tg末端序列的突变的人端粒酶rna基因在引入癌细胞后,将转录出突变的hterc组分,能够与细胞内的htert蛋白结合而形成具有催化活性的端粒酶,从而对相应的人染色体端粒3’末尾两个碱基为tc和/或tg的遗传缺陷端粒进行修复,使之变成tt结尾,进而在内源端粒酶作用下将端粒有效延伸,经pot1和tin2信号通路而降低端粒酶的异常表达,引导某(些)端粒已缩短至遗传缺陷区域的衰老细胞、潜在癌变细胞或已癌变细胞进入pcd凋亡,以进一步确认tc和/或tg遗传缺陷端粒对细胞癌变的作用。
[0189]
实施例3.1创建hterc基因单碱基突变体
[0190]
基于ncbi数据库中hterc基因全长为451bp,无内含子,直接从基因组dna中获得人源hterc序列,作为野生型基因(wt),其序列如seqidno.11所示。用重叠pcr方法将hterc序列中第-54位脱氧腺嘌呤核苷酸(a)突变成脱氧鸟嘌呤核苷酸(g),定义为hterc基因突变体i(ti,seqidno.12)。将第-54位脱氧腺嘌呤核苷酸(a)突变成脱氧胞嘧啶核苷酸(c),定义为hterc基因突变体ii(tii,seqidno.13)。并将它们克隆到t-载体中。具体实施如下:
[0191]
a:人全基因组dna制备。采用takaraminibestwholebloodgenomicdnaextractionkit试剂盒,制备全基因组dna,4℃保存,待用。
[0192]
b:重叠延伸pcr方法,定点突变所用引物为:
[0193]
1)tf:5
’‑
ttccatttttaaggtagtcgagg-3’(seqidno.no.14);
[0194]
2)tr:5
’‑
taaaaggcaacaaaaagcggaag-3’(seqidno.15);
[0195]
3)mf-i:5
’‑
gtctaaccctgactgagaagg-3’(seqidno.16);
[0196]
4)mr-i:5
’‑
ccttctcagtcagggttagac-3’(seqidno.17)。
[0197]
第一轮pcr:反应体系20ul(2.0ul10xpcrbufferwithmg2 ,1.6ul10mmdntp,2.0ul2.0umprimertf,2.0ul2.0umprimermr-i,5.0ul10ng/ulgenomicdna,0.2ul5u/ulpfupolymerase和7.2ulddh2o)。反应程序:95℃/5min,(95℃/30sec,58℃/30sec,72℃/60sec)x30个循环,72℃/10min,8℃停止。用1%琼脂糖凝胶电泳pcr产物。然后,用takaraminibestagarosegeldnaextractionkit回收376bp目标片段,并标记为pd1,稀释定量成10ng/ul。同时,用primertr替换primertf,primermf-i替换primermr-i,其余完全相同,回收639bp目标片段,并标记为pd2,稀释定量成10ng/ul。第二轮pcr:反应体系20ul(2.0ul10xpfubufferwithmg2 ,1.6ul10mmdntp,2.0ul2.0umprimertf,2.0ul2.0umprimertr,0.5ul10ng/ulpd1,0.5ul10ng/ulpd2,0.2ul5u/ulpfupolymerase和11.2ulddh2o)。反应程序:(95℃/30sec,62℃/30sec,72℃/60sec)x30个循环,72℃/10min,8℃停止。用1%琼脂糖凝胶电泳pcr产物。然后,用takaraminibestagarosegeldnaextractionkit回收967bp目标片段,并标记为td-i。同时,用1.0ul50ng/ulgenomicdna替换0.5ul10ng/ulpd1和0.5ul10ng/ulpd2,获得野生型目标基因片段,并标记为wd。用pmd18-t分别克隆td-i和wd。将目标片段
[0198]
td-i或wd进行3’末端加a反应,反应体积10ul(2.0ul10xpcrbufferwithmg2 ,1.01mmdatp,7.8ultd-i或wd目标片段,0.2ul5u/ulrtaq,),72℃保温15分钟。然后,根据takarapmdtm18-tvectorcloningkit的指导书进行连接、克隆,用takaraminibestplasmidpurificationkit提取dna样品,并进行测序确认无误后,分别标记为:pmd18/ td-i和pmd18/ wd。
[0199]
c:构建pmd18/ hterc(mt-i)转移载体。所用引物为:
[0200]
1)ecorter:
[0201]5’‑
gcgtgaattctccctttataagccgac-3’(seqidno.18);
[0202]
2)bamhnosp:
[0203]5’‑
caggatccaaaaaaaaatgggggctcacaagcc-3’(seqidno.19)。
[0204]
pcr反应体系20ul(10ul2xgcbuffer,3.2ul10mmdntp,2.0ul2.0umecorter,2.0ul2.0umbamhnosp,1uldnafrompmd18/td-i,0.2ul5u/ulpfupolymerase,1.6ulddh2o);pcr反应程序:95℃/2min,(95℃/30sec,60℃/30sec,72℃/60sec)x30个循环,72℃/10min,8℃停止。用1%琼脂糖凝胶电泳pcr产物,用takaraminibestagarosegeldnaextractionkit回收扩增片段。再将回收的扩增片段和pmdtm18-tvectordna分别用bamhi,ecori进行双酶切。酶切反应体系25ul(2.5ul10xbuffert,10ul60ng/uldna,0.6ul5u/ulbamhi,1.2ul5u/ulecori,3.25ddh2o)。反应条件:37℃保温2小时。用takaraminibestdnafragmentpurificationkit进行纯化。然后,根据takarapmdtm18-tvectorcloningkit的指导书进行连接、克隆,再用takaraminibestplasmidpurificationkit提取dna样品,并进行测序确认,获得hterc
基因的单碱基突变i的转移载体,标记为:pmd18/ hterc(mt-i)。
[0205]
d:用引物mf-ii:5
’‑
gtctaaccctcactgagaagg-3’(seqid no.20)替代mf-i:5
’‑
gtctaaccctgactgagaagg-3’(seq idno.16);mr-ii 5
’‑
ccttctcagtgagggttagac-3’(seq idno.21)替代引物mr-i,5
’‑
ccttctcagtcagggttagac-3’(seqid no.17),其余重复步骤b和步骤c,获得hterc基因的单碱基突 变ii的转移载体,并标记为:pmd18/ hterc(mt-ii)。
[0206]
e:用步骤b中构建的pmd18/ wd替代pmd18/ td-i,其余重复步 骤c,获得hterc基因的野生型的转移载体,并标记为:pmd18/ hterc(wt)。
[0207]
实施例3.2构建hterc基因突变体的重组真核表达载体
[0208]
利用真核表达载体pcdna3.1(-)和实施例3.1中获得的mt-i、mt-ii 和wt转移载体,通过bamhi和ecori双酶切,将hterc目标基因 插入pcdna3.1(-)真核表达载体,具体实施如下:
[0209]
用takara minibest plasmid purification kit制备pcdna3.1(-) (invitrogen,life technology)和pmd18/ hterc(mt-i)的质粒dna。 双酶切pmd18/ hterc(mt-i)dna反应体系:(5ul 10x buffer k,12ul180ng/ul pmd18/ hterc(mt-i)dna,2.5ul 5u/ul bamhi,2.5ul 5u/ulecori,28ul ddh2o);反应液37℃保温2小时后,用1%琼脂糖凝胶电 泳酶切产物。然后,用takara minibest agarose gel dna extraction kit 回收hterc(mt-i)目标片段(577bp)。同时,双酶切pcdna3.1(-)dna 反应体系:(5ul 10x buffer k,40ul 50ng/ul pcdna3.1(-)dna,2.5ul 5u/ulbamhi,2.5ul 5u/ul ecori);反应液37℃保温2小时后,直接用takaraminibest dna fragment purification kit纯化酶切样品,获得线性 pcdna3.1(-)dna。然后,将hterc(mt-i)目标片段与线性pcdna3.1(-) dna样品,用t4连接酶连接。连接体系10ul(1ul 10x t4 ligase buffer, 5ul 30ng/ul回收的线性pcdna3.1(-)dna片段,1ul 40ng/ul回收的 hterc(mt-i)目标片段,1ul 5u/ul t4 dna ligase),16℃保温12小时后, 按常规方法进行连接片段的转化、筛选、酶切鉴定以及测序鉴定,分别 获得携带目标基因的真核表达载体:pcdna3.1(-)/ hterc(mt-i)。
[0210]
用pmd18/ hterc(mt-ii)或pmd18/ hterc(wt)分别替代 pmd18/ hterc(mt-i),其余完全相同,重复上述方法。分别获得携带 目标基因的真核表达载体:pcdna3.1(-)/ hterc(mt-ii)和 pcdna3.1(-)/ hterc(wt)。(图4)
[0211]
实施例3.3构建人端粒酶启动子转移载体
[0212]
克隆htertpro所用引物:1)htertpro-f: 5
’‑
acatacgtacacgcacctgttcccag-3’(seq id no.22);2) htertpro
–
r:5
’‑
tcctgaattcgcgggggtggccggggcca-3
’ꢀ
(seq id no.23);pcr反应体系20ul(10ul 2x gc buffer,3.2ul 10mmdntp,2.0ul 2.0um htertpro-f,2.0ul 2.0um htertpro-r,0.5ul50ng/ul genomic dna,0.2ul 5u/ul rtaq polymerase,5.7ul ddh2o);反应 条件:95℃/2min,(95℃/30sec,61℃/30sec,72℃/60sec)x 30个循环,72℃ /10min,8℃停止。用1%琼脂糖凝胶电泳pcr产物,用takara minibestagarose gel dna extraction kit回收505bp扩增片段。再根据takarapmdtm18-t vector cloning kit的指导书,进行连接、克隆,并进行测 序确认无误后,将人端粒酶启动子(htertpro)转移载体标记为: pmd18/ htertpro。
[0213]
实施例3.4构建hterc基因突变体的ad5复制缺陷型腺病毒穿梭 载体
[0214]
a:用takara minibest plasmid purification kit制备删去hcmv腺 病毒载体
pca13/-hcmv和质粒pmd18/ htertpro的dna样品,用 hindiii和ecori双酶切方法将htertpro片段转入pca13/-hcmv质 粒。双酶切反应体系1:(2.5ul 10x buffer r,8ul pmd18/ htertpro dna, 0.8ul 5u/ul hindiii,1.25ul 5u/ul ecori,12.45ul ddh2o);反应液37℃ 保温2小时后,用1%琼脂糖凝胶电泳酶切产物。然后,用takaraminibest agarose gel dna extraction kit回收519bp目标片段。同时, 双酶切反应体系2:(2.5ul 10x buffer r,8ul pca13/-hcmv dna,0.8ul5u/ul hindiii,1.25ul 5u/ul ecori,12.45ul ddh2o);反应液37℃保温2 小时后,直接用takara minibest dna fragment purification kit纯化 酶切样品。最后,将519bp片段与双酶切、纯化的pca13/-hcmv dna 样品,用t4连接酶连接。连接体系10ul(1ul 10x t4 ligase buffer,1.5ul 回收的载体pca13/-hcmv片段,6.5ul回收的htertpro目标片段,1ul5u/ul t4 dna连接酶),16℃保温2小时后,按常规方法进行连接片段 的转化、筛选、酶切以及测序鉴定,获得目标重组质粒: pca13/ htertpro。
[0215]
b:用takara minibest plasmid purification kit制备 pca13/ htertpro和pmd18/ hterc(mt-i)质粒的dna,用bamhi 和ecori双酶切方法将hterc(mt-i)片段转入pca13/ htertpro载 体。双酶切pmd18/ hterc(mt-i)dna反应体系:(5ul 10x buffer k, 12ul 180ng/ul pmd18/ hterc(mt-i)dna,2.5ul 5u/ul bamhi,2.5ul5u/ul ecori,28ul ddh2o);反应液37℃保温2小时后,用1%琼脂糖 凝胶电泳酶切产物。然后,用takara minibest agarose gel dnaextraction kit回收hterc(mt-i)目标片段(577bp)。同时,双酶切 pca13/ htertpro dna反应体系:(5ul 10x buffer k,40ul 50ng/ulpca13/ htertpro dna,2.5ul 5u/ul bamhi,2.5ul 5u/ul ecori);反应 液37℃保温2小时后,直接用takara minibest dna fragmentpurification kit纯化酶切样品,获得线性pca13/ htertpro dna。最 后,将hterc(mt-i)目标片段与酶切、线性pca13/ htertpro dna 样品,用t4连接酶连接。连接体系10ul(1ul 10x t4 ligase buffer,5ul30ng/ul回收的线性pca13/ htertpro dna片段,1ul 40ng/ul回收的 hterc(mt-i)目标片段,1ul 5u/ul t4 dna连接酶),16℃保温2小时后, 按常规方法进行连接片段的转化、筛选、酶切以及测序鉴定,获得目标 基因穿梭载体:pca13/ htertpro hterc(mt-i)。
[0216]
用pmd18/ hterc(mt-ii)或pmd18/ hterc(wt)替代 pmd18/ hterc(mt-i),其余完全相同,重复上述方法。分别获得目标 基因穿梭载体:pca13/ htertpro hterc(mt-ii)和 pca13/ htertpro hterc(wt)。(图5)
[0217]
实施例3.5用admax系统包装ad5复制缺陷型重组腺病毒
[0218]
a:通过脂质体介导将目标基因转移载体 pca13/ htertpro hterc(mt-i)与人类腺病毒血清5型(e1/e3缺失) 骨架载体pbhg11共转染至hek293细胞,获得目标重组腺病毒。具体 方法:在转染前一天,在60mm平皿中以7.5
×
105个人胚肾293细胞铺 板,用5%dmem,置于37℃、5%co2细胞培养箱内培养。转染前3-4 小时(细胞回合率最好小于70%),更换成新鲜培养液,加入5ml无血 清培养基,确保细胞快速生长。然后,将目标基因转移载体 pca13/ htertpro hterc(mt-i),病毒骨架载体pbhg11和脂质体lipofectamine 2000(购自invitrogen公司)加入293细胞(具体操作 省略),共培养7-14天后出现病毒空斑,经过3次病毒空斑纯化后收集 细胞,于-80℃和37℃之间反复冻融3次,然后4℃、5000rpm离心10分 钟,去除细胞碎片,将包装好的重组腺病毒颗粒分装后-80℃,保存备用。
[0219]
b:重组腺病毒的鉴定
[0220]
应用qiagen dna blood mini kit,根据产品说明书,提取重组腺病 毒dna。
[0221]
再通过pcr扩增目标基因表达盒(htertpro hterc,1002bp), 上游引物target-f:5
’‑
attacatacgtacacgcacct-3’(seq id no. 24);下游引物target-r:5
’‑
aacgggaaagcgaactgcat-3’(seqid no.25)。反应条件:95℃/3min,(95℃/30sec,60℃/30sec,72℃/60sec)x 30个循环,72℃/10min,8℃停止。扩增产物用1%琼脂糖凝胶电泳。 用takara minibest agarose gel dna extraction kit回收1002bp目标 片段,经常规t-克隆后,送生工生物工程(上海)有限公司测序,确认 测序序列中hterc基因第-54号位点为鸟嘌呤脱氧核糖核苷酸“g”。并 将重组腺病毒定名为:ad5 hterc(mt-i)。
[0222]
用pca13/ htertpro hterc(mt-ii)替代 pca13/ htertpro hterc(mt-i),其余完全重复上述步骤a和步骤b, 确认测序序列中hterc基因第-54号位点为胞嘧啶脱氧核糖核苷酸“c”, 并将重组腺病毒定名为:ad5 hterc(mt-ii)。
[0223]
用pca13/ htertpro hterc(wt)替代 pca13/ htertpro hterc(mt-i),其余完全重复上述步骤a和步骤b, 确认测序序列中hterc基因第-54号位点为腺嘌呤脱氧核糖核苷酸“a”, 并将重组腺病毒定名为:ad5 hterc(wt)。
[0224]
c:重组腺病毒的扩增、纯化、及滴度测定
[0225]
基于常用重组腺病毒扩增方法,将hek293细胞接种于75cm2培养 瓶,加入20ml 10%fbs的dmem,培养至80-90%的细胞融合度时, 换成含15ml 2%fbs dmem。取0.5ul经病毒空斑纯化、鉴定正确的病 毒保存液,小心加入培养瓶,水平十字形慢慢晃动3次。在37℃5%co2 培养箱中培养48小时后收集细胞沉淀,加入5ml 5%dmem悬浮沉淀,
ꢀ‑
20℃至37℃反复冻融3次,600g离心20分钟后去沉淀取上清。按上 述步骤反复扩增至需要的病毒量,用hplc离子交换柱纯化病毒颗粒, 并测定纯化样品的物理滴度(vp/ml)和感染性滴度(tcid50/ml)。结 果:ad5 hterc(mt-i)为5.3
×
10
11
vp/ml与1.58
×
10
10
tcid50/ml; ad5 hterc(mt-ii)为2.69
×
10
11
vp/ml与7.94
×
109tcid50/ml;和 ad5 hterc(wt)为2.3
×
10
11
vp/ml与6.3
×
109tcid50/ml;
[0226]
实施例3.6用hterc野生型真核表达的重组体,测试重组体的基因 表达盒
[0227]
基于hela细胞基因组dna存在端粒3’末尾含有和遗传缺陷端粒, 以及很高的端粒酶活性,突变型重组载体对细胞生长有严重干扰(见: 实施例7)。所以,测试重组体的基因表达盒选用野生型的重组真核表达 载体。具体实施如下:
[0228]
a:确定g418最佳筛选浓度。取生长状态良好的hela细胞,用0.25% 胰酶消化,制备成细胞悬浮液,按1000个/ml接种在24孔培养板中,培 养6小时,吸去培养基,pbs洗涤一次,每孔中加入不同浓度的筛选培 养基(按100ug/ml、200ug/ml、300ug/ml、400ug/ml、500ug/ml、 600ug/ml、700ug/ml、800ug/ml、900ug/ml、1000ug/ml的梯度浓度用 培养基稀释g418制成筛选培养基)。每3天更换一次筛选培养基。以10-14 天内,将细胞全部杀死的浓度确定为最佳筛选浓度,即400ug/ml g418。
[0229]
b:用重组载体pcdna3.1(-)/ hterc(wt)转染hela细胞,获得稳 定转染的单细胞系。挑选生长状态良好的hela细胞,在转染前一天铺 六孔板,待细胞汇合度达到90%-95%时进行转染(effectene细胞转染 按试剂盒说明书)。转染24小时后将细胞用胰酶消化,以1:10的比例传 代,贴壁24小时候开始g418筛选,浓度为400ug/ml,十四天后绝大部 分细胞
死亡,极少数细胞形成克隆。在超净台内,弃去培养皿内的培养液,胰酶消化细胞,用200ul移液枪挑取单克隆,加入已预先加好培养基的24孔板内,待细胞贴壁后加入g418筛选培养基继续培养2-5天,细胞长满后,0.25%胰酶消化,转入6孔板内继续培养,获得稳定转染的hela细胞株。
[0230]
c:用trizol方法提取稳定转染的hela细胞株总rna,通过rt-pcr方法检测目标基因的表达,确定重组基因的表达正确。(图4)
[0231]
实施例3.7测试重组hterc基因突变体对hela细胞株的生长作用
[0232]
a:mtt方法测定hela细胞的生长。
[0233]
用构建的重组真核表达载体pcdna3.1(-)/ hterc(mt-i),pcdna3.1(-)/ hterc(mt-ii)和pcdna3.1(-)/ hterc(wt),另增加一个空载体pcdna3.1(-)/(空)为阴性对照,分别转染hela细胞。(按effectene试剂盒说明书操作)。首先,挑选生长状态良好的hela细胞,于转染前一天铺六孔板,待细胞汇合度达到90%-95%时进行转染(effectene细胞转染按试剂盒说明书)。转染24小时后,用胰酶消化收集,调整单细胞悬液密度为10
4-105个/ml。按200μl/孔(1
×
104个细胞)接种于96孔培养板内,每组细胞接种5个孔。置37℃、5%co2培养箱中培养。从第24h开始,每隔24h随机取出1块96孔板,每孔加入20ul5mg/mlmtt液,继续培养4h后,吸出孔内培养液,向每孔加入150uldmso。将培养板置于微孔板振荡器上振荡10min,在酶联免疫检测仪检测各孔570nm的od值。结果:pcdna3.1(-)/ hterc(mt-i)和pcdna3.1(-)/ hterc(tii)质粒的hela细胞生长速度显著减缓,阳性对照pcdna3.1(-)/ hterc(wt)和阴性对照pcdna3.1(-)生长较快。(图7)
[0234]
b:hoechst33342染色观察细胞凋亡特征
[0235]
hoechst33342染色观察细胞的形态,突变的pcdna3.1(-)/ hterc(mt-i)和pcdna3.1(-)/ hterc(tii)转染细胞一部分核膨大,有部分细胞出现染色质浓缩,高荧光状态呈现出细胞凋亡的特征。但未突变的阳性对照pcdna3.1(-)/ hterc(wt)和阴性对照pcdna3.1(-)均呈现正常细胞状态(图8)。
[0236]
实施例3.8测试重组hterc基因突变体对人乳腺癌实体瘤生长的抑制作用
[0237]
取人乳腺癌bcap-37经培养、传代细胞系,用3000离心3min,pbs悬浮至3x106/0.1ml/mouse,i.h.接种于裸鼠腋下,一次性接种20只。待10日后瘤体生长至3-4mm。挑选成瘤好的、瘤体相对均匀一致的15只,随机分成3组,分别以10ul1x106tcid50/ml的剂量将ad5 hterc(mt-i);ad5 hterc(mt-ii)和ad5 hterc(wt)单点注入给药;每周给药一次,连续三次之,给药第32天处死裸鼠,将肿瘤组织剥离下来称重,结果:hterc基因单碱基突变样本显著抑制人乳腺癌bcap-37实体瘤的生长,抑瘤率分别为38%和30.12%(图9)。将称重后的瘤体组织放入液氮中速冻,保存在-80℃中。
[0238]
实施例3.9测试重组hterc基因突变体对人乳腺癌端粒酶的抑制作用
[0239]
a:用trap方法,检测将实施例8中获得的实体瘤样本。取实体瘤100mg,放入预冷的研钵中,用液氮捣碎,加入200ul裂解液(10mmtris-hclph7.5,1mmegta,0.1mmaebsf,5mmβ-巯基乙醇,1%np-40,0.25mmnadoc,150mmnacl,1mmmgcl2,10%甘油),充分匀浆;将匀浆液移入1.5ml的ep管中,冰浴25min;再12,000g,4℃离心20min,将上清移取至1.5ml的ep管中,充分混匀,取5μl,用bradford方法进行蛋白质浓度测定,其余均匀分装到8个100μl的ep管中,置于-80℃保存,待用。
[0240]
b:端粒酶反应与pcr扩增。ts引物:5
’‑
ggatccaatccgtcgagcagagtt-3’(seqidno.26);cx引物:5
’‑
cccttacccttacccttaccctta-3’(seqidno.27)。反应体系50ul(20mmtris-hclph8.5,2.0mmmgcl2,50mmkcl,0.01%tritonx-100,1mmegta,1mmdtt,5ngbsa,50umdntps,2umts引物,2.5ul100ng总蛋白/ul端粒酶提取液,depch2o至50ul),20℃恒温孵育20min,80℃灭活处理10分钟。再加入0.5ul20mmcx引物和0.5ul5u/ultaqdna聚合酶,执行pcr反应:95℃/3min,(95℃/30sec,68℃/45sec)x30个循环,72℃/10min,8℃停止。
[0241]
c:用10%聚丙烯胺凝胶电泳分离pcr产物。
[0242]
制备10%非变性聚丙烯酰胺凝胶20ml(5.46ml,37%acr-bis36:1,12.4mlddh2o,2ml10
×
tbe,125ul10%aps,15ultemed),用0.5xtbe电泳缓冲液,电压17.5v/cm,垂直电泳预跑1小时,再取15ulpcr产物与1.5ul10x上样缓冲液进行上样,继续电泳1小时。银染:将凝胶置10%乙酸固定30min,蒸馏水漂洗3次,每次5min;用0.2g/l硫代硫酸钠浸泡1min,蒸馏水漂洗3次,每次30s;将凝胶放入0.2%硝酸银染液浸泡30min,用蒸馏水漂洗15s;然后,显色液中显色10min左右,放入10%乙酸固定10min。用genetool软件,进行相对定量分析。结果:hterc基因单碱基突变实体瘤样本相对端粒酶活性仅占野生型样本的21.6%(mt-i)和28.3%(mt-ii)呈现显著下降。(图10)
[0243]
实施例3.10观察重组hterc基因突变体对人乳腺癌细胞的结构变化利用常用组织石蜡切片和schiff试剂染色方法,观察实施例8获得的实体瘤组织样本,尤其是细胞核的变化。取实体瘤组织切成4x4mm的组织放入faa固定剂中固定,经一系列脱水、浸蜡、包埋、切成2-3um薄片,固定在载玻片上,经脱蜡、schiff试剂染色、封片。在10x100倍的普通显微镜下观察、拍照,记录结果:a:ad5 hterc(mt-i);b:ad5 hterc(mt-ii)与c:对照ad5 hterc(wt)样本相比,细胞核浓缩变小,大小不均一,有较多细胞核固缩导致的致密浓染的现象,细胞间接触变得松散,呈现出明显细胞凋亡的现象。(图11)
[0244]
实施例3.11检测重组hterc基因突变体对人乳腺癌细胞的程序性凋亡作用
[0245]
细胞程序性细胞凋亡的基因组dna片段由大约180bp的多聚体组成,在琼脂糖凝胶电泳分离上呈现出典型的“梯形”特征,这可明显区别于其它死亡细胞的dna降解特征。具体实施如下:
[0246]
采用takaraminibestuniversalgenomicdnaextractionkit制备实体瘤样本基因组dna,并用nanodrop2000进行定量。将用1%琼脂糖凝胶电泳分析特定单碱基突变hterc基因对人乳腺癌bcap-37实体瘤细胞总dna,结果呈现出几乎完全相同的特征。说明hterc基因单碱基突变引发了癌细胞的程序性凋亡。(图12)
[0247]
本实施例表明,使用本发明的重组端粒酶rna基因突变体,通过对tc和/或tg遗传缺陷端粒的修复,能显著降低它们的高端粒酶活性,明显抑制癌变细胞的生长,并将它们逆转进入正常的细胞程序性凋亡。这一实施例进一步证实了癌变细胞中tc或tg遗传缺陷端粒的存在,更重要地是验证了tc和/或tg遗传缺陷端粒的存在与驱动细胞癌变的直接关系,说明tc和/或tg遗传缺陷端粒作为细胞早期癌变的生物标志物的可靠性,可用于泛癌种超早期筛查,以及它又是细胞早期癌变的驱动因子,可作为创新药物的研发崭新靶点,也可以开发出本发明的泛癌种超早期筛查的伴随干预、预防和治疗的相关药物。
[0248]
本领域普通技术人员将理解,可对本发明进行许多变化和/或变动而不背离广泛
描述的本发明的精神或范围。上文列举的仅仅是本发明的若 干个具体实施例。除了本方法以外,还可以特异针对端粒3’末端最后一 个碱基实施化学荧光标记方法,或采用dna芯片检测技术等进行类似的 遗传缺陷端粒的检测,从而达到相同的目的。显然,本发明不限于以上 实施例,还涉及针对人基因组dna端粒3’末尾重复序列为tc或tg遗传 缺陷端粒而达到相同目的的其它任何方式、变形技术、变形方法、变形 途径等均属于本发明的范围均属于本专利范畴。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。