用于放射氟化的前体
1.分案申请
2.本技术为申请号2017800710479、申请日2017年11月17日、题为“用于放射氟化的前体”的分案申请。
技术领域
3.本发明涉及用于放射氟化(radiofluorierung)的前体以及用于所述前体的放射氟化的方法。
背景技术:
4.在医学诊断中,使用短寿命的、放射性标记的化合物,即所谓的放射性示踪剂,其生理和生化性质允许对人体中的代谢过程进行非侵入性断层扫描检测。通过使用正电子发射计算机辅助的断层扫描(pet)的现代断层扫描方法,可以量化代谢过程,并且可以通过所述放射性示踪剂的手段从外部检测放射性诊断剂的生物分布。放射性示踪剂(例如2-脱氧-2-[
18
f]氟-d-葡萄糖([
18
f]-fdg))的断层扫描检测允许肿瘤的早期诊断,该肿瘤与正常组织在葡萄糖代谢方面显著不同。通过在药理学上令人感兴趣的化合物的基础上开发新的放射性示踪剂,近年来各种临床图像的非侵入性诊断的新可能性已经开启了。
[0005]
近年来,在通过成像方法进行诊断的总市场中,正电子发射计算机辅助断层扫描(pet)的全球份额已经爆发性地增加。其中最大的份额是以[
18
f]氟化物作为放射性探针,因为其以f-18标记的糖衍生物([
18
f]-fdg)的形式,通过pet的手段使肿瘤的精确定位可视化,高达毫米范围,并允许肿瘤扩散的精确定位。然而,已经显示,通常被称为核医学的“主力”的[
18
f]-fdg仅用于检测原发性器官限制性前列腺癌(bouvez et al.,ejnmmi research 2016,6,40)。因此,为了更大程度地检测前列腺肿瘤和表达psma(前列腺特异性膜抗原)的转移灶,已经开发了新的放射性示踪剂,例如[
18
f]-dcfpyl(式1)和[
18
f]f-psma-1007(式2),其可用于检测前列腺特异性膜抗原(psma)。
[0006][0007]
在式1和2中可以看出,所述放射性示踪剂是多功能分子,因为它们具有大量的游离官能团,例如-oh、-conh、-cooh。通常地,具有许多游离官能团的分子不适合用
18
f直接标记。官能团通常与[
18
f]氟阴离子反应,通常使得hf产生。因此,不再有可用于成功放射性标记的活性氟化物。此外,高极性化合物在无水溶剂中的溶解度大大降低。而且,在水性溶剂中,[
18
f]氟阴离子没有被充分活化,因此在放射化学中使用的是所谓的“裸阴离子”,其中在有机溶液中,反离子(例如四正丁基铵盐)的正性中心(positive center)被非极性烃链屏蔽。
[0008]
因此,使用这些多功能分子的现有技术是插入保护基团或插入已经预先放射性标记的辅基(prosthetic group)(两种都是两阶段反应),使得只有合成引入的离去基团(leaving group)(在这种情况下为三甲基三氟甲磺酸铵)可与四正丁基碳酸氢铵活化的[
18
f]氟阴离子反应。出于辐射防护的原因,这些放射性示踪剂通常在所谓的“热室”中用自动合成模块制备,该自动合成模块使用一次性材料,例如盒(cassettes)(特别是经灭菌的盒)和试剂。使用这些系统,通常无法以经济有效的方式实现昂贵的多阶段合成路线。
[0009]
然而,保护基团的插入总是不利的,因为它们必须通过酸或碱昂贵地脱保护。因此,在这里我们也讨论了两阶段反应:第一阶段:用
18
f标记。第二阶段:用酸或碱脱保护。使用保护基团以及酸和碱通常都会导致大量的副产物,这些副产物必须从所需的
18
f标记物质中分离出来。这通常通过hplc(高效液相色谱法)的手段以高设备成本完成,因此耗时且昂贵。经由辅基的合成也需要至少两阶段来进行。首先,用
18
f标记辅基,然后将其与目标分子连接。
[0010]
时间因素在放射性药物中起重要作用,因为
18
氟阴离子的半衰期仅为109分钟,因此,合成时间和运输途径的任何延长都会对将获得的患者剂量的量产生直接影响。
[0011]
因此,已知有两种制备
18
f-dcfpyl的方式:第一种方式是通过受保护的前体进行两阶段合成(分别为ravert et al.,j.label compd.radiopharm 2016,59,439-50;方案1,或bouvet et al.,ejnmmi research,2016,6:40)。
br、-f或-i,并且r1、r2和r3彼此相同或不同,且各自为未取代的或取代的c
1-c6烷基;和
[0022]
(b)在活化盐存在的条件下使前体和[
18
f]氟阴离子反应生成放射氟化化合物,其中取代基y被[
18
f]氟化物取代,并且其中活化盐具有通式为n
(r4r5r6r7)的阳离子,其中r4、r5、r6和r7彼此相同或不同,且各自为未取代的或取代的c
1-c6烷基。
[0023]
根据本发明,可以在常规合成模块上提供在活化盐存在的条件下使前体和[
18
f]氟阴离子生成放射氟化化合物的反应完全自动化,随后进行柱纯化。在这里,放射氟化化合物的放射化学产率可以超过40%。
具体实施方式
[0024]
术语“c
1-c6烷基”涉及具有1-6个碳原子的直链或支链的饱和脂肪烃基。c
1-c6烷基的实例是甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、正己基。
[0025]
术语“取代的c
1-c6烷基”涉及如上定义的c
1-c6烷基,其具有一个或多个取代基,所述取代基选自由以下项组成的组:nh2、nh(c
1-c4烷基)、n(c
1-c4烷基)2、卤素、c
1-c4烷基、oh、o(c
1-c4烷基)、no2、cn、co2h或co2(c
1-c4烷基),其中每个前述c
1-c4烷基未被取代或被至少一个卤素原子取代。术语“卤素”涉及氟、氯、溴和碘。
[0026]
在本发明中,术语前体涉及可以在不使用保护基团的情况下通过放射氟化转化成放射化学化合物(即放射氟化化合物)的化合物。前体不具有羧酸盐基团,特别是没有羧酸盐阴离子-coo-。因此,前体也不具有与阳离子(例如阳离子螯合物或季铵阳离子)形成盐形式的羧酸盐基团。
[0027]
然而,前体可具有一个或多个羧基-cooh。利用根据本发明的方法,具有一个或多个羧基的前体可以在不使用保护基团的情况下转化为放射氟化化合物。前体和放射氟化化合物的不同之处仅在于取代基y被[
18
f]氟取代。前体的取代基y和放射氟化化合物的[
18
f]氟取代基处于相同的位置。
[0028]
优选地,取代基y是-n
(r1r2r3),其中r1、r2和r3彼此相同或不同,且各自为未取代的或取代的c
1-c6烷基。优选地,r1、r2和r3相同并且是甲基或丁基,其中特别优选甲基。在这种情况下,带有前体的芳环或杂芳环的基团-n
(r1r2r3)是三甲基季铵基团。与前体的基团-n
(r1r2r3)有关的阴离子可以是任何阴离子。例如,阴离子可选自包括以下项的组:氟、碘、溴、氯、磺酸盐、硫酸盐、磷酸盐、烷基硫酸盐、芳基硫酸盐或羧酸盐阴离子。优选地,作为阴离子,可以提供羧酸盐阴离子,例如cf3coo-、ch3coo-、c2h5coo-或hcoo-。
[0029]
如果基团-n
(r1r2r3)中的r1、r2和r3各自为甲基,则优选地,基团-n
(r1r2r3)的阴离子选自由以下项组成的组:三氟乙酸根(cf3coo-)、乙酸根(ch3coo-)、丙酸根(c2h5coo-)或甲酸根(hcoo-)。
[0030]
前体的芳环或杂芳环和因此放射化学化合物的芳环或杂芳环优选为单环或两个或更多个环的稠环系统,优选单环。杂芳环的杂原子优选地选自包n、o和s的组。优选地,杂原子是n。杂芳环可以具有一个或多个杂原子。优选地,杂芳环具有一个或两个杂原子,优选一个或两个氮原子。在这种情况下,环可以是吡啶环、哒嗪环、嘧啶环或吡嗪环。
[0031]
带有取代基y的优选的芳环或杂芳环如通式vi所示:
[0032][0033]
其中x是c-r8或n,y是-n
(r1r2r3)、-no2、-cl、-br、-f或-i,其中r1、r2和r3如上定义,而r8各自独立地是与间隔基团相连的键a2、氢或未取代的或取代的c
1-c6烷基,条件是只有一个残基r8是与间隔基团相连的键a2,而剩余的每一个r8彼此相同或不同,且各自代表氢或未取代的或取代的c
1-c6烷基。
[0034]
特别优选的芳环或杂芳环具有作为取代基y的基团-n
(r1r2r3),并且键a2位于基团-n
(r1r2r3)的对位,如通式via所示:
[0035][0036][0037]
其中x是c-r8或n,r1、r2和r3如上定义,而r8各自独立地是氢或未取代的或取代的c
1-c6烷基。
[0038]
带有作为取代基y的基团-n
(r1r2r3)的更优选的芳环或杂芳环,在基团-n
(r1r2r3)的对位上具有键a2,如通式vib所示:
[0039][0040]
其中x是c-r8或n,r1、r2和r3如上定义,而r8是氢或未取代的或取代的c
1-c6烷基。优选地,x是n且r1、r2和r3如上定义。
[0041]
带有作为取代基y的基团-n
(r1r2r3)的芳环或杂芳环的特别优选的实施例如式vic所示:
[0042][0043]
其中me代表甲基。在式vic中,关于通式vi,x是n;r1、r2和r3是甲基;位于基团-n
(r1r2r3)对位的r8为a2,而剩余的r8为氢。
[0044]
带有取代基y的优选的芳环或杂芳环如通式vid所示:
[0045][0046]
其中,
[0047]-x各自为c-r8或n,条件是至多两个x部分(moiety)为n,而剩余的每一个x部分为c-r8;和
[0048]-y是-n
(r1r2r3)、-no2、-cl、-br、-f或-i,其中r1、r2和r3如上定义,而r8各自独立地是与间隔基团相连的键a2、氢或未取代的或取代的c
1-c6烷基,条件是只有一个残基r8是与间隔基团相连的键a2,而剩余的每一个r8彼此相同或不同,且各自代表氢或未取代的或取代的c
1-c6烷基。
[0049]
键合单元能够结合肽或肽模拟物。例如,放射化学化合物可以通过键合单元与肽或肽模拟物的功能单元特异性连接。前体的键合单元和因此放射化学化合物的键合单元可以是通式i的键合单元:
[0050][0051]
其中a1是键合单元通过其与间隔基团连接的键,并且m和n彼此相同或不同,且各自是从0至10的整数。因此,m和n各自独立地可以是0、1、2、3、4、5、6、7、8、9或10。优选地,m=1且n=1。式i中所示的键合单元能够结合蛋白质psma(前列腺特异性膜抗原)。psma是在哺乳动物的前列腺中表达的蛋白质。对于前列腺癌,与健康的前列腺相比,它表达的程度更高。
[0052]
前体的间隔基团和因此放射化学化合物的间隔基团优选是通式ii或通式iii的间隔基团:
[0053][0054]
其中a1是间隔基团通过其与键合单元连接的键,a2是间隔基团通过其与前体或放射化学化合物的芳环或杂芳环连接的键,r9是氢或未取代的或取代的c
1-c6烷基,而z是未取代的或单取代的或多取代的烃基。优选地,r9是氢。
[0055]
在本发明的一个实施例中,z是式vii的基团:
[0056][0057]
其z基团具有式vii所示的含义的式iii的间隔基团为式iiia:
[0058][0059]
其中r9如上定义。优选地,r9是氢。
[0060]
根据本发明,在活化盐存在的条件下,使前体和[
18
f]氟阴离子反应生成放射氟化化合物。在这里,取代基y被[
18
f]氟化物取代。活化盐用于活化[
18
f]氟阴离子。活化盐具有通式为n
(r4r5r6r7)的阳离子,其中r4、r5、r6和r7彼此相同或不同,且各自为未取代的或取代的c
1-c6烷基。已经表明,这种活化盐不仅适合于使所述前体反应成放射氟化化合物,还适合于其他前体的放射氟化。优选地,r4、r5、r6和r7各自是未取代的c
1-c6烷基,更优选是丙基、丁基或戊基,特别优选地每个都是正丁基。优选地,活化盐具有阴离子,所述阴离子选自包括以下项的组:碳酸氢根(hco
3-)、硫酸氢根(hso
4-)、草酸根、磷酸根和甲苯磺酸根。更优选碳酸氢根和磷酸根。特别优选磷酸根。通式为n
(r4r5r6r7)的阳离子和阴离子以化学计量比存在于活化盐中。在优选的实施例中,活化盐是四正丁基碳酸氢铵或四正丁基磷酸铵,其中特别优选四正丁基磷酸铵。在下文中,具有四正丁基铵阳离子的活化盐也称为tba。
[0061]
到目前为止,使用具有通式为n
(r4r5r6r7)的阳离子的活化盐来活化[
18
f]氟阴离子还是未知的,其中r4、r5、r6和r7彼此相同或不同,且各自是未取代的或取代的c
1-c6烷基,而硫酸氢根、草酸根、磷酸根和甲苯磺酸根作为阴离子。这些活化盐不限于使所述前体生成放射氟化化合物的反应,还可用于其他前体的放射氟化。优选地,在硫酸氢盐、草酸盐、磷酸盐和甲苯磺酸盐中的r4、r5、r6和r7各自是未取代的c
1-c6烷基,更优选丙基、丁基或戊基,特别优选地每个都是正丁基。更优选碳酸氢盐和磷酸盐。特别优选磷酸盐。在优选的实施例中,活化盐是四正丁基碳酸氢铵或四正丁基磷酸铵,其中特别优选四正丁基磷酸铵。
[0062]
优选地,活化盐在极性溶液中,特别优选在具有水或含水混合溶剂的溶液中。混合溶剂可以是例如含有醇(如乙醇)的水。醇类添加剂用于稳定溶液。活化盐可以0.001至0.1m溶液提供,例如,特别是以0.075m溶液提供。
[0063]
放射氟化化合物例如是[
18
f]-dcfpyl(参见式1,同上)或[
18
f]f-psma-1007(参见式2,同上)。优选用于制备[
18
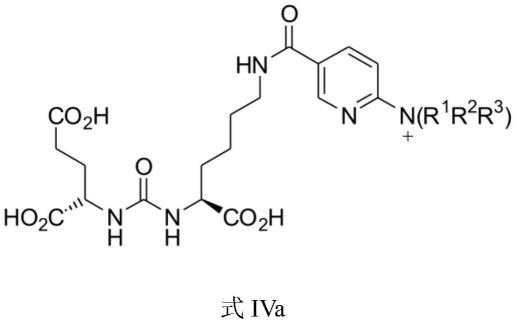
f]-dcfpyl的前体是通式iv的化合物:
[0064][0065]
其中取代基y选自由以下项组成的组:-n
(r1r2r3)、-no2、-cl、-br、-f或-i,且r1、r2和r3彼此相同或不同,且各自为未取代的或取代的c
1-c6烷基。式iv中所示的前体的键合单元对应于式i中所示的键合单元,其中m和n各自为1。
[0066]
更优选的用于制备[
18
f]-dcfpyl的前体是通式iva的化合物:
[0067][0068][0069]
其中r1、r2和r3彼此相同或不同,且各自为未取代的或取代的c
1-c6烷基。特别优选的r1、r2和r3各自为甲基。其中r1、r2和r3各自为甲基的式iva的前体如式ivb所示。
[0070][0071]
优选的用于制备[
18
f]f-psma-1007的前体是通式v的化合物:
[0072][0073]
其中取代基y选自由-n
(r1r2r3)、-no2、-cl、-br、-f或-i组成的组,且r1、r2和r3彼此相同或不同,且各自为未取代的或取代的c
1-c6烷基。式v中所示的前体的键合单元对应于式i中所示的键合单元,其中m和n各自为1。
[0074]
更优选的用于制备[
18
f]f-psma-1007的前体是通式va的化合物:
[0075][0076]
其中r1、r2和r3彼此相同或不同,且各自为未取代的或取代的c
1-c6烷基。特别优选的是,r1、r2和r3各自为甲基。其中r1、r2和r3各自为甲基的式va前体如式vb所示。
[0077][0078]
优选地,前体在非质子极性溶剂中提供,例如乙腈、二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmaa)、n-甲基-2-吡咯烷酮(nmp)、二甲基亚砜(dmso)或它们的混合物。
[0079]
在步骤(b)中使用的[
18
f]氟阴离子可通过已知方法制备。例如,通过用能量为9.6mev的质子照射富含至少97%的h
218
o,在回旋加速器中制备[
18
f]氟阴离子。将如此获得的水性[
18
f]氟化物溶液固定在阴离子交换柱(qma)上,并通过相转移催化剂(ptc)转移到反应容器中,相转移催化剂为例如冠醚、季铵盐或碱盐或碱土盐。作为ptc,优选使用[2,2,2]-穴醚(或k222)、四正丁基磷酸铵、氢氧化物、草酸盐、甲苯磺酸盐或任选其他冠醚(例如18-冠-6)。在共沸脱水后,将前体溶解在有机溶剂中并加入到干燥的反应混合物中。有机溶剂可以是非质子极性溶剂,例如乙腈、二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmaa)、n-甲基-2-吡咯烷酮(nmp)、二甲基亚砜(dmso)或其混合物。优选使用二甲基亚砜作为溶剂。
[0080]
步骤(b)优选在封闭的反应容器中、在升高的温度下、在热反应方案下进行。根据本发明的方法的步骤(b)优选在非质子极性溶剂中进行,例如乙腈、二甲基甲酰胺(dmf)、二甲基亚砜(dmso)或其混合物。优选使用二甲基亚砜作为溶剂。优选地,该方法在1至8的ph值下进行。优选地,ph值在4至8的范围内,特别优选为5。然而,该方法也可以在高于8的ph值下进行,但是产量会降低。本发明人惊奇地发现,仅在4至8的ph值范围内才产生更少的副产物,并且可以实现极高的标记产率。
[0081]
根据本发明的方法的步骤(b)优选在1至60分钟的时间段内进行,更优选在3至30分钟,特别优选在8至20分钟的时间段进行。
[0082]
根据本发明的方法的步骤(b),优选在低于100℃的温度下进行,更优选在室温至95℃的温度下进行,甚至更优选在室温至90℃的温度下进行,而特别优选在70至90℃的温度下进行。
[0083]
根据本发明的方法的步骤(b),也可以作为微波辅助反应进行。为此,将瓦数为50w至150w优选75w至85w的微波辐射到特定的封闭反应容器上。
[0084]
为了确定标记产率和放射性副产物,可以使用薄层色谱法(tlc)和高效液相色谱
法(hplc)。
[0085]
方案3说明了根据本发明的方法的优选实施例。在这里,在[
18
f]氟阴离子和活化盐存在的条件下,式iv的前体经反应成为[
18
f]-dcfpyl:
[0086][0087]
应该注意的是,前体可具有另一种阴离子而不是cf3coo-阴离子。优选地,活化盐是tba,特别优选tba磷酸盐。
[0088]
方案3a说明了根据本发明的方法的优选实施例。在这里,在[
18
f]氟阴离子和活化盐存在的条件下,式iva的前体经反应成为[
18
f]-dcfpyl:
[0089][0090]
应该注意的是,前体可具有另一种阴离子而不是cf3coo-阴离子。优选地,活化盐是tba,特别优选tba磷酸盐。
[0091]
在一个优选的实施例中,在[
18
f]氟阴离子和tba(优选tba磷酸盐)作为活化盐存在的条件下,用于制备[
18
f]-dcfpyl的式ivb前体如方案3b所示进行反应。
[0092][0093]
应该注意的是,前体可具有另一种阴离子而不是cf3coo-阴离子。
[0094]
方案4说明了根据本发明的方法的优选实施例。在这里,在[
18
f]氟阴离子和活化盐存在的条件下,式v的前体经反应成为[
18
f]f-psma-1007:
[0095][0096]
应该注意的是,前体可具有另一种阴离子而不是cf3coo-阴离子。优选地,活化盐是tba,特别优选tba磷酸盐。
[0097]
方案4a说明了根据本发明的方法的优选实施例。在这里,在[
18
f]氟阴离子和活化盐存在的条件下,式va的前体经反应生成[
18
f]f-psma-1007:
[0098][0099]
应该注意的是,前体可具有另一种阴离子而不是cf3coo-阴离子。优选地,活化盐是tba,特别优选tba磷酸盐。
[0100]
在一个特别优选的实施例中,在[
18
f]氟阴离子和tba(优选tba磷酸盐)作为活化盐存在的条件下,用于制备[
18
f]f-psma-1007的式vb前体如方案4b所示进行反应。
[0101][0102]
应该注意的是,前体可具有另一种阴离子而不是cf3coo-阴离子。
[0103]
根据本发明,进一步提供了用于制备放射氟化化合物的前体。前体具有带有取代基y的芳环或杂芳环、可以与肽或肽模拟物结合的键合单元以及连接芳环或杂芳环与键合单元的间隔基团。键合单元带有至少一个第二取代基,所述第二取代基选自由-oh、-conh和-cooh组成的组,其中键合单元通过键a1与间隔基团连接,而间隔基团通过键a2与芳环或杂芳环连接。在这里,取代基y选自由-n
(r1r2r3)、-no2、-cl、-br、-f或-i组成的组,r1、r2和r3彼此相同或不同,且各自为未取代的或取代的c
1-c6烷基。上文已经根据本发明的方法描述了根据本发明的前体的更多细节。
[0104]
根据本发明,进一步提供了根据本发明的前体在制备放射氟化化合物中的用途,该放射氟化化合物具有带有[
18
f]氟作为第一取代基的芳环或杂芳环、可以与肽或肽模拟物结合的键合单元,以及连接芳环或杂芳环与键合单元的间隔基团。键合单元可以带有至少一个第二取代基,所述第二取代基选自由-oh、-conh和-cooh组成的组,其中键合单元通过键a1与间隔基团连接,而间隔基团通过键a2与芳环或杂芳环连接。
[0105]
本发明允许一阶段(one-stage)合成所示的放射氟化化合物。一方面,这缩短了合成时间。另一方面,所达到的标记产率可以是文献中已知的两阶段方法的两倍以上。此外,一阶段合成的反应产物更易于纯化,由此可以避免在设备方面使用昂贵的hplc。可以通过柱子(所谓的spe柱)非常容易地纯化放射氟化化合物并且时间更少。此外,优选在gmp环境(gmp=良好操作规范)中避免浓酸和碱,因为在gmp领域中经常使用可腐蚀的不锈钢。根据本发明的方法的简单性允许根据本发明的放射氟化化合物的自动合成,例如借助于共同合成模块上的一次性盒和试剂盒。可以在合成过程中通过spe柱进行纯化,以便在合成结束时可以提供根据本发明的放射氟化化合物的即用型溶液。
[0106]
根据本发明的前体带有至少一个第二取代基,即至少一个未保护的oh、conh和/或cooh基团,目标化合物即放射氟化化合物也带有。之前不预期带有未保护的oh、conh和cooh基团的化合物的放射性标记确实行得通。实施本发明可以实现自动合成的显著简化,从而实现放射氟化化合物的制备的显著简化,因为不再需要以前的至少两阶段的合成,而可以在一阶段中完成,节省大量时间。节省时间允许显著提高产率,并因此允许放射氟化化合物更容易和更高的可用性。因此,从放射合成中获得更多活性,并因此,当使用在放射合成中
制备的放射氟化化合物作为放射性示踪剂时,可以检查更多患者。
[0107]
本发明借助于实施例详细解释,实施例不旨在限制本发明。
[0108]
实施例1
[0109]
式ivb前体的合成
[0110][0111]
起始化合物vi的合成如文献中所述(ravert et al.,j.label compd.radiopharm 2016,59,439-50;bouvet et al.,ejnmmmi research,2016,6:40)。在23.5ml三氟乙酸、0.62ml三异丙基硅烷和0.62ml水的混合物中,溶解2.48g起始化合物xx,并在室温下搅拌3小时。随后,在冰浴冷却和剧烈搅拌下,将反应混合物逐滴添加至241ml mtb醚中。用玻璃料(frit)吸去沉淀的白色固体,用100ml mtb醚洗涤两次。分离出白色固体的1.82g(84%)式ivb前体(=5-((s)-5-羧基-5-(3-((s)-1,3-二羧基-丙基)脲基)戊基-氨基甲酰基)-n,n,n-三甲基吡啶-2-氨基-2,2,2-三氟乙酸酯)。
[0112]
实施例2
[0113]
式vb前体的合成
[0114][0115]
起始化合物viii的合成如文献中所述(cardinale et al.,j.nucl.med.2016,接收出版;wo2015/062370a1)。将0.2mmol起始化合物viii在3.5ml二甲基甲酰胺中摇晃30分钟。之后,加入109mg n,n,n-三甲基-5-((2,3,5,6-四氟苯氧基)-羰基)吡啶-2-氨基氯
(olberg et al.,j.med.chem.2010,53,1732-1740)和0.042ml三乙胺。将反应混合物摇晃2小时,然后过滤树脂,用dmf洗涤三次,并用二氯甲烷洗涤三次。对于裂解和脱保护,将树脂与4ml三氟乙酸、0.11ml三异丙基硅烷和0.11ml水的混合物一起摇晃90分钟。随后,过滤混合物,并将滤液滴加到40ml mtb醚中。将混合物离心,吸出上清液,并用mtb醚洗涤剩余物三次。用hplc进行纯化。分离出白色固体的172mg(72%)式vb前体(=5-((s)-4-羧基-1-((s)-4-羧基-1-(4-((s)-1-((s)-5-羧基-5-(3-((s)-1,3-二羧基-丙基)-脲基)戊基氨基)-3-(萘-2-基)-1-氧代丙烷-2-基氨基甲酰基)-苄基氨基)-1-氧代丁烷-2-基氨基)-1-氧代丁烷-2-基氨基甲酰基)-n,n,n-三甲基吡啶-2-氨基-2,2,2-三氟乙酸盐)。
[0116]
实施例3
[0117]
式ivb前体在四正丁基碳酸氢铵存在的条件下生成[
18
f]-dcfpyl的反应
[0118]
7.5mg在1ml dmf中的式ivb前体、1ml 0.075m四正丁基碳酸氢铵(tba-hco3)和[
18
f]氟阴离子的反应混合物在ph值为约8.5、温度为75℃下反应14分钟。获得47.9%的[
18
f]-dcfpyl。另外,可以检测放射性副产物。[
18
f]氟化物的比例为28.6%。
[0119]
实施例4
[0120]
式ivb前体在四正丁基甲苯磺酸铵存在的条件下生成[
18
f]-dcfpyl的反应
[0121]
7.5mg在1ml dmf中的式ivb前体、750μl 0.075m四正丁基甲苯磺酸铵(tba甲苯磺酸盐)和[
18
f]氟阴离子的反应混合物在ph值为约5.0、温度为75℃下反应14分钟。检测到37.4%的[
18
f]-dcfpyl和30.9%的[
18
f]氟化物。另外,检测到放射性副产物,其比例为约30%。
[0122]
实施例5
[0123]
式ivb前体在四正丁基磷酸铵存在的条件下生成[
18
f]-dcfpyl的反应
[0124]
2.5mg在1.5ml dmf中的式ivb前体、750μl 0.075m四正丁基磷酸铵(tba磷酸盐)和[
18
f]氟阴离子的反应混合物在ph值为约4.7、温度为85℃下反应10分钟。在这些条件下,前体几乎定量转化为[
18
f]-dcfpyl(97.0%)。副产物可以减少到少于2%,仅能检测到痕量的剩余的[
18
f]氟化物。
[0125]
实施例6
[0126]
式ivb前体在四正丁基硫酸氢铵存在的条件下生成[
18
f]-dcfpyl的反应
[0127]
7.5mg在1ml dmf中的式ivb前体、750μl 0.075m四正丁基硫酸氢铵(tba硫酸氢盐)和[
18
f]氟阴离子的反应混合物在ph值为约1.7、温度为75℃下反应14分钟。获得15.6%的[
18
f]-dcfpyl,而[
18
f]氟化物的比例为73.4%。因此,用[
18
f]氟阴离子进行的标记相对较差。
[0128]
实施例3至6表明,在一阶段法中,在tba作为活化盐存在的条件下,式ivb前体与[
18
f]氟阴离子的反应导致放射氟化化合物[
18
f]-dcfpyl的产率相对较高。实施例5表明,在存在tba磷酸盐的微酸性ph范围内,仅产生痕量的副产物,并且可以实现极高的标记产率。
[0129]
实施例7
[0130]
式vb前体在四正丁基碳酸氢铵存在的条件下生成[
18
f]-psma-1007的反应
[0131]
10mg在1ml乙腈和600μl dmf的混合物中的式vb前体、750μl 0.075m tba碳酸氢盐和[
18
f]氟阴离子的反应混合物在ph值为约7、温度为120℃下孵育10分钟。除了37.9%的游离[
18
f]氟化物外,还检测到59.3%的[
18
f]f-psma-1007。可以检测到痕量的放射性副产物。
[0132]
实施例8
[0133]
式vb前体在四正丁基碳酸氢铵存在的条件下生成[
18
f]-psma-1007的反应
[0134]
2.5mg在1.5ml dmf中的式vb前体、750μl 0.075m tba碳酸氢盐和[
18
f]氟阴离子的反应混合物在ph值约7、温度为85℃下孵育10分钟。除了8.2%的游离[
18
f]氟化物外,还检测到90.7%的[
18
f]-psma-1007。可以检测到痕量的放射性副产物。
[0135]
实施例9
[0136]
式vb前体在四正丁基磷酸铵存在的条件下生成[
18
f]-psma-1007的反应
[0137]
2.5mg在1.5ml dmf中的式vb前体、750μl 0.075m tba磷酸盐和[
18
f]氟阴离子的反应混合物在ph值约4.7、温度为85℃下孵育10分钟。定量形成期望的产物[
18
f]-psma-1007(99.6%)。仅可以检测到痕量的游离[
18
f]氟化物。
[0138]
实施例7至9表明,在一阶段法中,在tba作为活化盐存在的条件下,式vb前体与[
18
f]氟阴离子的反应导致放射氟化化合物[
18
f]-psma-1007的产率相对较高。实施例9表明,在存在tba磷酸盐的微酸性ph范围内,可以实现定量标记产率。
[0139]
实施例10
[0140]
式vb前体在四正丁基碳酸氢铵存在的条件下通过合成模块ge tracerlab
(r)
mx
fdg
生成[
18
f]-psma-1007的全自动反应
[0141]
使用耐溶剂的活塞,建立类似于在ge tracerlab
(r)
mx
fdg
上的fdg合成盒的[
18
f]-psma-1007合成盒,并开发合成序列(synthesis sequence)。详细地,合成按照以下步骤进行:在qma柱上浓缩[
18
f]氟化物,用0.750ml tba碳酸氢盐洗脱,随后在95℃下干燥15分钟,用3mg在2ml dmf中的式vb前体在85℃下进行放射性标记14分钟,spe纯化和重新配制。可以获得放射化学产率》40%的[
18
f]-psma-1007。放射化学纯度》95%。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。