1.本发明属于催化剂技术领域,具体涉及一种氮掺杂生物炭包埋钴基催化剂的制备方法,以及其在外加电场条件下处理废水,尤其是抗生素类废水的应用。
背景技术:
2.随着科技的快速发展,人民对生活质量的追求越来越高,产生的废水也越来越难处理,如抗生素类废水、印染废水、含有消毒副产物的废水、苯酚废水等。其中抗生素类废水由于其原料利用率低,提炼纯度低,废水中残留抗菌素含量高,成为一个污水处理的难题。常见的污水处理方法有生物法、物理法和化学法,其中生物法虽然处理成本较低,但受外界环境影响较大。物理法(沉淀、过滤、膜分离)对污染物只进行了分离作用,并未将污染物降解为小分子物质或矿化。化学法因其适应性强而被广泛用于处理抗生素废水,其中高级氧化法是一种既高效又廉价的处理难降解抗生素的方法。
3.高级氧化技术是一种将光、电、声和催化剂与氧化剂结合处理难降解有机污染物的技术,其最大的特点是在反应过程中会产生大量的活性自由基与非自由基,主要的活性自由基有
·
oh和so4·-,非自由基有1o2与高价金属等。以往通过超声、紫外和加热活化过硫酸盐产生活性物质的方式效率较低,需要复杂系统。因此有必要寻找一种高效和环境友好的活化方式来激活氧化过程中的过硫酸盐。相比于上述活化方法,通过非均相过渡金属催化剂活化过硫酸盐可能是更加简便有效的方法。
4.迄今为止,已经探索了许多金属基催化剂来活化过硫酸盐(ps),包括co基、fe基、mn基催化剂等,在一众催化剂中,由于co基催化剂具有价廉、金属变价产生能量较大、活化性能较佳等优势,成为极具潜力的ps活化剂。目前已经探索了许多钴基催化剂来活化ps,包括零价钴、co3o4、碳包覆的钴基纳米复合材料(co@rbc)等,在这些催化剂中,co@rbc纳米复合材料因其独特的异质结构和电子构型在催化反应过程中具有明显的优势而引起了广泛的关注。更重要的是,co@rbc纳米复合材料解决了传统co基催化剂由于其金属裸露在外,很容易浸出,导致出现催化剂活性下降、循环使用率低的问题。然而,仍然存在活化ps能力不足,电子转移速率满,金属高低价态转化效率低等问题。因此,有必要寻求一种更加有效的技术来加速催化剂中的电子转移速率以及金属高低价态转化。
5.为了进一步增强催化剂活化过硫酸盐降解抗生素的能力,发现在原料中加入含氮物质,n作为储电子罐,可以有效将金属表面电子转移到生物炭中,从而加快催化剂的电子转移速率。而在实践中,研究人员发现不同批次制备得到的氮掺杂生物炭包埋钴基催化剂催化性能不稳定,持续催化能力差异较大。因此,亟需寻求一种氮掺杂生物炭包埋钴基催化剂的制备方法。
技术实现要素:
6.本发明的目的在于提供一种氮掺杂生物炭包埋钴基催化剂的制备方法,以及其在外加电场条件下处理废水的应用,旨在获得结构稳定,性能优异的氮掺杂生物炭包埋钴基
催化剂。根据本发明方法制备的氮掺杂生物炭包埋钴基催化剂可有效将金属表面电子转移到生物炭中,从而加快催化剂的电子转移速率,并且制备方法简单,在外加电场下具有抗生素降解效果显著、高催化活性、高电子传输效率与几乎零流失催化剂的特点。
7.一方面,本发明涉及一种氮掺杂生物炭包埋钴基催化剂的制备方法,该方法包括如下步骤:
8.s1.将金属硝酸盐和有机配体溶液混合,制备mof-co前驱液;
9.s2.将生物质预处理后浸泡于所述前驱液,浸渍饱和后,取出晾干,得到mof-co/bc前驱体;
10.s3.在惰性氛围下,对所述mof-co/bc前驱体进行热处理,冷却至室温,得到氮掺杂生物炭包埋钴基催化剂n-co@rbc。
11.另一方面,本发明涉及一种由上述方法制备的氮掺杂生物炭包埋钴基催化剂,所述催化剂由外层氮掺杂生物炭薄膜包埋钴。
12.另一方面,本发明还涉及所述氮掺杂生物炭包埋钴基催化剂在污水处理中的应用。
附图说明:
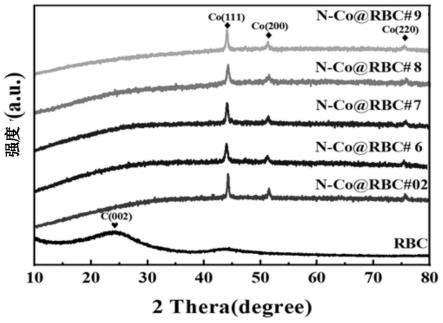
13.图1为不同催化剂n-co@rbc#02、n-co@rbc#6、n-co@rbc#7、n-co@rbc#8、n-co@rbc#9和纯生物炭rbc的xrd图谱。
14.图2为不同催化剂n-co@rbc#02、n-co@rbc#6、n-co@rbc#7、n-co@rbc#8、n-co@rbc#9和纯生物炭rbc的拉曼光谱。
15.图3为不同催化剂n-co@rbc#02、n-co@rbc#6、n-co@rbc#7、n-co@rbc#8、n-co@rbc#9和纯生物炭rbc的电化学阻抗图谱。
16.图4为在0.6g/l催化剂(n-co@rbc#02、n-co@rbc#6、n-co@rbc#7、n-co@rbc#8、n-co@rbc#9)、50mmol
·
l-1
na2so4、1mmol
·
l-1
pds、ph为6.5、和10ma/cm2电流密度下,测得的所述催化剂活化pds对sd的去除率曲线图。
17.图5为在0.6g/l催化剂(n-co@rbc#01、n-co@rbc#02、n-co@rbc#03)、50mmol
·
l-1
na2so4、ph为6.5、10ma/cm2电流密度、1mmol
·
l-1
pds下,测得的未加入尿素的催化剂活化pds对sd的去除率曲线图。
18.图6为在0.6g/l n-co@rbc#8、50mmol
·
l-1
na2so4、ph为6.5下,分别:仅电场(10ma/cm2电流密度)、仅pds(1mmol
·
l-1
)、pds 电场条件下sd的去除率曲线图。
19.图7为在0.6g/l n-co@rbc#8、50mmol
·
l-1
na2so4、ph为6.5、10ma/cm2电流密度下,不同浓度pds(0.3mmol
·
l-1、0.5mmol
·
l-1
、1mmol
·
l-1
、1.5mmol
·
l-1pds)的条件下sd的去除率曲线图。
20.图8为在50mmol
·
l-1
na2so4、ph为6.5、10ma/cm2电流密度、1mmol
·
l-1
pds下,不同催化剂浓度(0.2g/l、0.4g/l、0.6g/l、0.8g/l)的条件下sd的去除率曲线图。
21.图9为在0.6g/l n-co@rbc#8、50mmol
·
l-1
na2so4、ph为6.5、1mmol
·
l-1
pds下,不同电流密度(5ma/cm2、10ma/cm2、15ma/cm2)的条件下sd的去除率曲线图。
22.图10为在0.6g/l n-co@rbc#8、50mmol
·
l-1
na2so4、10ma/cm2电流密度、1mmol
·
l-1
pds下,不同ph(4.5、5.5、6.5、7.5、8.5)条件下sd的去除率曲线图。
23.图11为在0.6g/l n-co@rbc#8、50mmol
·
l-1
na2so4、10ma/cm2电流密度、1mmol
·
l-1
pds和10mm浓度的nacl、nahco3、nah2po4、nano3存的条件下sd的去除率曲线图。
具体实施方式
24.下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
25.本发明所述“生物质”(简称“bc”)是指经加工得到产物,例如生物燃料如乙醇,或牲畜饲料,或用于造纸和制浆工业产品的纤维素的植物材料。这样的植物材料可以包括完整植株、或植物的部分,例如茎、叶、枝条、枝、根和块茎等。
26.本发明涉及一种氮掺杂生物炭包埋钴基催化剂的制备方法,其特征在于,包括如下步骤:
27.s1.将母液a和母液b混合,制备mof-co前驱液,其中所述母液a为金属硝酸盐溶液,所述母液b为有机配体溶液;
28.s2.将生物质预处理后浸泡于所述mof-co前驱液,浸渍饱和后,取出晾干,得到mof-co/bc前驱体;
29.s3.在惰性氛围下,对所述mof-co/bc前驱体进行热处理,冷却至室温,得到氮掺杂生物炭包埋钴基催化剂n-co@rbc。
30.根据本发明所述的方法,所述生物质包括玫瑰花、玉米秸秆、树叶等;优选玫瑰花。
31.在某一具体的实施方案中,将新鲜玫瑰花晾干,完全浸泡至盐酸水溶液中进行酸洗和/或水洗,直到溶液澄清,之后室温晾干备用。
32.在某些具体的实施方案中,所述盐酸水溶液的浓度为10%、20%或30%。清洗的次数可以是两次、三次、四次、五次、六次、或七次,以确保bc表面的残存的清洗物质盐酸彻底清洗干净,否则很容易污染bc表面。盐酸易溶于水所以一次超纯水清洗可能仍有残留,用水重复多次清洗达到基本完全去除。
33.根据本发明所述方法,将所述母液a与所述母液b充分混合,得到mof-co前驱液,所述母液a与所述母液b的体积比为(0.5~1.5):1,优选为(0.8~1.2):1,最优选为1:1。
34.根据本发明所述方法,所述金属硝酸盐溶液是将三价钴盐溶于n,n-二甲基甲酰胺(dmf)配制而成;所述有机配体溶液是将对苯二甲酸(pta)与尿素溶于n,n-二甲基甲酰胺(dmf)配制而成。
35.根据本发明所述方法,所述三价钴盐选自六水合硝酸钴、六水合氯化钴、或四水合醋酸钴,优选为六水合硝酸钴。
36.进一步地,所述母液a的摩尔浓度为0.83~3.75mol/l;所述母液b的摩尔浓度为0.17~0.75mol/l;所述尿素的摩尔浓度为0.03~0.2mol/l。
37.根据本发明所述方法,步骤s2中,所述预处理后的bc在所述mof-co前驱液中于常温静置24-48h。
38.根据本发明所述方法,步骤s3中,所述热处理是将所述mof-co/bc前驱体以4~6℃/min的升温速率升至720~870℃,优选以4℃/min的升温速率升至750-850℃,最优选以4
℃/min的升温速率升至800℃。
39.本发明还提供了一种由上述方法制备的氮掺杂生物炭包埋钴基催化剂,所述催化剂由外层氮掺杂生物炭薄膜包埋钴,掺杂氮能同时增大催化剂比电容,促进催化剂石墨化程度和导电性,且掺杂氮中的吡啶氮能稳定包埋钴中的零价钴,从而显著促进其催化性能。此外,在外加电场作用下,催化剂被极化成粒子电极,利于保持催化剂性能稳定。并且催化剂本身具有很强的磁性,因此采用磁分离法易于催化剂回收利用。
40.根据本发明所述方法制备的氮掺杂生物炭包埋钴基催化剂,其表面生物炭层不仅作为电子传输通道能加速催化反应的进行,而且将内层的钴与复杂的外部环境隔离开来,能有效的减少金属钴的浸出,从而实现对抗生素的高效降解,并显著提高催化剂的抗干扰能力,使其在复杂多变的环境中都能起到很好的作用,非常适用于水体情况复杂的工业废水处理。
41.由此,本发明还提供了上述氮掺杂生物炭包埋钴基催化剂在污水处理中的应用。
42.根据本发明所述应用,所述氮掺杂生物炭包埋钴基催化剂用于在电场作用下活化pds降解含抗生素的废水中的抗生素。本发明提供的氮掺杂生物炭包埋钴基催化剂作为一种优异的三维粒子电极,具体地,co0和co
2
在活化pds失去电子分别变为co
2
和co
3
,同时外加电场可以原位将co
2
和co
3
还原为co0和co
2
,加速金属离子高低价态循环,进而保证反应能够高效稳定地进行。
43.进一步地,所述电场密度为3~18ma/cm2恒定电流,优选为5~15ma/cm2恒定电流,更优选为10ma/cm2恒定电流。室温搅拌反应50~70min,在充分混合的状态下加快体现内传质效率,并提供氧气。
44.进一步地,所述pds的浓度为0.5~2mmol/l;优选为0.5~1.5mmol/l;更优选为1mmol/l;且调节所述含抗生素的废水的初始ph为2.5~10.5;优选为4.5~8.5;更优选为6.5。
45.根据本发明所述应用,利用本发明所述氮掺杂生物炭包埋钴基催化剂的降解方法对多种抗生素的讲解均有较好的效果,尤其对于使用量大降解难的抗生素磺胺嘧啶(sd)也有很好地降解作用。具体地,所述抗生素包括四环素(tc)、土霉素(otc)、金霉素(ctc)、左旋氧氟沙星(lvf)或磺胺嘧啶(sd)中的一种或多种。
46.在研究中发现,仅仅通过引入外源电场,仍无法满足一些极难降解抗生素的去除,如磺胺嘧啶(sd)。经研究,外加电场下,根据本发明方法制备的氮掺杂生物炭包埋钴基催化剂能在较高浓度无机阴离子(100mmol
·
l-1
)以及宽泛的ph内对磺胺嘧啶废水具有优良的去除效率,具有独特的抗干扰能力以及宽泛的ph适应能力,在抗生素废水处理领域具有广泛的应用前景。
47.本发明所述催化剂作为一种优异的三维粒子电极,由于其表面生物炭层,以及氮掺杂后增强的比电容、石墨化程度和稳定更多的零价钴,一方面加速了催化剂中的电子转移速率,另一方面也提高了参与pds活化的零价钴含量,同时在电场作用下,加速金属高低价态转化效率,从而明显提升了对有机污染物的降解反应速率。
48.此外,本发明中pds成本低,pds为固体颗粒,便于运输,因而在实际工程应用中可操作性强。而本发明提供的氮掺杂生物炭包埋钴基催化剂的制备方法操作简单,具备可持续使用能力卓越,制备成本低等优点。
49.试剂来源:
50.过硫酸盐(k2s2o8,pds≥99.0%),对苯二甲酸(pta,≥99.9%)二甲基亚砜(dmso,≥99.9%),左氧氟沙星(lvf,≥99.0%),环丙沙星(cip,≥99.0%),罗丹明b(rhb,≥99.0%),四环素(tc,≥99.0%),土霉素(otc,≥99.0%),磺胺甲恶唑(smx,≥99.0%),磺胺嘧啶(sd,≥99.0%)从阿拉丁生化技术有限公司(中国上海)获得。甲醇(meoh,≥97.0%),叔丁醇(tba,≥99.0%),对苯醌(bq,≥99.0%),糠醇(ffa,≥99.0%),腐殖酸(ha,98%),六水合物硝酸钴(co(no3)2·
6h2o,99.0%),硫酸钠(na2so4≥99.0%),盐酸(hcl,90%),氢氧化钠(naoh,≥99%),氯化钠(nacl,≥99%),磷酸二氢钠(nah2po4,≥99%)硝酸钠(nano3≥99.0%)、碳酸氢钠(na2hco3,≥90%)购自国药集团化学试剂沈阳有限公司(中国沈阳)。所有化学品至少具有分析等级,并按正确的方式使用。玫瑰是从当地的花卉市场购买的。本实验中使用的所有溶液均使用蒸馏水制备。实验方法:
51.反应装置由方形玻璃反应容器(100ml)、直流电源和磁力搅拌器组成。镀铂钛(pt/ti)和不锈钢分别用作主电极的阳极和阴极。电极尺寸为4.0cm
×
2.5cm,两块电极之间的距离为50mm。将电极水平放入100ml水溶液中,电极的实际有效面积为8.0cm2。将电极连接到直流电源(ps-6403d,longwei,china)以获得恒定电流。首先,在反应器中加入一定量催化剂,持续搅拌30min以达到吸附-解吸平衡,然后加入一定浓度pds开始氧化反应。定时取出部分溶液,通过滤膜分离催化剂粉末。在紫外-可见分光光度计(persee-t6,北京)上在254nm处分析降解的sd浓度。
52.实施例1:
53.bc的预处理:
54.将新鲜玫瑰花晾干,完全浸泡至浓度为10%盐酸水溶液中进行酸洗,清洗两次后更换超纯水再次清洗,直到溶液澄清,之后室温晾干备用。
55.制备mof-co前驱液:
56.将0.01mol六水合硝酸钴溶于8ml dmf得到母液a。将0.01mol pta与0.002mol尿素溶于40ml dmf得到母液b。将母液a与母液b按体积比为0.8:1混合搅拌均匀,得到mof-co前驱液。
57.制备mof-co/bc前驱体:
58.将预处理后的bc置于上述mof-co前驱液中,并使其完全浸没,室温下静置48h,干燥,得到mof-co/bc前驱体。
59.制备n-co@rbc催化剂:
60.将上述mof-co/bc前驱体,在n2氛围下,以每分钟4℃升至750℃,保温3h后自然冷却至室温,即获得催化剂n-co@rbc#1。
61.实施例2:
62.bc的预处理:
63.将新鲜玫瑰花晾干,完全浸泡至浓度为20%盐酸水溶液中进行酸洗,清洗三次后更换超纯水再次清洗,直到溶液澄清,之后室温晾干备用。
64.制备mof-co前驱液:
65.将0.02mol四水合醋酸钴溶于10ml dmf得到母液a。将0.02mol pta与0.004mol尿素溶于60ml dmf得到母液b。将母液a与母液b按体积比为0.8:1混合搅拌均匀,得到mof-co
前驱液。
66.制备mof-co/bc前驱体:
67.将预处理后的bc置于上述mof-co前驱液中,并使其完全浸没,室温下静置48h,干燥,得到mof-co/bc前驱体。
68.制备n-co@rbc催化剂:
69.将上述mof-co/bc前驱体,在n2氛围下,以每分钟4℃升至850℃,保温1h后自然冷却至室温,即获得催化剂n-co@rbc#2。
70.实施例3:
71.bc的预处理:
72.将新鲜玫瑰花晾干,完全浸泡至浓度为30%盐酸水溶液中进行酸洗,清洗三次后更换超纯水再次清洗,直到溶液澄清,之后室温晾干备用。
73.制备mof-co前驱液:
74.将0.03mol六水合氯化钴溶于12ml dmf得到母液a。将0.03mol pta与0.004mol尿素溶于58ml dmf得到母液b。将母液a与母液b按体积比为1:1混合搅拌均匀,得到mof-co前驱液。
75.制备mof-co/bc前驱体:
76.将预处理后的bc置于上述mof-co前驱液中,并使其完全浸没,室温下静置48h,干燥,得到mof-co/bc前驱体。
77.制备n-co@rbc催化剂:
78.将上述mof-co/bc前驱体,在n2氛围下,以每分钟6℃升至850℃,保温1h后自然冷却至室温,即获得催化剂n-co@rbc#3。
79.实施例4:
80.bc的预处理:
81.将新鲜玫瑰花晾干,完全浸泡至浓度为20%盐酸水溶液中进行酸洗,清洗三次后更换超纯水再次清洗,直到溶液澄清,之后室温晾干备用。
82.制备mof-co前驱液:
83.将0.03mol六水合硝酸钴溶于10ml dmf得到母液a。将0.03mol pta与0.006mol尿素溶于58ml dmf得到母液b。将母液a与母液b按体积比为1.2:1混合搅拌均匀,得到mof-co前驱液。
84.制备mof-co/bc前驱体:
85.将预处理后的bc置于上述mof-co前驱液中,并使其完全浸没,室温下静置48h,干燥,得到mof-co/bc前驱体。
86.制备n-co@rbc催化剂:
87.将上述mof-co/bc前驱体,在n2氛围下,以每分钟5℃升至870℃,保温1h后自然冷却至室温,即获得催化剂n-co@rbc#4。
88.实施例5:
89.bc的预处理:
90.将新鲜玫瑰花晾干,完全浸泡至浓度为20%盐酸水溶液中进行酸洗,清洗两次后更换超纯水再次清洗,直到溶液澄清,之后室温晾干备用。
91.制备mof-co前驱液:
92.将0.03mol六水合硝酸钴溶于10ml dmf得到母液a。将0.03mol pta与0.006mol尿素溶于58ml dmf得到母液b。将母液a与母液b按体积比为1:1混合搅拌均匀,得到mof-co前驱液。
93.制备mof-co/bc前驱体:
94.将预处理后的bc置于上述mof-co前驱液中,并使其完全浸没,室温下静置48h,干燥,得到mof-co/bc前驱体。
95.制备n-co@rbc催化剂:
96.将上述mof-co/bc前驱体,在n2氛围下,以每分钟3℃升至720℃,保温1h后自然冷却至室温,即获得催化剂n-co@rbc#5。
97.实施例6:
98.bc的预处理:
99.将新鲜玫瑰花晾干,完全浸泡至浓度为20%盐酸水溶液中进行酸洗,清洗两次后更换超纯水再次清洗,直到溶液澄清,之后室温晾干备用。
100.制备mof-co前驱液:
101.将0.02mol六水合硝酸钴溶于10ml dmf得到母液a。将0.02mol pta与0.002mol尿素溶于56ml dmf得到母液b。将母液a与母液b按体积比为1:1混合搅拌均匀,得到mof-co前驱液。
102.制备mof-co/bc前驱体:
103.将预处理后的bc置于上述mof-co前驱液中,并使其完全浸没,室温下静置48h,干燥,得到mof-co/bc前驱体。
104.制备n-co@rbc催化剂:
105.将上述mof-co/bc前驱体,在n2氛围下,以每分钟4℃升至800℃,保温3h后自然冷却至室温,即获得催化剂n-co@rbc#6。催化剂的物相组成(xrd图谱)、电化学阻抗(eis)和石墨化程度(拉曼光谱)分别如图1、2和3所示。比电容和co0的含量如表1所示。
106.实施例7:
107.bc的预处理:
108.将新鲜玫瑰花晾干,完全浸泡至浓度为20%盐酸水溶液中进行酸洗,清洗两次后更换超纯水再次清洗,直到溶液澄清,之后室温晾干备用。
109.制备mof-co前驱液:
110.将0.02mol六水合硝酸钴溶于10ml dmf得到母液a。将0.02mol pta与0.004mol尿素溶于56ml dmf得到母液b。将母液a与母液b按体积比为1:1混合搅拌均匀,得到mof-co前驱液。
111.制备mof-co/bc前驱体:
112.将预处理后的bc置于上述mof-co前驱液中,并使其完全浸没,室温下静置48h,干燥,得到mof-co/bc前驱体。
113.制备n-co@rbc催化剂:
114.将上述mof-co/bc前驱体,在n2氛围下,以每分钟4℃升至800℃,保温2h后自然冷却至室温,即获得催化剂n-co@rbc#7。催化剂的物相组成(xrd图谱)、电化学阻抗(eis)和石
墨化程度(拉曼光谱)分别如图1、2和3所示。比电容和co0的含量如表1所示。
115.实施例8:
116.bc的预处理:
117.将新鲜玫瑰花晾干,完全浸泡至浓度为20%盐酸水溶液中进行酸洗,清洗两次后更换超纯水再次清洗,直到溶液澄清,之后室温晾干备用。
118.制备mof-co前驱液:
119.将0.02mol六水合硝酸钴溶于10ml dmf得到母液a。将0.02mol pta与0.006mol尿素溶于56ml dmf得到母液b。将母液a与母液b按体积比为1:1混合搅拌均匀,得到mof-co前驱液。
120.制备mof-co/bc前驱体:
121.将预处理后的bc置于上述mof-co前驱液中,并使其完全浸没,室温下静置48h,干燥,得到mof-co/bc前驱体。
122.制备n-co@rbc催化剂:
123.将上述mof-co/bc前驱体,在n2氛围下,以每分钟4℃升至800℃,保温2h后自然冷却至室温,即获得催化剂n-co@rbc#8。催化剂的物相组成(xrd图谱)、电化学阻抗(eis)和石墨化程度(拉曼光谱)分别如图1、2和3所示。比电容和co0的含量如表1所示。
124.实施例9:
125.bc的预处理:
126.将新鲜玫瑰花晾干,完全浸泡至浓度为20%盐酸水溶液中进行酸洗,清洗两次后更换超纯水再次清洗,直到溶液澄清,之后室温晾干备用。
127.制备mof-co前驱液:
128.将0.02mol六水合硝酸钴溶于10ml dmf得到母液a。将0.02mol pta与0.008mol尿素溶于56ml dmf得到母液b。将母液a与母液b按体积比为1:1混合搅拌均匀,得到mof-co前驱液。
129.制备mof-co/bc前驱体:
130.将预处理后的bc置于上述mof-co前驱液中,并使其完全浸没,室温下静置48h,干燥,得到mof-co/bc前驱体。
131.制备n-co@rbc催化剂:
132.将上述mof-co/bc前驱体,在n2氛围下,以每分钟4℃升至800℃,保温2h后自然冷却至室温,即获得催化剂n-co@rbc#9。催化剂的物相组成(xrd图谱)、电化学阻抗(eis)和石墨化程度(拉曼光谱)分别如图1、2和3所示。比电容和co0的含量如表1所示。
133.对比例1:
134.bc的预处理:
135.将新鲜玫瑰花晾干,完全浸泡至浓度为20%盐酸水溶液中进行酸洗,清洗两次后更换超纯水再次清洗,直到溶液澄清,之后室温晾干备用。
136.制备mof-co前驱液:
137.将0.01mol六水合硝酸钴溶于10ml dmf得到母液a。将0.01mol pta溶于56ml dmf得到母液b。将母液a与母液b按体积比为1:1混合搅拌均匀,得到mof-co前驱液。
138.制备mof-co/bc前驱体:
139.将预处理后的bc置于上述mof-co前驱液中,并使其完全浸没,室温下静置48h,干燥,得到mof-co/bc前驱体。
140.制备n-co@rbc催化剂:
141.将上述mof-co/bc前驱体,在n2氛围下,以每分钟4℃升至800℃,保温2h后自然冷却至室温,即获得催化剂n-co@rbc#01。
142.对比例2:
143.bc的预处理:
144.将新鲜玫瑰花晾干,完全浸泡至浓度为20%盐酸水溶液中进行酸洗,清洗两次后更换超纯水再次清洗,直到溶液澄清,之后室温晾干备用。
145.制备mof-co前驱液:
146.将0.02mol六水合硝酸钴溶于10ml dmf得到母液a。将0.02mol pta溶于56ml dmf得到母液b。将母液a与母液b按体积比为1:1混合搅拌均匀,得到mof-co前驱液。
147.制备mof-co/bc前驱体:
148.将预处理后的bc置于上述mof-co前驱液中,并使其完全浸没,室温下静置48h,干燥,得到mof-co/bc前驱体。
149.制备n-co@rbc催化剂:
150.将上述mof-co/bc前驱体,在n2氛围下,以每分钟4℃升至800℃,保温2h后自然冷却至室温,即获得催化剂n-co@rbc#02。催化剂的物相组成(xrd图谱)、电化学阻抗(eis)和石墨化程度(拉曼光谱)分别如图1、2和3所示。比电容和co0的含量如表1所示。
151.对比例3:
152.bc的预处理:
153.将新鲜玫瑰花晾干,完全浸泡至浓度为20%盐酸水溶液中进行酸洗,清洗两次后更换超纯水再次清洗,直到溶液澄清,之后室温晾干备用。
154.制备mof-co前驱液:
155.将0.03mol六水合硝酸钴溶于10ml dmf得到母液a。将0.03mol pta溶于56ml dmf得到母液b。将母液a与母液b按体积比为1:1混合搅拌均匀,得到mof-co前驱液。
156.制备mof-co/bc前驱体:
157.将预处理后的bc置于上述mof-co前驱液中,并使其完全浸没,室温下静置48h,干燥,得到mof-co/bc前驱体。
158.制备n-co@rbc催化剂:
159.将上述mof-co/bc前驱体,在n2氛围下,以每分钟4℃升至800℃,保温2h后自然冷却至室温,即获得催化剂n-co@rbc#03。
160.筛选出对比例2和实施例6-9所制备的催化剂,测定其各种价态钴含量(摩尔百分比)和比电容,结果如下表1所示:
161.表1各催化剂样品中各种价态钴含量(摩尔百分比)和比电容
162.催化剂co0(%)co
2
(%)co
3
(%)比电容(f.g-1
)n-co@rbc#020.060.510.475.00
×
10-2
n-co@rbc#60.090.500.4510.17
×
10-2
n-co@rbc#70.110.600.3312.00
×
10-2
1pds后,调节ph为6.5,在电流密度分别为5ma/cm2、10ma/cm2、15ma/cm2条件下,在室温下均匀搅拌60min,即完成在不同电流密度下利用n-co@rbc#8活化pds对sd的去除。测试结果见图9,本测试中sd在60min的去除率分别为86.40%、99.75%与79.67%。
175.ph值的影响
176.将5ml的200mg/l磺胺嘧啶(sd)以及95ml超纯水加入到反应容器中,置于搅拌器上搅拌均匀;然后向反应容器中加入0.6g/l n-co@rbc#8、50mmol
·
l-1
na2so4与0.5mmol
·
l-1
pds后,调节电流密度为10ma/cm2,在ph分别为4.5、5.5、6.5、7.5与8.5的条件下,在室温下均匀搅拌60min,即完成在不同初始ph下利用n-co@rbc#8活化pds对sd的去除。测试结果见图10,本测试中sd在60min的去除率分别为86.28%、92.68%、99.75%、78.32%与65.67%。
177.干扰离子的影响
178.将5ml的200mg/l磺胺嘧啶(sd)以及95ml超纯水加入到反应容器中,置于搅拌器上搅拌均匀;然后向反应容器中加入0.6g/l 30n-co@rbc、50mmol
·
l-1
na2so4与0.5mmol
·
l-1
pds后,调节ph为6.5、电流密度为10ma/cm2,再分别加入100mmol
·
l-1
的nacl、nahco3、nah2po4与nano3,在室温下均匀搅拌60min,即完成在不同干扰离子下利用30n-co@rbc活化pds对sd的去除。测试结果见图11,本测试中sd在60min的去除率分别为94.50%、64.00%、94.50%与84.37%。
179.以上内容是结合具体的优选实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只限于这些说明。对于本发明所属领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。