含i型聚酮合酶基因的质粒的制备方法
技术领域
1.本发明涉及一种含i型聚酮合酶(pks)基因的质粒的制方法。
背景技术:
2.已知由放线菌和丝状菌等微生物产生的天然化合物是具有多种结构和生物活性的有用物质。目前,通过解读基因组,可以很容易地确定用于生物合成有用物质的基因簇。此外,还明确了存在大量用于生物合成人类未利用的有用物质的基因簇。在微生物的次级代谢产物中,重要研究了用于对具有产业重要性的聚酮类化合物和肽类化合物进行生物合成的基因簇。例如,可以列举出i型聚酮合酶(polyketidesynthase;pks),其用于在放线菌产生的二级代谢产物中临床应用的erythromycin、fk-506(tacrolimus)、rapamycin及avermectin等大环内酯类化合物的生物合成。
3.考虑到通过传统的化学方法生产聚酮化合物等的难度、以及野生型细胞中的聚酮通常的低生产性,寻找用于生产聚酮化合物的改良或替代方法成为备受瞩目的焦点。由于这些原因,通过将生物合成所需的基因簇从原始菌株引入到其他细胞中来尝试化合物的异源生产。实际上,异源表达的传统方法大多限定于小的基因簇(<40kb),而很多pks基因簇都比它大得多,其为从数千碱基到100千碱基以上的dna。
4.作为组装dna的方法,已知有金门(golden gate)法、吉布森(gibson)法等连接多个dna片段的方法(非专利文献1)。
5.golden gate法是准备一个或两个末端包含由iis型限制性核酸内切酶识别的碱基序列的多个dna片段,并用iis型限制性核酸内切酶和dna连接酶进行处理的方法。可以通过由iis型限制性核酸内切酶切割产生的粘性末端(sticky end),来使多个dna片段杂交,接着利用dna连接酶连接缺口,来制造出具有所需碱基序列的dna片段。通过设计由iis型限制性核酸内切酶识别的碱基序列的类型和排列,可以将由iis型限制性核酸内切酶切割的多个dna片段连接起来以使限制性核酸内切酶的识别位点消除,从而制造出具有所需碱基序列的dna片段。报告了一种通过利用golden gate法来将具有所需碱基序列的dna片段导入克隆载体的方法等(专利文献1和非专利文献1)。
6.gibson法是准备以相邻连接的dna片段的各自末端部的连接区域重叠15~80个碱基对(bp)左右的方式(以成为相同碱基序列的方式)进行设计的多个dna片段,用5'核酸外切酶、dna聚合酶和dna连接酶进行处理的方法。通过5'核酸外切酶从dna片段末端部分地产生单链dna。所产生的单链dna在重复的碱基序列部分中杂交。然后,通过dna聚合酶填充间隙,并通过dna连接酶来连接缺口,从而可以制造出具有所需碱基序列的dna片段。
7.在gibson法中,由于不需要包含由限制性核酸内切酶识别的碱基序列等,因此没有碱基序列的限制,并且也适用于长dna片段的构建(非专利文献2、专利文献2以及专利文献3)。
8.现有技术文献
9.专利文献
10.专利文献1:美国专利申请公开第2010/0291633号说明书
11.专利文献2:美国专利第7723077号说明书
12.专利文献3:日本特表2011-512140号公报
13.非专利文献
14.非专利文献1:plos one,2009年,4(5),e5553
15.非专利文献2:化学和生物,2016年,vol.54,no.10,pp.740~746
技术实现要素:
16.发明所要解决的问题
17.据说在golden gate法中,可一次性连接的dna片段的数量约为20左右,如果超过该数量,效率会降低。如pks基因簇这样的序列是重复序列及gc含量高的碱基序列,因此错误连接的频率存在提高的倾向,因此很难一次性地准确连接多个dna片段。
18.在gibson法中,当组装包含重复序列或gc含量高的碱基序列的pks基因簇的情况下,由于单链dna杂交的准确性降低,因此错误连接的频率会变得极高。实际上,其受限于最多15个片段的组装规模。
19.本发明的目的在于高效地合成聚酮合酶基因。
20.用于解决问题的方法
21.本发明提供一种包含以下i型聚酮合酶基因的质粒及其制备方法、制备pks酶的方法。
22.〔1〕一种制备包含编码pks的dna在内的质粒的方法,其包括以下工序:在枯草芽孢杆菌感受态细胞中导入包含编码i型聚酮合酶(pks)的dna串联重复序列在内的dna构建体的工序。
23.〔2〕根据〔1〕所述的方法,其包括以下工序:通过用typeii限制性核酸内切酶切割多个dna片段前体,来制备两端具有粘性末端的多个dna片段的工序;通过连接多个所述dna片段,来制备所述dna构建体的工序。
24.〔3〕根据〔2〕所述的方法,其包括以下工序:通过将包含不同种类的多个所述dna片段的溶液进行混合以使得各溶液中的dna片段的摩尔浓度比为0.8~1.2,来制备编码i型聚酮合酶(pks)的dna串联重复序列的工序。
25.〔4〕根据〔2〕或〔3〕所述的方法,其中,所述dna片段前体的gc含量为65%以下。
26.〔5〕根据〔1〕至〔4〕中任一项所述的方法,其中,所述导入工序是将所述dna构建体与枯草芽孢杆菌感受态细胞共培养的工序。
27.〔6〕根据〔1〕至〔5〕中任一项所述的方法,其中,其还包括从导入了所述dna构建体的枯草芽孢杆菌中回收质粒dna的工序。
28.〔7〕一种包含编码i型聚酮合酶(pks)的dna在内的质粒,其中,其通过根据〔1〕至〔6〕中任一项所述的方法而得到。
29.〔8〕一种制备pks酶的方法,其包括以下工序:用质粒转化宿主细胞的工序;培养转化后的所述宿主细胞的工序;其中,所述质粒是通过根据〔1〕至〔6〕中任一项所述的方法而得到的、且包含编码i型聚酮合酶(pks)的dna在内的质粒。
30.〔9〕根据〔8〕所述的方法,其中,所述宿主细胞是streptomyces属细菌。
31.发明效果
32.可以提供一种准确且有效地对编码如pks这样的巨大基因簇的长链dna进行组装的方法。
附图说明
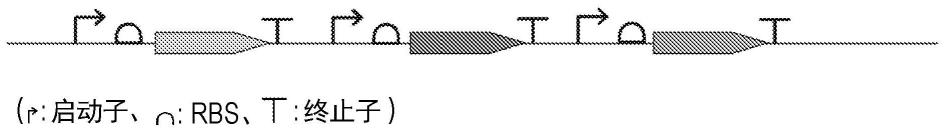
33.图1是示出连接3个debs pks基因编码序列,并在其各自的前面插入启动子、rbs,而在其后面插入终止子序列的靶序列的概略图。
34.图2示出了在使用大肠杆菌的比较例1中得到的质粒所包含的pks基因的限制性核酸内切酶降解产物的电泳结果。
35.图3示出了在使用枯草芽孢杆菌的实施例1中得到的质粒所包含的pks基因的限制性核酸内切酶降解产物的电泳结果。图3中的检查示出了获得所需的debs pks基因编码序列。
具体实施方式
36.以下,对本发明具体实施方式详细进行说明。但是,本发明并不限于以下实施方式。
37.本实施方式所涉及的制备包含编码pks的dna在内的质粒的方法,其包括以下工序:在枯草芽孢杆菌感受态细胞中导入包含编码i型聚酮合酶(有时仅仅记载为“pks”或“i型pks”)的dna串联重复序列在内的dna构建体的工序。
38.在本说明书中,所谓包含编码pks的dna(下面,有时记载为“pks基因”。)串联重复序列在内的dna构建体,是指在枯草芽孢杆菌细胞内能够转换成包含1个或多个pks基因在内的质粒的构建体。例如,通过将上述dna构建体与枯草芽孢杆菌感受态细胞共培养,来将上述dna构建体导入到枯草芽孢杆菌细胞内。接下来,在枯草芽孢杆菌细胞内,利用包含编码pks的dna在内的重复单元的同源性,使上述dna构建体环状化,并将其转换成包含编码1个或多个pks的dna在内的质粒。因此,可以从该枯草芽孢杆菌中获得包含编码pks的dna在内的质粒。
39.作为枯草芽孢杆菌感受态细胞的制备方法,优选使用anagnostopoulou,c.and spizizen,j.,j.bacteriol.,81,741-746(1961)中记载的方法。
40.加入到枯草芽孢杆菌感受态细胞中的dna构建体溶液的液量并没有特别限制。相对于枯草芽孢杆菌感受态细胞的培养液的液量,dna构建体溶液的液量优选为1/20至等量的范围内,更优选为半量。
41.编码pks的dna串联重复序列在形成质粒时,将编码pks的dna中的至少1个、优选地将编码1个pks的dna整合到质粒中。作为从枯草芽孢杆菌中纯化质粒的方法,可以使用公知的方法。
42.通过上述方法而得到的质粒具有编码目标pks的dna,这可以通过限制性核酸内切酶切割而产生的片段的大小模式、pcr法、碱基序列确定法来确认。
43.上述dna构建体只要是包含编码i型聚酮合酶(pks)的dna(例如,编码pks中包含的结构域的dna)在内的构建体即可,而pks的类型并没有特别限定。此外,dna构建体的大小也没有特别限定。编码pks的dna(例如,编码pks中包含的结构域的dna)的类型不仅可以是具
有微生物等天然来源序列的dna,也可以是具有人工设计序列的dna等,并没有特别限制。在天然来源序列中,为了表达对应的氨基酸,不同的生物物种主要使用一个密码子,但在进行异源表达时,优选使用与宿主的密码子使用频率相匹配的人工设计序列。可能影响异源表达结果的其他因素可以列举出gc含量(碱基序列内鸟嘌呤和胞嘧啶的总含量)、重复序列等。重复序列会降低遗传稳定性,存在错误杂交的风险,并阻碍重复片段的合成。因此,在异源表达的情况下,编码pks的dna优选地与密码子使用量和gc含量相关联进行地优化。然而,这些要求通常很难同时得到最佳满足。例如,作为优化密码子的结果,可能会导致非常重复的dna序列或高gc含量。
44.在本发明中,编码pks的dna的gc含量为30~70%。编码pks的dna的gc含量优选为70%以下、68%以下、65%以下、60%以下。通过使用本发明所涉及的方法,即使编码pks的dna的gc含量为50%以上、52%以上、55%以上、58%以上、60%以上,也可以高效率地合成目的质粒。在本发明中,编码pks的dna优选地优化密码子,以使得其不会出现20bp以上的碱基序列的重复。优选地避免编码pks的dna中gc含量出现极端差异。例如,最高和最低的50bp长度间的gc含量的差异优选为52%以下。最好尽量减少均聚物。最好尽可能地使分散在编码pks的dna内的小重复序列的数量/长度最小化。重复序列的长度小,且5~10bp的重复数的总和优选为150以下、120以下、100以下、80以下、60以下。通过使用本发明,编码pks的基因中所含的5~10bp长度的重复数可以为40以上、45以上、50以上。
45.本发明中得到的包含pks基因的质粒通过与特定的宿主组合,可以高效地产生聚酮。在一个优选的实施方式中,本发明可以利用天然产生的pks基因实质上缺失的、且进行了基因操作的宿主细胞。这些宿主细胞可以用包含各种pks基因的质粒进行转化,以产生活性聚酮。本发明提供在生长周期的适当阶段大量产生的产物。这样产生的聚酮取决于聚酮的类型,并且可以作为治疗剂用于治疗多种疾病。例如,已发现由本发明的质粒转化而成的宿主所产生的一些聚酮可用于免疫抑制剂、抗肿瘤剂、以及治疗病毒、细菌和寄生虫感染。通过重组产生聚酮的能力也提供了一种表征pks及其作用机制的强力工具。
46.更优选地,用于重组目标聚酮的宿主细胞可以来源于可由本发明的质粒转化而成的任何生物。因此,本发明的宿主细胞可以来源于原核生物或真核生物。然而,优选的宿主细胞由放线菌构成,更优选为streptomyces属的宿主。使用streptomyces属的宿主的最大优点在于,与使用大肠杆菌的异源表达生产相比,生产效价高,并且存在i型pks的活性表达所必需的翻译后修饰体系。具体而言,可以列举出s.albus、s.ambofaciens、s.avermitilis、s.azureus、s.cinnamonensis、s.coelicolor、s.curacoi、s.erythraeus、s.fradiae、s.galilaeus、s.glaucescens、s.hygroscopicus、s.lividans、s.parvulus、s.peucetius、s.rimosus、s.roseofulvus、s.thermotolerans、s.violaceoruber等,优选s.albus。
47.上述宿主细胞可以通过使用标准技术(例如同源重组)使来源于该宿主细胞的天然产生的pks基因缺失来进行基因操作。
48.编码pks的dna可以是天然型的,也可以是改变了密码子用法的dna,还可以是改变了1个或2个以上氨基酸的dna。在一个优选实施方式中,streptomyces pks包括3个开放阅读框(orf1、orf2、orf3)的产物。pks包含酮合成酶(ks)结构域、酰基转移酶(at)结构域、酰基载体蛋白(acp)结构域这3种结构域,通过这3种结构域可以延长聚酮链。pks还可以具有
酮还原酶(kr)结构域、脱水酶(dh)结构域、烯醇还原酶(er)结构域等与主链修饰相关的结构域。作为由pks制备的化合物,可以列举出6-脱氧红霉内酯b(6-deb)、富伦菌素、榴菌素、曲霉素、6-甲基水杨酸、土霉素、四环素、红霉素、甘油霉素、七尾霉素、美达霉素、柔红霉素、酪氨酸、卡波霉素、螺旋霉素、阿维菌素、莫能菌素、无活菌素、克拉霉素、利福霉素、脂霉素和杀念菌素。
49.质粒包含与编码所需pks的dna可操作地连接的控制序列。用于本发明的合适的表达系统包括在真核生物宿主细胞和原核生物宿主细胞中发挥作用的系统。然而,如上所述,原核生物系统是优选的,而且与streptomyces属细菌兼容的系统尤其重要。用于这种系统的控制序列包括启动子、核糖体结合位点、终止子、增强子等。有用的启动子在streptomyces属的宿主细胞中发挥功能,例如,可以列举出pgapdh、perme、pkaso等,但并不限定于此。
50.选择标记也可以包含在质粒中。已知各种标记在转化细胞系的选择中是有用的,而且,通常,当细胞在适当的选择培养基中生长时,其表达包括赋予转化细胞上可选择的表型的基因。例如,这些标记包括赋予质粒以抗生素抗性或敏感性的基因。或者,一些聚酮本身是有颜色的,其特征在于提供一种用于对由本发明的构建物成功转化而成的细胞进行选择与生俱来的(built-in)标记。
51.质粒可以包含在宿主细胞中发挥功能的功能序列。作为功能序列,例如,可以列举出质粒复制起点序列、编码用于将质粒整合在宿主基因组中的酶的序列、接合(conjugation)起始点序列等。
52.可以通过将控制序列、选择标记以及功能序列等事先嵌入到dna构建体的重复单位中,来使其包含在质粒中。
53.将本发明的质粒导入适当的宿主细胞中的方法对于本领域的技术人员来说是公知的,而且代表性地包括使用cacl2或2价阳离子和如dmso那样的其他药剂。质粒也可以通过电穿孔导入宿主细胞中。一旦表达pks,则可鉴定聚酮产生菌落,而且可使用公知的技术进行分离。本发明的质粒可以使用细菌间的接合转移导入到宿主细胞中。在本发明的一个优选实施方式中,将包含编码pks的dna在内的区域从本发明的质粒转移到大肠杆菌的质粒中以改造质粒骨架(亚克隆),接着通过接合(conjugation)从大肠杆菌转移到streptomyces属微生物中,由此实施向宿主细胞的导入。由此,将编码pks的dna整合到如streptomyces属微生物那样的宿主细胞基因组中。
54.在一个实施方式中,本发明的质粒可以包含枯草芽孢杆菌、大肠杆菌和streptomyces属微生物中的复制起点序列、用于从大肠杆菌到streptomyces属微生物的接合转移的接合起始点序列(orit)以及编码将streptomyces属微生物整合到基因组中所需的整合酶的序列。这样的质粒从枯草芽孢杆菌中回收后,可以在无需进行用于改造质粒骨架的亚克隆的情况下,转化大肠杆菌,然后通过接合从大肠杆菌转移至streptomyces属微生物。
55.本发明的另一个优选实施方案是包含编码大模块pks的dna在内的质粒。例如,6-脱氧红霉素b合酶(debs)催化作为红霉素糖苷配基的6-脱氧红霉素b的生物合成。三个开放阅读框编码debs多肽,而且在saccharopolyspora erythraea基因组的ery簇中跨越32kb。该基因由分别被称为“模块”的、6个重复单元组织而成。各模块编码一组活性位点,该活性
位点在聚酮生物合成期间催化附加单体在生长链上的缩合。每个模块可包含酰基转移酶(at)、β-酮酰基载体蛋白合酶(ks)、酰基载体蛋白(acp)、以及还原活性位点的亚群(β-酮酰还原酶(kr)、脱水酶(dh)、烯酰还原酶(er))。模块内的还原位点的数量与各缩合循环中的β-酮还原的程度相对应。
56.dna构建体例如可以通过用dna连接酶连接两个末端为粘性末端的多个dna片段而得到。具体而言,作为dna片段,例如,可以列举出具有末端的片段,该末端利用粘性末端的碱基序列的互补性,能够在相互保持顺序的状态下重复连接。该粘性末端的结构只要是除回文结构(palindrome)以外的结构即可,还包括5'末端突出、3'末端突出的形状的不同,并没有特别限制。但是,在制备dna片段时,优选地通过限制性核酸内切酶的消化来制备突出末端。作为限制性核酸内切酶,当使用用于可识别特定的序列并在其附近制作任意序列的突出末端的酶时,由于dna片段的粘性末端在各连接位点会有所不同,因此可保持其连接顺序。通过适当地设计各dna片段的粘性末端的序列,可以得到各dna片段以预定的顺序进行排列的dna构建体。作为这些限制性核酸内切酶的例子,除了通常的分子生物学中使用的限制性核酸内切酶以外,还可以列举出人工限制性核酸内切酶的talen或znf、或者crispr-cpf1等可生成粘性末端的crispr技术的相关酶等,优选使用如aari、alwni、bbsi、bbvi、bcodi、bfuai、bgli、bsai、bsaxi、bsmai、bsmbi、bsmfi、bspmi、bspqi、btgzi、draiii、foki、pflmi、sfani、sfii等这样的typeii限制性核酸内切酶。通过使用nebeta
tm tools,可以确定最佳的粘性末端。粘性末端的碱基数优选为3~6,更优选为3~4。1个dna片段中所含的碱基数优选为1~5kb。也可以是2kb以上、3kb以上、4kb以上。对于用于生成粘性末端的1个dna片段的剪切,优选利用1种限制性核酸内切酶进行切割。未必需要通过同一种的限制性核酸内切酶的消化来得到所有的dna片段,但使用的限制性核酸内切酶的类型的总数越少越好,优选为3种以下,更优选为2种以下,进一步优选为1种。
57.例如,dna片段可以通过包含以下工序的方法来进行制备:通过typeii限制性核酸内切酶切割dna片段前体(通过typeii限制性核酸内切酶的切割而产生对应的dna片段的dna片段。例如,也可以是质粒)来制备两端具有粘性末端的多个dna片段的工序;以及通过连接多个dna片段来制备dna构建体的工序。
58.通过连接多个dna片段来制备dna构建体的工序例如可以是通过dna连接酶连接多个dna片段来制备dna构建体的工序。
59.通过连接多个dna片段来制备dna构建体的工序可以是通过混合包含不同类型的多个前dna片段在内的溶液来制备编码i型聚酮合酶(pks)的dna串联重复序列的工序。进行混合以使得所有dna片段的摩尔浓度比为0.8~1.2,优选为0.9~1.1,更优选为0.95~1.05,进一步优选为约1.0。dna片段的混合也可以根据需要在存在合成用质粒的情况下进行,从而使dna片段合成。
60.作为dna连接酶,可以列举出t4噬菌体和大肠杆菌的dna连接酶。
61.dna构建体除了编码pks的dna(例如,编码pks中包含的结构域的dna)以外,还可以包含其他基因。例如,当dna构建体包含pks基因的情况下,除了pks基因以外,为了1)修饰最终的聚酮产物、2)将聚酮输送到细胞外、或3)赋予对抗生素聚酮的抗性,优选地还包含其他非pks基因。换言之,pks基因的部分是靶簇的核心生物合成部分,但许多情况下也需要上述非pks基因。
62.作为dna片段的类型,为3~60(种),优选为5~50(种),更优选为8~40(种),进一步优选为10~30(种)。
63.各dna片段的gc含量可以为65%以下。优选地,不会出现20bp以上的碱基序列的重复。
64.例如,当编码pks的dna由按照p、q、r、s的顺序连接了4个结构域的p-q-r-s进行表示的情况下,dna构建体包括由-(p-q-r-s)
n-表示的串联重复序列(n为2以上的整数)。优选地,在dna构建体的各orf(p-q-r-s)的5'末端或3'末端上附加在宿主细胞中发挥功能的启动子、核糖体结合序列(rbs序列)、增强子等控制序列。此外,也可以在dna构建体的各orf(p-q-r-s)的5'末端或3'末端上附加在宿主细胞中发挥功能的功能序列、以及选择标记等序列。此外,dna构建体优选地包括对枯草芽孢杆菌有效的复制起点。
65.通过将枯草芽孢杆菌感受态细胞与dna构建体一起培养,来将dna构建体摄入至枯草芽孢杆菌内,从而形成包含编码pks的dna在内的质粒。
66.编码pks的dna串联重复序列在形成质粒时,将包含编码串联重复序列pks的dna在内的重复单元(包含orf、根据需要的控制序列、在宿主细胞中发挥功能的功能序列以及选择标记等。)整合到质粒中。
67.优选地,在质粒中附加在宿主细胞中发挥功能的启动子、核糖体结合序列(rbs序列)、增强子等控制序列。此外,优选地,质粒中包含对枯草芽孢杆菌有效的复制起点。例如,在枯草芽孢杆菌的情况下,具有θ型复制机制,具体而言,可以列举出ptb19(imanaka,t.,etal.,j.gen.microbioi.,130,1399-1408.(1984))和pls32(tanaka,t and ogra,m.febs lett.,422,243-246.(1998)、pamβ1(swinfield,t.j.,etal.,gene,87,79-90.(1990))等质粒中含有的复制起点等序列。
68.终止子只要是在宿主细胞中发挥功能的终止子即可,并没有特别限定,例如可以优选地列举出来源于fd噬菌体的终止子(fd-ter)、来源于t4噬菌体的终止子(t4-ter)及来源于t7噬菌体的终止子(t7-ter)等。其中,从上述稳定化效果更佳的观点出发,特别优选为来源于fd噬菌体的终止子。
69.可以使用公知的核糖体结合序列(rbs)。
70.在本发明的一个实施方式中,可以培养将包含编码pks的dna在内的质粒导入宿主细胞的转化子,并通过该培养物得到聚酮。所谓“培养物”,是指培养上清、培养细胞、培养菌体、或细胞或菌体的破碎物中的任一种。培养本发明的转化子的方法可以按照宿主培养中使用的常规方法来进行。
71.培养本发明的转化子的培养基只要是含有宿主可同化的碳源、氮源、无机盐类等且能够有效地培养转化子的培养基即可,也可以使用天然培养基、合成培养基中的任一种。作为碳源,可以列举出葡萄糖、半乳糖、果糖、蔗糖、棉子糖、淀粉等碳水化合物、乙酸、丙酸等有机酸、乙醇、丙醇等醇类。作为氮源,可以列举出氨、氯化铵、硫酸铵、醋酸铵、磷酸铵等无机酸或有机酸的铵盐或其他含氮化合物。除此之外,也可以使用蛋白胨、肉提取物、玉米浆、各种氨基酸等。作为无机物,可以列举出磷酸二氢钾、磷酸氢二钾、磷酸镁、硫酸镁、氯化钠、硫酸亚铁、硫酸锰、硫酸铜、碳酸钙等。
72.培养通常在温度28~38℃、振荡培养或通气搅拌培养等好氧的条件下进行。ph的调整通过使用无机或有机酸、碱溶液等来进行。
73.在上述培养条件下培养时,可以高收率地生产聚酮。
74.培养后,当在菌体内或细胞内生产聚酮的情况下,可以通过实施均质器处理等来使菌体或细胞破碎,从而提取该表达产物。另一方面,当将聚酮输送到菌体外或细胞外的情况下,直接使用培养液、或通过离心分离等去除菌体或细胞。然后,通过硫酸铵沉淀提取等从所述培养物中提取该表达产物,根据需要进一步使用各种色谱等进行分离纯化。
75.实施例
76.下面,根据实施例对本发明进行说明,但本发明并不限定于这些实施例。
77.实施例1和比较例1
78.《dna片段的设计》
79.作为靶序列,设计了3个debs pks基因排列的序列(图1)。3个debs pks基因编码序列从saccharopolyspora erythraea的红霉素基因簇中获得。对于这3个debs pks基因编码序列,为了在宿主(大肠杆菌)中表达,制备了密码子优化后的dna。
80.密码子优化前的gc含量(%):
81.eryai:74.2%
82.eryaii:74.0%
83.eryaiii:73.9%
84.密码子优化后的gc含量(%):
85.eryai(序列号1):61.7%
86.eryaii(序列号2):61.5%
87.eryaiii(序列号3):61.1%
88.密码子优化后的3个debs pks基因编码序列如序列号1~3所示。将用于在宿主(大肠杆菌)中表达的控制序列(启动子和rbs序列)追加在各cds之前。将终止子(t)和间隔序列追加在各cds之后(图1)。通过修改同义密码子,删除了bsai和aari的限制位点。
89.将靶序列分割为28个dna片段(各~1.1kb),在各片段的两侧邻接bsai限制位点。28个dna片段如序列号4~31所示。
90.为了确认正确的组装,用genious软件模拟了1pot golden gate反应。
91.《dna组装》
92.序列号4~31所示的dna片段由twist biosciences订购,并作为标准克隆载体的克隆dna提供。
93.《《大肠杆菌中的golden gate组装(比较例1)》》:
94.在最适合用于大规模组装的反应条件下的1pot golden gate反应中,连接使用28个dna片段。1pot golden gate实验方法参照acs synth.biol.2018、7、11、2665-2674。
95.具体按照以下顺序来实施。
96.1)用uv分光光度计(thermofisher nanodrop)测量所有的dna片段的浓度,并调整为制备等摩尔的dna片段混合物。以1:1和2:1的插入片段:载体的比率添加目标载体(pet-24 )。
97.2)使用反应容量为25μl的1.5μl的bsai-hfv2限制性核酸内切酶和0.5μl的连接酶进行组装。
98.3)长dna组装的热循环方案使用了以下程序。
99.(37℃下5分钟
→
16℃下5分钟)
×
30循环,接着60℃下5分钟
100.4)将2μl的组装反应混合物转化为neb 10-beta电感受态e.coli细胞。
101.5)从得到的菌落中提取质粒以进行限制模式的验证。
102.在以下条件下验证了在提取的质粒中是否正确地制备了靶序列。结果如图2所示。
103.《debs pks assembly with golded gate and e.coli的实验条件》
104.分取得到的质粒100-500ng,并与2μl的10
×
cutsmart缓冲液、0.5μl的bamhi(10单位)以及无核酸酶水(最大20μl)进行混合,在37℃下孵育1小时,将靶序列分解为图2所示的片段。
105.将含1μg/ml的溴化乙锭(sigma)的100ml的1
×
tae缓冲液在120v下进行30分钟电泳,并将凝胶染色20分钟。用长波长紫外线(366mm)照射凝胶,并使分解的dna片段可视化。
106.《《枯草芽孢杆菌中的组装(实施例1)》》
107.1)将所有的dna片段(序列号4~31)在大肠杆菌中扩增,并进行再提取。
108.2)用uv分光光度计(thermofisher nanodrop)测量所有的dna片段(序列号4~31)的浓度,并调整为制备等摩尔的片段混合物。
109.3)利用dnase(lucigen corporation)进行处理,并去除混入的线状dna。
110.4)相对于各dna片段,浓度标准化为100ng/μl。
111.5)将500ng的各dna片段集中放于管中,用bsai-hfv2限制性核酸内切酶(neb)进行处理。
112.6)为了纯化用限制性核酸内切酶消化的dna,进行苯酚氯仿处理、丁醇处理、乙醇沉淀。
113.7)使用透析管进行凝胶提取,从消化的质粒混合物中去除靶片段,并将得到的消化后的靶片段用乙醇沉淀法进行纯化。
114.8)将消化后的片段与消化的载体(pet-24载体)、1μl的t4 dna ligase(takara bio)以及连接酶缓冲液混合,在37℃下孵育3小时后,完成连接,并得到包含编码debs pks的dna串联重复序列在内的dna构建体。
115.9)将包含所述dna构建体的dna连接酶溶液与枯草芽孢杆菌感受态细胞混合,将细胞在37℃下进行短时间的孵育后铺展在四环素选择板上。
116.10)菌落增殖一段时间后,从板上拾起转化子,并在37℃下于2ml lb中增殖一晚。
117.11)质粒提取按照以往的方案进行,在与《debs pks assembly with golded gate and e.coli的实验条件》相同的条件下验证是否按照预想的那样对得到的dna进行了组装(图3)。
118.《结果》
119.大肠杆菌中的golden gate法
120.如果是用pet-24载体组装的debs,则用bamhi进行处理时,可以看到图2左侧所示的图案,但即使反复尝试用大肠杆菌进行组装,在一系列的反应条件和浓度下也不会成功(图2)。
121.组装
122.如果是用pet-24载体组装的debs,则用bamhi进行处理时,可以看到图3右侧所示的图案。绿色的复选标记表示这样的模式,在枯草芽孢杆菌中的debs pks簇的组装成功,并
以高效率进行了证实(图3)。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
