1.本发明涉及高分子材料领域,进一步地说,是涉及一种生物基聚酯弹性体、制备方法及形变诱导结晶的聚酯弹性体。
背景技术:
2.所谓生物基材料是利用可再生生物质为原料,包括农作物及其废弃物,如秸秆等,通过生物、化学以及物理等方法制造的新材料,具有绿色环保、节能减排、原料可再生等优势,有的品类还具有良好的生物降解特性。原油储量正逐年减少。以石油为基础的相关产业和产品的发展受到极大限制的同时,不可降解的石油基塑料也严重的破坏着我们赖以生存的自然环境。因此,在全球石油资源供给日趋紧张,环保问题日益突出,对低碳经济发展需求日益强烈的严峻形势下,可再生资源为基础的生物基材料迅速发展成为必然趋势。生物基材料具有传统高分子材料不具备的绿色、环境友好、原料可再生以及可生物降解的特性。其制品既包括应用范围广泛的生产、生活用品,如包装材料、一次性日用品等,也包括技术含量高、附加值高的药物控制释放材料和骨固定材料及人体组织修复材料等生物医用材料。随着产业化进程的不断深入,生物基材料在人类能源、环境、社会发展、医药保健等方面发挥举足轻重的作用,成为各国研究和推广的热点。
3.生物降解高分子材料的降解影响因素:外部的环境与内部的材料结构均会很大程度上影响生物降解材料的降解速率以及降解程度。材料的结构是高分子材料降解性能的决定性因素。由于微生物的增值,以及酶催化水解都需要在水介质中进行,材料的亲水性能对其降解性能的影响极为重要。一般来说,含有亲水基团的高分子材料能使水分子更好地浸润材料表面,只有在材料保持湿润的情况下才能使微生物黏附于材料表面,并良好增殖。相比于芳香族聚合物,脂肪族聚合物具有更强的分子链运动能力,因而更易降解。高分子材料晶区与非晶区具有不同的结晶速率。通常水分子很难渗透分子链紧密堆砌晶区,使酶催化水解的速度降低,而高分子晶粒的大小、形态及数量都会影响材料的降解速率。
4.橡胶在常温下是无定形的高弹态物质,当受到拉伸时会使大分子链沿应力方向取向形成结品;晶粒分散在无定形大分子中起到补强作用。具有自补强性的橡胶如天然橡胶、丁腈橡胶,在较低的温度下或较低的应变条件下可以产生结晶。自补强性橡胶拉伸强度、撕裂强度等机械强度高。对于天然橡胶来说,橡胶的高效增强在很大程度上归结于分子链在拉伸时的高度取向或由此而引发的诱导结晶,自补强性很大;平均分子量很大,因而赋予其较高的机械强度。同时,目前很多研究都是基于共混体系中加入填料或者聚酰胺体系来达到形变诱导结晶的目的,如聚酰胺体系,有代表性的是王莉莉(王莉莉,朱平,董侠,等.长碳链聚酰胺及其共聚物的拉伸诱导结晶[j].高分子学报,2020,51(1):12)合成的结晶性热塑性多嵌段共聚物,软硬段的比例决定材料的塑性和弹性性能,软段含量高的体系为弹性体.在弹性体体系中,软段拉伸诱导结晶的形成能够提高样品的模量,从而使应力快速增大,但是聚酰胺体系只在100℃附近有较好的应变诱导结晶的现象,限制了其应用。
[0005]
近些年生物基聚酯弹性体也得到了很大的发展。中国发明专利cn113136027a公开了一种丁烯二醇基聚酯弹性体的制备方法。按照该方法加入二元醇和二元酸制得出来的生物基聚酯弹性体本发明提供了一种丁烯二醇基聚酯弹性体及制备方法。但该发明合成的是一系列高分子量窄分布的丁烯二醇基聚酯弹性体,其侧重点在于弹性体的可降解性,并没有从分子结构上解决聚酯弹性体的形变诱导结晶性能。
[0006]
聚合物中用长碳链脂肪族单体可能会得到一种形变诱导结晶的生物基聚酯弹性体,但目前长碳链二元醇,由于其沸点较高难以挥发,因而不利于反应平衡向生成产物的方向移动,难以生成高分子量聚酯;且长碳链单体在共聚物中,亚甲基堆叠,因此在合成聚酯弹性体时,可能会合成结晶度很高的产物。
技术实现要素:
[0007]
为了解决现有技术中存在的技术问题,本发明提供了一种生物基聚酯弹性体、制备方法及形变诱导结晶的聚酯弹性体。
[0008]
本发明开发一种长碳链脂肪族单体合成聚酯弹性体的合成方法,使用长碳链脂肪族单体,属于生物基单体,来源广泛,合成的聚酯弹性体,长碳链的加入,降低酯基密度,提高共聚酯的降解稳定性,具有类聚乙烯的结构通过引入长碳链单体,以期获得类聚乙烯性能的聚酯弹性体,多元共聚破坏共聚酯本身的结晶,多个亚甲基堆叠又会形成一部分尺寸较小的微晶,但这部分结晶是无法表现出来的,只有在应力作用下,其共聚物达到应变诱导结晶现象,拓宽了生物基聚酯弹性体单体的范围实现常温下的长碳链脂肪族聚酯的应变诱导结晶。同时,与其它应力增强不同的是,其过程是可回复的,应力作用下,分子链进行规整的排列,应力消失后,分子链又会自动蜷曲回复。应力并没有破坏分子链的结构,只是改变了内部的排列方式。
[0009]
本发明所用的短链二元醇在缩聚条件下是挥发性的,在终缩聚阶段可以通过脱除二元醇提高分子量可以保证生成高分子量,通过长碳链二元酸将长碳链的结构引入聚酯弹性体中,制得的聚酯弹性体可以作为形变诱导结晶制品使用,本发明合成路线简单,制备方法简单,反应装置要求不高,较为合适目前的传统的生产线进行生产,单体来源广泛。
[0010]
本发明的目的之一是提供一种生物基聚酯弹性体。
[0011]
所述生物基聚酯弹性体是一种无规聚合物,其结构式为:
[0012][0013]
以结构式中各聚合单元的总摩尔数为1,
[0014]
a,b,c,d,e分别为0~0.5摩尔分数;a,b,c,d,e不同时为0;优选地,c,d,e为0,a,b不0,a的摩尔分数为0.01~0.45,优选为0.1~0.4,b的摩尔分数为0.05~0.5,优选为0.1~0.4;
[0015]
u,v,w,x分别为0~0.5摩尔分数;u,v,w,x不同时为0;优选地,x不为0,u、v、w中有1
个不为0,x的摩尔分数为0.01~0.2,优选为0.04~0.15,u v w的摩尔分数为0.2~0.48,优选为0.3~0.45;
[0016]
m,n分别为0~0.1摩尔分数,m,n不同时为0;优选地,m,n中有1个为0,m,n中之一的摩尔分数为0.05~0.1;
[0017]
10≤rn≤18,优选地,12≤rn≤18;重复的rn可以相同或不同,rn不同时,x为rn的总摩尔分数之和。
[0018]
本发明的一种优选的实施方式中,
[0019]
所述生物基聚酯弹性体由包括二元酸、二元醇、阻聚剂、抗氧剂、催化剂在内的原料制备而得;优选地,
[0020]
所述二元酸为1,4-丁二酸、1,6-己二酸、1,10-癸二酸、衣康酸中的一种或两种和hooc-rn-cooh中的至少一种,其中:10≤rn≤18,在二元醇中没有丁烯二醇时,二元酸必须包括衣康酸;更优选地,12≤rn≤18;所述hooc-rn-cooh占二元酸的摩尔分数不低于8%,优选为8~35%;
[0021]
所述二元醇为丁烯二醇、乙二醇、1,3-丙二醇,1,4-丁二醇,1,5-戊二醇,1,6-己二醇中的两种或三种,其中:在二元酸中没有衣康酸时,所述二元醇必须包括丁烯二醇。
[0022]
本发明的一种优选的实施方式中,
[0023]
所述阻聚剂可采用现有技术中常规的阻聚剂,优选为酚类阻聚剂、醚类阻聚剂、醌类阻聚剂或芳胺类阻聚剂;更优选为对苯二酚、对叔丁基邻苯二酚、对羟基苯甲醚、苯醌、二苯胺、对苯二胺中的至少一种;进一步优选为一种或两种;
[0024]
所述抗氧剂可采用现有技术中常规的抗氧剂,抗氧剂优选为磷酸或亚磷酸化合物;更优选为磷酸、亚磷酸、磷酸酯、亚磷酸酯、磷酸苯酯、亚磷酸苯酯中的至少一种;进一步优选为一种或两种;
[0025]
所述催化剂为钛系催化剂及其溶液、锑系催化剂及其溶液、锗系催化剂及其溶液、有机锡类催化剂的至少一种;优选为钛酸四丁酯及其溶液、二丁基锡二月桂酸酯、辛酸亚锡、二醋酸二丁基锡中的至少一种。
[0026]
本发明的一种优选的实施方式中,
[0027]
二元醇与二元酸的摩尔比为(1.02~1.8):1;优选为(1.05~1.3):1;
[0028]
在二元醇中没有丁烯二醇时,衣康酸占二元酸的摩尔百分数3~30%;优选为5~20%,更优选为10~20%;
[0029]
在二元酸中没有衣康酸时,1,4-丁烯二醇占二元醇的摩尔百分数3~30%;优选为5~20%,更优选为10~20%;,
[0030]
阻聚剂的质量为反应加入的物料总质量的0.01~0.2%;优选为0.01~0.08%;
[0031]
抗氧剂的质量为反应加入的物料总质量的0.01~0.2%;优选为0.01~0.08%;
[0032]
催化剂的质量为反应加入的物料总质量的0.05%~1%;优选为0.2~0.5%。
[0033]
本发明的目的之二是提供一种生物基聚酯弹性体的制备方法,包括以下步骤:
[0034]
(1)将二元酸、二元醇、阻聚剂、抗氧剂按照所述用量混合,进行酯化反应;
[0035]
(2)加入催化剂,经预缩聚、终缩聚,缩聚反应完成前通入保护性气体保护,得到所述生物基聚酯弹性体。
[0036]
本发明的一种优选的实施方式中,
[0037]
步骤(1),
[0038]
酯化反应结束前1~1.5小时通入保护性气体;所述保护性气体优选为氮气;如过早通入氮气,流动的气体会在高温时带走还没来得及反应的小分子的二元醇,因此只在反应结束前1~1.5小时通入。
[0039]
酯化反应在带有机械搅拌的反应装置中进行;搅拌速度为180~250r/min;
[0040]
酯化反应温度为170~200℃;
[0041]
酯化反应时间为5~7h;
[0042]
反应至体系澄清透亮时结束,在实验室中,此时尾接瓶出水量达到理论出水量的90~95%,其中理论出水量=二元酸的摩尔数*2*18。
[0043]
本发明的一种优选的实施方式中,
[0044]
步骤(2),
[0045]
预缩聚温度为200~220℃;预缩聚压力为10~15kpa;预缩聚时间为1~2h;在预缩聚阶段关闭氮气,这个阶段的主要目的是为了去除过量加入的二醇单体;
[0046]
终缩聚温度为220~240℃;终缩聚压力为20~50pa;终缩聚时间为8~20h;缩聚反应后脱除体系中的小分子和少部分水,然后冷却至室温,制得所述生物基聚酯弹性体。
[0047]
本发明的目的之三是提供一种上述方法制得的生物基聚酯弹性体。
[0048]
本发明的目的之四是提供一种可以形变诱导结晶的聚酯弹性体。
[0049]
所述可以形变诱导结晶的聚酯弹性体用过氧化物类交联剂硫化后制备而得。
[0050]
本发明的一种优选的实施方式中,
[0051]
所述过氧化物类交联剂为橡胶工业常用的过氧化物类交联剂,优选为过氧化二异丙苯、过氧化二苯甲酰、双叔丁基过氧化异丙苯中的至少一种;更优选为其中的一种或两种;和/或,
[0052]
所述过氧化物类交联剂的质量为所述生物基聚酯弹性体总质量的0.1~3%;优选为0.1~1%。
[0053]
现有技术中存在的一些问题:
[0054]
(1)现有聚酯高分子由于分子链规整性,很容易结晶,主要为塑料;通过多元共聚破坏结晶后,虽然可以得到无定形的弹性体材料,但是需要加入补强填料来提高强度,无法直接使用。而补强填料多为不可降解的碳黑和白炭黑,不利于聚酯弹性体制品的生物降解性。
[0055]
(2)若在聚酯弹性体结构中引入长碳链单体,长碳链单体本身会发生结晶,无法通过多元共聚来破坏结晶,很容易合成得到长碳链的聚酯塑料。
[0056]
与现有技术相比,本发明的有益效果:
[0057]
(1)本发明引入了含有长碳链的二元酸单体,制备得到了具有可拉伸结晶性能的聚酯弹性体,不需要引入补强填料,可通过在拉伸过程中产生的结晶来获得高的拉伸强度。
[0058]
(2)本发明对长碳链单体的含量进行控制,使聚酯弹性体既可以具有拉伸结晶性能,又不会因为其用量过大而导致结晶程度太高,变成塑料。
[0059]
(3)本发明首次提出了可拉伸结晶的聚酯弹性体,拓宽了聚酯弹性体的应用。
附图说明
[0060]
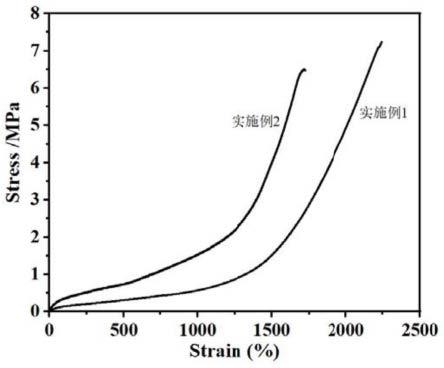
图1为实施例1~2制得的聚酯弹性体的应力-应变曲线图;
[0061]
图2为对比例1制得的聚酯弹性体的应力-应变曲线图;
[0062]
图3为实施例1、对比例1制得的聚酯弹性体的傅里叶红外光谱图;
[0063]
图4为实施例1、对比例1制得的聚酯弹性体的dsc图;
[0064]
图5为实施例2制得的聚酯弹性体的核磁谱图。
具体实施方式
[0065]
下面结合具体附图及实施例对本发明进行具体的描述,有必要在此指出的是以下实施例只用于对本发明的进一步说明,不能理解为对本发明保护范围的限制,本领域技术人员根据本发明内容对本发明做出的一些非本质的改进和调整仍属本发明的保护范围。
[0066]
实施例中所用原料均为常规市购原料。
[0067]
dsc测试:dsc测试(常规,现有技术通用):于氮气气氛下,将样品以10℃/min速率从25℃升温至150℃,并保持5min;随后将样品以10℃/min速率由150℃降温至-150℃,并保持10min;再以10℃/min速率从-150℃升温至150℃。从第二段升温曲线中,读取所得样品的tg、tm值,实验测试结果见表1。
[0068]
gpc测试(常规,现有技术通用):以聚苯乙烯作为标定物,四氢呋喃为流动相,测定所得样品的相对分子质量与分布,实验测试结果见表1。
[0069]
拉伸测试:制作中间部分长为20mm,宽为1mm的哑铃型样条,设置量程为5000n,拉伸速率为50mm/min。
[0070]
实施例1
[0071]
将乙二醇(0.327mol)、1,3-丙二醇(0.164mol)、1,4-丁烯二醇(0.055mol)、1,4-丁二酸(0.409mol)、1,15-十五烷二酸(0.045mol)、亚磷酸(总质量的0.01%),对苯二酚(总质量的0.04%)加入到100ml带有机械搅拌的四颈烧瓶中,先将体系温度升至180℃,置换空气后常压反应5h,通入氮气反应1h,至澄清透明,用时约6h。酯化过后,升温到220℃,在氮气氛围加入催化剂钛酸四丁酯(总质量的0.2%),减压至10kpa将反应多余的二醇抽出,这个过程持续约1h。随后保持温度,减压至20pa,反应12h左右,有明显的爬杆现象,反应结束,温度降80℃,取出,得到生物基聚酯弹性体。
[0072]
在室温下,二氯甲烷溶解得到生物基聚酯弹性体20g,加入0.02g的双叔丁基过氧化异丙苯,充分搅拌溶解后,室温蒸发溶剂12h,60℃恒温烘箱静置6h后进行硫化测试。制备的聚酯弹性体结构如下:
[0073][0074]
其中,a=0.327,b=0.164,m=0.055,u=0.409,x=0.045。
[0075]
实施例2
[0076]
将乙二醇(0.165mol)、1,3-丙二醇(0.33mol)、1,4-丁烯二醇(0.055mol)、1,4-丁二酸(0.315mol)、1,12-十二烷二酸(0.135mol)、亚磷酸(总质量的0.01%),对苯二酚(总质
量的0.04%)加入到100ml带有机械搅拌的四颈烧瓶中,先将体系温度升至180℃,置换空气后常压反应5h,通入氮气反应1h,至澄清透明,用时约6h。酯化过后,升温到220℃,在氮气氛围加入催化剂钛酸四丁酯(总质量的0.2%),减压至10kpa将反应多余的二醇抽出,这个过程持续约1h。随后保持温度,减压至20pa,反应12h左右,有明显的爬杆现象,反应结束,温度降80℃,取出,得到生物基聚酯弹性体。
[0077]
在室温下,二氯甲烷溶解得到生物基聚酯弹性体20g,加入0.02g的双叔丁基过氧化异丙苯,充分搅拌溶解后,室温蒸发溶剂12h,60℃恒温烘箱静置6h后进行硫化测试。制备的聚酯弹性体结构如下:
[0078][0079]
其中,a=0.165,b=0.33,m=0.055,u=0.315,x=0.135。
[0080]
实施例3
[0081]
将乙二醇(0.165mol)、1,3-丙二醇(0.33mol)、1,4-丁烯二醇(0.055mol)、1,4-丁二酸(0.405mol)、1,15-十五烷二酸(0.045mol)、亚磷酸(总质量的0.02%),对苯二酚(总质量的0.04%)加入到100ml带有机械搅拌的四颈烧瓶中,先将体系温度升至200℃,置换空气后常压反应4h,通入氮气反应1h,至澄清透明,用时约5h。酯化过后,升温到220℃,在氮气氛围加入催化剂辛酸亚锡(总质量的0.5%),减压至10kpa将反应多余的二醇抽出,这个过程持续约2h。随后保持温度,减压至40pa,反应16h左右,有明显的爬杆现象,反应结束,温度降80℃,取出,得到生物基聚酯弹性体。
[0082]
在室温下,二氯甲烷溶解得到的生物基聚酯弹性体20g,加入0.15g的双叔丁基过氧化异丙苯,充分搅拌溶解后,室温蒸发溶剂12h,60℃恒温烘箱静置6h后进行硫化测试。制备的聚酯弹性体结构如下:
[0083][0084]
其中,a=0.165,b=0.33,m=0.055,u=0.405,x=0.045。
[0085]
实施例4
[0086]
将乙二醇(0.275mol)、1,3-丙二醇(0.275mol)、1,4-丁二酸(0.36mol)、1,18-十八烷二酸(0.045mol)、衣康酸(0.045mol)亚磷酸(总质量的0.08%),对苯二酚(总质量的0.06%)加入到100ml带有机械搅拌的四颈烧瓶中,先将体系温度升至180℃,置换空气后常压反应5h,通入氮气反应1h,至澄清透明,用时约6h。酯化过后,升温到220℃,在氮气氛围加入催化剂钛酸四丁酯(总质量的0.2%),减压至20kpa将反应多余的二醇抽出,这个过程持续约1h。随后保持温度,减压至50pa,反应18h左右,有明显的爬杆现象,反应结束,温度降80℃,取出,得到生物基聚酯弹性体。
[0087]
在室温下,二氯甲烷溶解得到的生物基聚酯弹性体20g,加入0.1g的双叔丁基过氧化异丙苯,充分搅拌溶解后,室温蒸发溶剂12h,60℃恒温烘箱静置6h后进行硫化测试。制备
的聚酯弹性体结构如下:
[0088][0089]
其中,a=0.275,b=0.275,u=0.36,x=0.045,n=0.045。
[0090]
实施例5
[0091]
将乙二醇(0.15mol)、1,3-丙二醇(0.298mol)、1,4-丁烯二醇(0.087mol)、1,4-丁二酸(0.323mol)、1,12-十二烷二酸(0.071mol)、1,18-十八烷二酸(0.071mol)、亚磷酸(总质量的0.02%),对苯二酚(总质量的0.01%)加入到100ml带有机械搅拌的四颈烧瓶中,先将体系温度升至180℃,置换空气后常压反应6h,通入氮气反应1h,至澄清透明,用时约7h。酯化过后,升温到200℃,在氮气氛围加入催化剂钛酸四丁酯(总质量的0.4%),减压至15kpa将反应多余的二醇抽出,这个过程持续约1h。随后升高温度至240℃,减压至50pa下,反应16h左右,有明显的爬杆现象,反应结束,温度降80℃,取出,得到生物基聚酯弹性体。
[0092]
在室温下,二氯甲烷溶解得到的生物基聚酯弹性体20g,加入0.06g的双叔丁基过氧化异丙苯,充分搅拌溶解后,室温蒸发溶剂12h,60℃恒温烘箱静置6h后进行硫化测试。制备的聚酯弹性体结构如下:
[0093][0094]
其中,a=0.15,b=0.298,u=0.323,x1=0.071,x2=0.071,m=0.087。
[0095]
实施例6
[0096]
与实施例4的区别为:乙二醇(0.346mol)、1,3-丙二醇(0.173mol)、1,4-丁二酸(0.337mol)、1,18-十八烷二酸(0.066mol)、衣康酸(0.079mol);
[0097]
其他均与实施例4相同,得到生物基聚酯弹性体。
[0098]
在室温下,二氯甲烷溶解得到生物基聚酯弹性体20g,加入0.02g的双叔丁基过氧化异丙苯,充分搅拌溶解后,室温蒸发溶剂12h,60℃恒温烘箱静置6h后进行硫化测试。制备的聚酯弹性体结构如下:
[0099][0100]
其中,a=0.346,b=0.173,u=0.337,x=0.066,n=0.079。
[0101]
对比例1
[0102]
与实施例1的区别为:1,15-十五烷二酸(0.045mol)替换为1,10-癸二酸(0.045mol);
[0103]
其他均与实施例1相同,反应后得到生物基聚酯弹性体。
[0104]
在室温下,二氯甲烷溶解样品20g,加入0.02g的双叔丁基过氧化异丙苯,充分搅拌溶解后,室温蒸发溶剂12h,60℃恒温烘箱静置6h后进行硫化测试。制备的聚酯弹性体结构如下:
[0105][0106]
其中,a=0.327,b=0.164,m=0.055,u=0.409,x=0.045。
[0107]
表1
[0108] tg(℃)tm(℃)mnmwpdi实施例1-41.52-5.655.5810.591.90实施例2-40.32-9.285.329.351.88实施例3-45.373.905.6510.901.93实施例4-47.347.354.637.791.68实施例5-34.966.375.228.321.59实施例6-10.2330.255.359.271.78对比例1-46.13-4.7611.462.41
[0109]
表1为实施例1~6、对比例1的tg、tm、mn、mw、pdi测试结果,从表1中数据可以看出,与对比例1相比,实施例1制备的聚酯弹性体具有更高的相对分子质量和更窄的相对分子质量分布,体系具有“高分子量、窄分布”的优势,其主要原因在于:饱和体系比较稳定,因此在高温熔融缩聚过程中,不容易发生副反应,进而能保证较窄的相对分子质量分布;实施例1~6制得的聚酯弹性体均具有更高的相对分子质量和更窄的相对分子质量分布。
[0110]
图1、图2分别为实施例1~2和对比例1制得的聚酯弹性体的应力-应变曲线图,对比例1的断裂伸长率只有180%;
[0111]
从图1~2中可以看出,与对比例1相比,实施例1~2达到应变1000%前是典型的弹性体应力应变曲线,应变达到1000%后,应力有明显的上升趋势,证明得到的是一种可以形变诱导结晶的聚酯弹性体。
[0112]
图3为实施例1和对比例1制得的聚酯弹性体的傅里叶红外光谱图,1725cm-1
为(c=o)碳基伸缩振动峰,2975cm-1
为(-ch
2-)的伸缩振动峰,证实合成了实施例1结构式的聚酯弹性体。
[0113]
图4为实施例1、对比例1制得的聚酯弹性体的dsc图,从图上可以看出,实施例1所得聚酯材料有一个低于室温的玻璃化转变温度,结晶温度低于室温,因此室温下是一种弹性体材料;与对比例1相比,实施例1所制备的聚酯弹性体有长碳链单体的加入,有尺寸较小的微晶产生,属于碳链堆叠的微晶,经拉伸后结晶更加完善。
[0114]
图5为实施例2制得的聚酯弹性体的核磁图,确证反应合成了预想的生物基聚酯弹性体。
[0115]
实施例1~6制备的聚酯弹性体,具有可拉伸结晶性能,不需要引入补强填料,可通过在拉伸过程中产生的结晶来获得高的拉伸强度,拓宽了聚酯弹性体的应用,且合成路线简单,制备方法简单,反应装置要求不高,较为合适目前的传统的生产线进行生产。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。