1.本发明属于囊泡制备技术领域,尤其涉及一种单一单链烷基酮酸囊泡的制备方法。
背景技术:
2.囊泡是由两亲分子的闭合双分子层包裹微水相而形成的有序聚集体,其中亲水性的极性头分别朝向囊泡内部和外部,与水相接触;疏水性的双分子层形成疏水微区,远离水相。由于囊泡独特的结构特点,其实际应用价值很高,在生物细胞膜模拟、药物或生物活性物质的传输控释、微反应器或材料合成等众多领域具有广阔的应用前景。
3.囊泡的制备方法一般可分为机械力制备法(超声、挤压等外力作用)和自发形成法(无需外力)。相比而言,自发形成法操作简单,所形成囊泡稳定性好,因此是近年来囊泡领域的研究热点。囊泡的自发形成可通过使用两种或两种以上表面活性剂复配、双尾链表面活性剂来实现,或通过改变环境条件、引入添加剂等。在无任何助剂的条件下,单一单链表面活性剂体系囊泡在较宽条件下的自发形成,仍然是巨大挑战。
4.脂肪酸是烷基羧酸类物质的统称,其分子结构简单,其皂盐是一种物美价廉的阴离子表面活性剂。在水溶液中,脂肪酸分子能够以质子化形式(酸型)和去质子化形式(皂型)存在,两者在合适条件下通过极性头基间的氢键作用形成“酸-皂”二聚体随后自组装形成囊泡聚集体。然而,传统脂肪酸溶液的临界囊泡浓度较高(如辛酸钠溶液cvc约为120-200mm),脂肪酸囊泡的形成ph范围(pka附近,~7-9)较窄,对二价阳离子其敏感(在毫摩尔级别的mg
2
环境中沉淀),限制了其更广阔的应用。
技术实现要素:
5.有鉴于此,本发明的目的在于提供一种单一单链烷基酮酸囊泡的制备方法,采用本发明的制备方法得到的单一单链烷基酮酸囊泡可在极稀溶液中自发形成,且制备简单,在较宽ph范围和较高盐浓度环境中保持稳定,在生物细胞膜模拟、药物或生物活性物质的传输控释、微反应器或材料合成等众多领域具潜在应用。
6.为了实现上述发明目的,本发明提供了以下技术方案:
7.本发明提供了一种单一单链烷基酮酸囊泡的制备方法,包括以下步骤:
8.1)将草酸酯溶液与烷基卤化镁溶液混合,在无氧环境、-78℃反应1~3h,得到烷基酮酸;
9.所述草酸酯溶液的质量浓度为0.1~0.2g/ml;
10.所述烷基卤化镁溶液的摩尔浓度为1~2mol;
11.2)将所述步骤1)得到的烷基酮酸溶解于水中,得到单一单链烷基酮酸囊泡。
12.优选的,所述步骤1)草酸酯溶液与烷基卤化镁溶液的体积比为50~60:20.4~54.5。
13.优选的,所述步骤1)草酸酯溶液中草酸酯包括草酸二乙酯、草酸二甲酯、草酰氯单
乙酯和草酰氯单甲酯中的一种或多种。
14.优选的,所述步骤1)烷基卤化镁溶液中烷基卤化镁包括己基溴化镁、己基氯化镁、辛基溴化镁、辛基氯化镁和癸基溴化镁中的一种或多种。
15.优选的,所述草酸酯溶液的溶剂为无水溶剂,所述无水溶剂包括无水四氢呋喃、无水乙醚和无水2-甲基四氢呋喃中的一种或多种。
16.优选的,所述步骤2)烷基酮酸的摩尔与水的体积比为0.1~500mmol:1l。
17.优选的,当所述烷基酮酸为α-酮辛酸时,所述草酸酯为草酸二乙酯,所述烷基卤化镁为己基溴化镁。
18.优选的,当所述烷基酮酸为α-酮癸酸时,所述草酸酯为草酸二甲酯,所述烷基卤化镁为辛基溴化镁。
19.优选的,当所述烷基酮酸为α-酮月桂酸时,所述草酸酯为草酸二乙酯,所述烷基卤化镁为癸基溴化镁。
20.优选的,所述步骤1)在无氧环境、-78℃反应1-3h后还包括,经盐酸溶液终止反应,使用有机溶剂进行萃取,得到的有机相经饱和氯化钠洗涤、无水硫酸镁干燥,旋蒸去除溶剂,将得到的产物分散于酸性水溶液、搅拌,去除溶剂后,依次经有机相萃取、水洗、饱和氯化钠洗、无水硫酸镁干燥、去除溶剂和重结晶,得到烷基酮酸。
21.本发明采用目视法和显微观察法等划分相区,绘制烷基酮酸/水相二元体系的相图,根据相图,选择烷基酮酸分子和水的比例,以制备囊泡溶液。
22.将本发明制备的烷基酮酸分子进行核磁共振氢谱(1hnmr)表征,结果显示此合成方法可成功制备得到烷基酮酸,制备方法简单。将本发明制备的烷基酮酸囊泡样品进行冷冻透射电子显微镜、负染色透射电子显微镜和相差显微镜表征,结果显示本发明的囊泡可在极稀溶液中自发形成(≥0.4mmol/l),可在较宽ph范围内保持稳定(2-10),可耐受较高mg
2
浓度(毫摩尔级别),这可归因于分子中酮羰基的作用。一方面,烷基酮酸是一种特殊脂肪酸,在pka(~2.7)附近质子化烷基酮酸和去质子化烷基酮酸通过羧酸极性头基间的氢键作用形成“酸-皂”二聚体,类似于双尾链表面活性剂,进一步自组装形成囊泡;另一方面,酮羰基之间可通过氢键形成“水桥结构”及与mg
2
较强结合作用增加氢键复合物在较宽ph范围和mg
2
环境中的稳定性,也是囊泡高稳定性的主要因素。
23.相比传统的复配表面活性剂、双尾链表面活性剂和传统的脂肪酸囊泡体系等,本发明的囊泡是由单一单链的烷基酮酸在水溶液中自发形成,因烷基酮酸独特的分子结构,其囊泡具有较低的临界形成浓度,较好的稳定性,且制备方法简单、成本低,可应用于生物细胞膜模拟、药物或生物活性物质的传输控释、微反应器或材料合成等众多领域。
附图说明
24.图1为实施例1制得的α-酮辛酸的核磁共振氢谱(1hnmr)图;
25.图2为实施例1制得的不同浓度α-酮辛酸囊泡的冷冻透射电子显微镜图。(a)c
α-酮辛酸
=2mm;(b)c
α-酮辛酸
=5mm;(c)c
α-酮辛酸
=85mm;(d)c
α-酮辛酸
=500mm;
26.图3为实施例1制得的α-酮辛酸囊泡在不同浓度氯化镁存在下的冷冻透射电子显微镜图:(a)c
mgcl2
=0mm;(b)c
mgcl2
=30mm;(c)c
mgcl2
=50mm;(d)c
mgcl2
=80mm;(e)α-酮辛酸囊泡在不同浓度氯化镁存在下的动态光散射粒径分布图;
27.图4为实施例1制得的α-酮辛酸囊泡在不同ph条件下的负染色透射电子显微镜图:(a)ph 2.0;(b)ph 5.1;(c)ph 7.2;(d)ph 10.5;
28.图5为实施例2制得的α-酮辛酸的核磁共振氢谱(1hnmr)图;
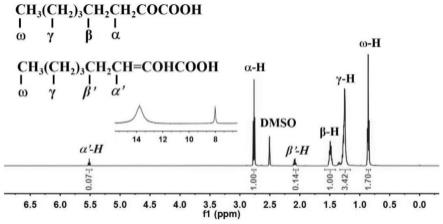
29.图6为实施例3制得的α-酮辛酸的核磁共振氢谱(1hnmr)图;
30.图7为实施例4制得的α-酮癸酸的核磁共振氢谱(1hnmr)图;
31.图8为实施例4制得的不同浓度α-酮癸酸囊泡的冷冻透射电子显微镜图。(a)c
α-酮癸酸
=0.4mm;(b)c
α-酮癸酸
=40mm;(c)c
α-酮癸酸
=200mm;
32.图9为实施例4制得的α-酮癸酸囊泡在不同浓度氯化镁存在下的相差显微镜图:(a)c
mgcl2
=0mm;(b)c
mgcl2
=10mm;(c)c
mgcl2
=30mm;
33.图10为实施例4制得的α-酮癸酸囊泡在不同ph条件下的冷冻透射电子显微镜图:(a)ph 2.0;(b)ph 7.3;(c)ph 10.0;
34.图11为实施例5制得的α-酮癸酸的核磁共振氢谱(1hnmr)图;
35.图12为实施例6制得的α-酮癸酸的核磁共振氢谱(1hnmr)图;
36.图13为实施例7制得的α-酮月桂酸的核磁共振氢谱(1h nmr)图;
37.图14为实施例7制得的α-酮月桂酸溶液的差示扫描量热图;
38.图15为实施例7制得的不同浓度α-酮月桂酸囊泡的(a)冷冻透射电子显微镜图和(b,c)相差显微镜图。(a)c
α-酮月桂酸
=1mm;(b)c
α-酮月桂酸
=40mm;(c)c
α-酮月桂酸
=100mm;
39.图16为实施例7制得的α-酮月桂酸囊泡在不同浓度氯化镁存在下的相差显微镜图:(a)c
mgcl2
=0mm;(b)c
mgcl2
=6mm;(c)c
mgcl2
=9mm;
40.图17为实施例7制得的α-酮月桂酸囊泡在不同ph条件下的冷冻透射电子显微镜图:(a)ph 2.5;(b)ph 6.5;(c)ph 10.2。
具体实施方式
41.本发明提供了一种单一单链烷基酮酸囊泡的制备方法,包括以下步骤:
42.1)将草酸酯溶液与烷基卤化镁溶液混合,在无氧环境、-78℃反应1~3h,得到烷基酮酸;
43.所述草酸酯溶液的质量浓度为0.1~0.2g/ml;
44.所述烷基卤化镁溶液的摩尔浓度为1~2mol;
45.2)将所述步骤1)得到的烷基酮酸溶解于水中,得到单一单链烷基酮酸囊泡。
46.本发明将草酸酯溶液与烷基卤化镁溶液混合,在无氧环境、-78℃反应1~3h,得到烷基酮酸;所述草酸酯溶液的质量浓度为0.1~0.2g/ml;所述烷基卤化镁溶液的摩尔浓度为1~2mol。
47.在本发明中,所述草酸酯溶液与烷基卤化镁溶液的体积比优选为50~60:20.4~54.5。在本发明中,所述草酸酯溶液的制备方法优选包括:将草酸酯溶解于无水溶剂中,置于-78℃下抽气5~10min,然后通入氮气10-15min,如此循环3次。在本发明中,所述草酸酯溶液中草酸酯优选包括草酸二乙酯、草酸二甲酯、草酰氯单乙酯和草酰氯单甲酯中的一种或多种。在本发明中,所述草酸酯溶液的溶剂优选为无水溶剂,所述无水溶剂优选包括无水四氢呋喃、无水乙醚和无水2-甲基四氢呋喃中的一种或多种。在本发明中,所述草酸酯溶液与烷基卤化镁溶液混合优选在氮气氛围下,将烷基卤化镁溶液滴加到草酸酯溶液中。在本
发明中,所述烷基卤化镁溶液中烷基卤化镁优选包括己基溴化镁、己基氯化镁、辛基溴化镁、辛基氯化镁和癸基溴化镁中的一种或多种。
48.本发明在无氧环境、-78℃反应1-3h后还优选包括,经盐酸溶液终止反应,使用有机溶剂进行萃取,得到的有机相经饱和氯化钠洗涤、无水硫酸镁干燥,旋蒸去除溶剂,将得到的产物分散于酸性水溶液、搅拌,去除溶剂后,依次经有机相萃取、水洗、饱和氯化钠洗、无水硫酸镁干燥、去除溶剂和重结晶,得到烷基酮酸。在本发明中,所述盐酸溶液的浓度优选为2mol/l,将所述盐酸溶液逐滴滴加到反应体系中,以终止反应。在本发明中,所述有机溶剂优选包括乙醚、四氢呋喃、二氯甲烷、己烷和氯仿中的一种或多种。在本发明中,所述酸性水溶液优选包括醋酸溶液、盐酸溶液或硫酸溶液中的一种或多种。在本发明中,所述搅拌的时间优选为12~24h。本发明优选使用己烷进行重结晶,次数优选为3次,得到烷基酮酸。
49.在本发明中,当所述烷基酮酸优选为α-酮辛酸时,所述草酸酯优选为草酸二乙酯,所述烷基卤化镁优选为己基溴化镁。在本发明中,当所述烷基酮酸优选为α-酮癸酸时,所述草酸酯优选为草酸二甲酯,所述烷基卤化镁优选为辛基溴化镁。在本发明中,当所述烷基酮酸优选为α-酮月桂酸时,所述草酸酯优选为草酸二乙酯,所述烷基卤化镁优选为癸基溴化镁。
50.本发明将得到的烷基酮酸溶解于水中,得到单一单链烷基酮酸囊泡。在本发明中,所述烷基酮酸的摩尔与水的体积比优选为0.1~500mmol:1l。在本发明中,所述烷基酮酸在水中自发形成烷基酮酸囊泡。在本发明中,所述烷基酮酸囊泡可在极稀溶液中形成,在ph值为2~10下保持稳定,具有良好的抗mg
2
稳定性(毫摩尔级别),所述烷基酮酸囊泡的粒径为几十纳米到几个微米。
51.下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
52.实施例1
53.烷基酮酸选择为α-酮辛酸。
54.1)α-酮辛酸的制备:
55.将5.58g草酸二乙酯溶解于50ml无水四氢呋喃中,然后置于-78℃的干冰/丙酮浴中,抽气5min,然后通入氮气10min,如此循环3次。然后将20.4ml 2mol/l己基溴化镁的四氢呋喃溶液在n2氛围下逐滴滴加至上述草酸二乙酯溶液中,搅拌反应1.5h。反应结束后,向反应体系中逐滴滴加2mol/l盐酸水溶液终止反应。用二氯甲烷(3
×
120ml)将产物从水相萃取至有机相,有机相用饱和氯化钠洗涤(100ml),无水硫酸镁干燥,旋蒸除去溶剂。将得到的上述产物分散在醋酸(hac,55ml)和盐酸(37%hcl,15ml)中,搅拌12h,旋蒸去除掉大部分溶剂,得到浓缩液。向浓缩液中加入水(50ml)和二氯甲烷(100ml),再用二氯甲烷(3
×
100ml)将产物从水相萃取至有机相,有机相先后用水洗(100ml)、饱和氯化钠洗(100ml),无水硫酸镁干燥,旋蒸除去溶剂。最后,在冰己烷中重结晶3次,得到白色粉末(质量4.05g,收率67%)。
56.图1为制得白色粉末在氘代dmso中的核磁共振氢谱图。图中位移值为2.76ppm处的三重峰对应于α-酮辛酸分子中与亲水性酮基直接相连的亚甲基上的氢原子(α-h),位移值为1.49ppm处的多重峰对应于α-酮辛酸分子中与亲水性酮基相连的β-亚甲基上的氢原子,插图中位移值为13.77ppm的宽峰对应于羧酸中的活泼氢。另外,在5.52ppm处的三重峰和
2.08ppm处的四重峰分别对应于α-酮辛酸的烯醇式互变异构体中的α
’‑
h和β
’‑
h,在8.02ppm处的宽峰对应于烯醇式异构体中羟基氢原子。从核磁共振氢谱图可以看出,所得产物即为α-酮辛酸。
57.2)α-酮辛酸囊泡的制备:
58.称取上述方法制备得到的α-酮辛酸,在室温下搅拌至完全溶解,配置成2-500mm不同浓度的水溶液(自然ph值条件下,ph值介于1.4-2.9之间),即得到系列囊泡样品。对其中浓度为2mm、5mm、85mm和500mm的α-酮辛酸水溶液进行cryo-tem表征,结果表明α-酮辛酸水溶液在浓度~2-500mm范围内自发形成了具中空结构的囊泡,囊泡粒径几十纳米到数微米之间,如图2所示。
59.选择浓度为200mm的α-酮辛酸溶液,测定其耐盐性:首先称取2.033g氯化镁溶解于10ml三次水中,制得浓度为2.0m的氯化镁储备液。在快速涡旋状态下,将0μl、15μl、25μl和40μl的氯化镁储备液分别加入1mlα-酮辛酸溶液中(最终镁离子浓度分别为0mm、30mm、50mm和80mm),以考察其耐盐性。
60.图3为200mmα-酮辛酸囊泡溶液在不同浓度氯化镁存在下的相差显微镜照片、外观照片和动态光散射粒径分布图。加入氯化镁前后,溶液外观没有明显变化,均泛微弱蓝光,即使放置1个月也没有出现沉淀,初步说明α-酮辛酸囊泡体系的耐盐稳定性。由于相差显微镜的分辨率较低,因此通过相差显微镜仅观察到粒径在微米级别的球形或椭球形囊泡(~0.6-10μm)。通过进一步的动态光散射表征(图3中e),可以看出囊泡溶液的粒径分布是多分散的,同时存在微米级别和纳米级别的囊泡;加入不同浓度的氯化镁前后,粒径分布无明显变化,表明α-酮辛酸囊泡可耐受的镁离子浓度大于80mm。
61.需指出,对于浓度介于65-160mm之间的α-酮辛酸囊泡溶液,室温放置2周后,溶液分为上下两相,上相为澄清透明的胶束相,下相为泛滥光的囊泡相,但对α-酮辛酸囊泡的耐盐稳定性无影响。
62.选择浓度为20mm的α-酮辛酸囊泡溶液,测定其ph稳定性:将2mnaoh溶液滴加至α-酮辛酸囊泡溶液,调节ph,分别得到ph值为2.0、5.1、7.2、10.5的α-酮辛酸溶液。图4为不同ph条件下α-酮辛酸溶液的负染色透射电子显微照片和外观照片。由图可知,α-酮辛酸可在ph~2-10范围内形成中空囊泡结构。在较高ph值时(ph偏离pk
a 2.7),囊泡数量较少,但依然有少量囊泡存在,具一定ph稳定性。
63.实施例2
64.烷基酮酸选择为α-酮辛酸。
65.1)α-酮辛酸的制备:
66.与实施例1的区别仅在于,将第一次萃取、洗涤、干燥、去除溶剂后的中间产物分散于30%稀硫酸水溶液中。最终,得到白色粉末(质量2.64g,收率44%)。
67.图5为制得白色粉末在氘代dmso中的核磁共振氢谱图,结果与实施例1一致,表明所得产物即为α-酮辛酸。
68.2)α-酮辛酸囊泡的制备:
69.α-酮辛酸囊泡的制备与实施例1相同。
70.实施例3
71.烷基酮酸选择为α-酮辛酸。
72.1)α-酮辛酸的制备:
73.与实施例1的区别仅在于,所用草酸酯为草酸二甲酯(4.49g),将第一次萃取、洗涤、干燥、去除溶剂后的中间产物分散于30%稀硫酸水溶液中。最终,得到白色粉末(质量2.51g,收率41.5%)。
74.图6为制得白色粉末在氘代dmso中的核磁共振氢谱图,结果与实施例1一致,表明所得产物即为α-酮辛酸。
75.2)α-酮辛酸囊泡的制备:
76.α-酮辛酸囊泡的制备与实施例1相同。
77.实施例4
78.烷基酮酸选择为α-酮癸酸。
79.1)α-酮癸酸的制备:
80.将5.84g草酸二甲酯溶解于60ml无水四氢呋喃中,然后置于-78℃的干冰/丙酮浴中,抽气5min,然后通入氮气10min,如此循环3次。然后将27.2ml 2mol/l辛基溴化镁的乙醚溶液在n2氛围下逐滴滴加至上述草酸二甲酯溶液中,搅拌反应2h。反应结束后,向反应体系中逐滴滴加2mol/l盐酸水溶液终止反应。用乙醚(3
×
150ml)将产物从水相萃取至有机相,有机相用饱和氯化钠洗涤(120ml),无水硫酸镁干燥,旋蒸除去溶剂。将得到的上述产物分散在醋酸(hac,66ml)和盐酸(37%hcl,18ml)中,搅拌16h,旋蒸去除掉大部分溶剂,得到浓缩液。向浓缩液中加入水(60ml)和乙醚(120ml),再用乙醚(3
×
120ml)将产物从水相萃取至有机相,有机相先后用水洗(120ml)、饱和氯化钠洗(120ml),无水硫酸镁干燥,旋蒸除去溶剂。最后,在冰己烷中重结晶3次,得到白色粉末(质量3.67g,产率40%)。
81.图7为制得白色粉末在氘代dmso中的核磁共振氢谱图。图中位移值为2.76ppm处的三重峰对应于α-酮癸酸分子中与亲水性酮基直接相连的亚甲基上的氢原子(α-h),位移值为1.49ppm处的多重峰对应于α-酮癸酸分子中与亲水性酮基相连的β-亚甲基上的氢原子,插图中位移值为13.84ppm的宽峰对应于羧酸中的活泼氢。另外,在5.51ppm处的三重峰和2.07ppm处的四重峰分别对应于α-酮癸酸的烯醇式互变异构体中的α
’‑
h和β
’‑
h,在8.03ppm处的宽峰对应于烯醇式互变异构体中羟基氢原子。从核磁共振氢谱图可以看出,所得产物即为α-酮癸酸。
82.2)α-酮癸酸囊泡的制备:
83.称取上述方法制备得到的α-酮癸酸,在室温下搅拌溶解,配置0.4-200mm不同浓度的水溶液(自然ph值条件下,ph值介于1.6-3.6之间),即得到系列囊泡样品。
84.对其中浓度为0.4mm、40mm和200mm的α-酮癸酸水溶液进行cryo-tem表征。图8为不同浓度α-酮癸酸溶液的显微照片和外观照片,结果表明α-酮癸酸在水溶液中自发形成了中空囊泡结构。在0.4mm极稀溶液中,囊泡粒径在50-400nm之间,大部分为球形单层单室囊泡;在40mm较高浓度溶液中,囊泡粒径、层数、曲率和室数呈多分散性,除球形单层单室囊泡外,出现大量多室、多层、负曲率的大囊泡和巨型囊泡;在200mm浓度溶液中,囊泡仍呈多分散性,多为曲率多变的多层囊泡。
85.需指出,对于浓度介于1.3-15mm之间的α-酮辛酸囊泡溶液,溶液底部有部分针状结晶和囊泡相共存;继续增加浓度至20-200mm,溶液均一、泛蓝光,为单一囊泡相。
86.选择浓度为40mm的α-酮癸酸囊泡溶液,测定其耐盐性:首先称取2.033g氯化镁溶
解于10ml三次水中,制得浓度为2.0m的氯化镁储备液。在快速涡旋状态下,将0μl、6μl和18μl的氯化镁储备液分别加入1.2mlα-酮癸酸溶液中(最终镁离子浓度分别为0mm、10mm和30mm),以考察其耐盐性。
87.图9为40mmα-酮癸酸囊泡溶液在不同浓度氯化镁存在下的相差显微镜照片和外观照片。加入氯化镁后,溶液仍泛蓝光,浊度略升高,但没有出现沉淀,初步说明α-酮癸酸囊泡体系的耐盐稳定性。进一步的相差显微镜观察发现,当加入的氯化镁浓度为10mm或20mm时,囊泡依然存在,粒径分布无明显变化;当加入氯化镁浓度为30mm时,囊泡和沉淀微晶共存,囊泡膜形状不规整,表明在氯化镁浓度为30mm时,部分囊泡发生破裂,但囊泡依然存在。这表明α-酮癸酸囊泡可耐受的镁离子浓度大约为30mm,镁离子浓度大于30mm时,溶液底部出现白色沉淀。由于相差显微镜的分辨率较低,因此通过相差显微镜仅观察到粒径在微米级别的球形或椭球形囊泡(~0.6-10μm),不能观察到纳米级别的囊泡。但α-酮癸酸囊泡溶液的粒径分布是多分散的,同时存在微米级别和纳米级别的囊泡。需指出,加入氯化镁后的α-酮癸酸囊泡溶液放置约24h后,出现沉淀,囊泡破坏消失,即α-酮癸酸溶液在约24h内可耐受镁离子浓度约为30mm。
88.选择浓度为30mm的α-酮癸酸囊泡溶液,测定其ph稳定性:将2mnaoh溶液滴加至α-酮癸酸囊泡溶液,调节ph,分别得到ph值为2.4、7.3、10.0的α-酮癸酸溶液。图10为不同ph条件下α-酮癸酸溶液的冷冻透射电子显微照片和外观照片。由图可知,α-酮癸酸可在ph~2-10范围内形成中空囊泡结构。在较高ph值时(ph偏离pk
a 2.7),囊泡数量较少,但依然有少量囊泡存在,具一定ph稳定性。
89.实施例5
90.烷基酮酸选择为α-酮癸酸。
91.1)α-酮癸酸的制备:
92.与实施例4的区别仅在于,所用草酸酯为草酸二乙酯(5.57g),所用烷基卤化镁为辛基氯化镁溶液(29.9ml,1.4mol/l)。最终,得到白色粉末(质量2.27g,收率32.1%)。
93.图11为制得白色粉末在氘代dmso中的核磁共振氢谱图,结果与实施例4一致,表明所得产物即为α-酮癸酸。
94.2)α-酮癸酸囊泡的制备:
95.α-酮癸酸囊泡的制备与实施例4相同。
96.实施例6
97.烷基酮酸选择为α-酮癸酸。
98.1)α-酮癸酸的制备:
99.与实施例4的区别仅在于,所用草酸酯为草酰氯单甲酯(4.66g)。最终,得到白色粉末(质量0.63g,收率8.9%)。
100.图12为制得白色粉末在氘代dmso中的核磁共振氢谱图,结果与实施例4一致,表明所得产物即为α-酮癸酸。
101.2)α-酮癸酸囊泡的制备:
102.α-酮癸酸囊泡的制备与实施例4相同。
103.实施例7
104.烷基酮酸选择为α-酮月桂酸。
105.1)α-酮月桂酸的制备:
106.将5.58g草酸二乙酯溶解于60ml无水四氢呋喃中,然后置于-78℃的干冰/丙酮浴中,抽气5min,然后通入氮气10min,如此循环3次。然后将42.0ml 1mol/l癸基溴化镁溶液在n2氛围下逐滴滴加至上述草酸二乙酯溶液中,搅拌反应3h。反应结束后,向反应体系中逐滴滴加2mol/l盐酸水溶液终止反应。用己烷(3
×
150ml)将产物从水相萃取至有机相,有机相用饱和氯化钠洗涤(120ml),无水硫酸镁干燥,旋蒸除去溶剂。将得到的上述产物分散在30%稀硫酸水溶液中,搅拌24h,旋蒸去除掉大部分溶剂,得到浓缩液。向浓缩液中加入水(60ml)和乙醚(120ml),再用乙醚(3
×
120ml)将产物从水相萃取至有机相,有机相先后用水洗(120ml)、饱和氯化钠洗(120ml),无水硫酸镁干燥,旋蒸除去溶剂。最后,在冰己烷中重结晶3次,得到白色粉末(0.74g,产率9%)。
107.图13为制得白色粉末在氘代dmso中的核磁共振氢谱图。图中位移值为2.76ppm处的三重峰对应于α-酮月桂酸分子中与亲水性酮基直接相连的亚甲基上的氢原子(α-h),位移值为1.49ppm处的多重峰对应于α-酮月桂酸分子中与亲水性酮基相连的β-亚甲基上的氢原子,插图中位移值为13.75ppm的宽峰对应于羧酸中的活泼氢。另外,在5.51ppm处的三重峰和2.07ppm处的四重峰分别对应于α-酮月桂酸的烯醇式互变异构体中的α
’‑
h和β
’‑
h,在8.01ppm处的宽峰对应于烯醇式异构体中羟基氢原子。从核磁共振氢谱图可以看出,所得产物即为α-酮月桂酸。
108.2)α-酮月桂酸囊泡的制备:
109.称取上述方法制备得到的α-酮月桂酸,室温下加入水搅拌,很难溶解,有大量沉淀不溶物,表明α-酮月桂酸溶液的krafft点在室温以上,进一步的差示扫描量热(dsc)结果表明,相转变温度约为34.7℃,如图14所示。因此,以下α-酮月桂酸溶液均在40℃条件下制备,配制1-100mm不同浓度的样品溶液,即可得到系列囊泡样品(ph值介于2.5-3.7之间)。
110.对其中浓度为1mm、40mm和100mm的α-酮月桂酸水溶液进行cryo-tem表征。图15为不同浓度囊泡溶液的冷冻透射电子显微镜图或相差显微镜图和外观状态图。当浓度为1mm时,溶液浊度较小,囊泡数量或粒径较小,因此采用分辨率较高的冷冻透射电子显微镜观察微观形貌,囊泡粒径在50-500nm之间;当浓度为40mm和100mm时,溶液泛蓝光,粘度增大,推测存在较大粒径或尺寸的囊泡,因此采用分辨率相对较低的相差显微镜即可观察到囊泡微观形貌,可观察到的囊泡粒径在微米级别。随浓度增大,囊泡粒径和数量明显增加。
111.选择浓度为50mm的α-酮月桂酸囊泡溶液,测定其耐盐性:首先称取0.5083g氯化镁溶解于10ml三次水中,制得浓度为0.5m的氯化镁储备液。在快速涡旋状态下,将0μl、9μl、13.5μl和18μl的氯化镁储备液分别加入0.75mlα-酮月桂酸溶液中(最终镁离子浓度分别为0mm、6mm、9mm和12mm),以考察其耐盐性。
112.图16为50mmα-酮月桂酸囊泡溶液在不同浓度氯化镁存在下的相差显微镜照片和外观照片。加入氯化镁后,溶液仍泛蓝光,浊度略升高,但没有出现明显沉淀,初步说明α-酮月桂酸囊泡体系的耐盐稳定性。进一步的相差显微镜观察发现,当加入的氯化镁浓度为6mm或9mm时,少量囊泡依然存在,囊泡膜形状不规整。当加入氯化镁浓度为12mm时,溶液底部出现不溶性凝聚物,发生明显相分离,囊泡完全破坏。需指出,加入氯化镁浓度为6mm或9mm的α-酮月桂酸囊泡溶液放置约24h后,溶液底部均出现不溶性凝聚物,发生明显相分离,囊泡完全破坏,即α-酮月桂酸溶液在约24h内可耐受镁离子浓度约为9mm。
113.选择浓度为32mm的α-酮月桂酸囊泡溶液,测定其ph稳定性:将2mnaoh溶液滴加至α-酮月桂酸囊泡溶液,调节ph,分别得到ph值为2.5、6.5、10.2的α-酮月桂酸溶液。图17为不同ph条件下α-酮月桂酸溶液的冷冻透射电子显微照片和外观照片。由图可知,α-酮月桂酸可在ph~2-10范围内形成中空囊泡结构。在较高ph值时(ph偏离pk
a 2.7),囊泡数量较少。
114.以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。