一种
δ
3型卡品酯的制备方法
技术领域
1.本发明属于药物合成技术领域,尤其涉及药物相关杂质合成技术领域,具体涉及盐酸头孢卡品酯的相关杂质δ3型卡品酯的制备方法。
背景技术:
2.盐酸头孢卡品酯(cefcapenepivoxil hydrochloride hydrate),化学名为2,2-二甲基丙酰氧基甲基(6r,7r)-7-[(2z)-2-(2-氨基噻唑-4-基)戊-2-烯酰氨基]-3-氨甲酰氧基甲基-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸酯一盐酸一水合物,其化学结构如下所示:
[0003][0004]
盐酸头孢卡品酯是日本盐野义制药株式会社开发的酯型口服第三代头孢烯类抗生素,1997年以商品名flomox首次在日本上市,片剂的规格有75mg和100mg两个规格,颗粒剂的规格为10%(50mg/0.5g)。盐酸头孢卡品酯原研制剂中颗粒剂已在国内上市。其适应症为浅表皮肤感染、深层皮肤感染、淋巴/淋巴结炎、慢性脓皮病,继发感染,如外伤/烧伤和手术伤口、乳腺炎、肛周脓肿,咽炎/喉炎继发感染、扁桃体炎(包括扁桃体周围脓肿、扁桃体周围脓肿)、急性支气管炎、肺炎、慢性呼吸道病变,膀胱炎、肾盂肾炎,尿道炎、宫颈炎,胆囊炎、胆管炎,前列腺炎,宫内感染、子宫附件炎,泪囊炎、睑腺炎、睑板腺炎,外耳炎、中耳炎、鼻窦炎,牙周炎、冠周炎、下颌炎症。盐酸头孢卡品酯在体外具有广泛的抗菌谱,从需氧和厌氧革兰氏阳性菌到革兰氏阴性菌,并且它还表现出对耐青霉素肺炎链球菌和耐氨苄西林流感嗜血杆菌的抗菌活性。
[0005]
药物的杂质是目前新药以及仿制药研发中重点关注的项目,药物中含有的杂质可能会直接影响到药物的疗效,并且杂质含量超过一定量,就有可能使药物的理化常数、外观形状发生改变,并且进一步影响药物的稳定性和安全性;药物杂质的检测和控制是目前药物研发过程中必不可少的项目。
[0006]
对于药物研发人员来说,主要工作不仅仅在于如何获得高质量的原料药(api)、开发高效的合成工艺,更重要的是研究原料药中杂质种类、来源以及如何控制工艺杂质的产生。通常研究人员会定向合成工艺中所产生的杂质,并开发高效的杂质合成路线,以便获得大量的杂质对照品,保证每批原料药质量研究工作(例如,杂质hplc定位、杂质含量测定等)的顺利开展。
[0007]
现有技术中已报道的盐酸头孢卡品酯的合成路线主要包括以下关键步骤:母核3位的氨甲酰化反应,母核4位的酯化反应,母核7位的侧链缩合反应和7号位侧链boc(叔丁氧羰基)基团脱除。从报道的工艺路线中可知,使用的母核主要有d-7-aca(羟甲基-7-氨基头
孢烷酸)和7-aca(7-氨基头孢烷酸),使用的侧链为bapa((z)-2-(2-叔丁氧羰基氨基噻唑-4-基)-2-戊烯酸)以及bapa活化酯。在酯化反应以及脱boc反应过程中,很容易产生杂质δ3型卡品酯。
[0008]
δ3型卡品酯作为盐酸头孢卡品酯的同分异构体,是盐酸头孢卡品酯质量控制中需要研究的重要杂质。盐酸头孢卡品酯及其中间体的正常构型与δ3构型为位置异构,它们的化学与物理性质相似,同时存在的情况下难以纯化分离。而确定盐酸头孢卡品酯的关键杂质δ3型卡品酯的制备方法,并提供合格的对照品,对盐酸头孢卡品酯的质量控制具有十分重要的积极作用。《盐酸头孢卡品酯3个代表性有关物质的合成工艺》(杨金光,潘必高等,chinese journal of new drugs,2019,28(21))中公开了一种合成δ3型卡品酯的方法,以boc-头孢卡品为原料,经异构化、酯化、脱保护反应制备得到。该方法中的起始物料boc-头孢卡品为非常见的化工中间体,难以采购;制备过程中需至少进行两次柱层析纯化,操作过程复杂,对人员和仪器设备要求较高,且得到的产物的收率和纯度都较低。
技术实现要素:
[0009]
为了解决上述问题,本发明提供了一种δ3型卡品酯的制备方法,该方法具有操作简单、成本低、纯度高、收率高、适合规模化生产的优点。
[0010]
为了实现上述目的,本发明采用如下技术方案:
[0011]
一种δ3型卡品酯的制备方法,包括以下步骤:
[0012]
s1、氮气保护下,在第一有机溶剂中,7-aca先与bsa发生反应,再在有机碱作用下发生异构化反应,然后在酸性条件下脱去保护基团,经析晶、纯化得到δ3型7-aca;
[0013]
s2、δ3型7-aca在碱溶液中水解,水解产物析晶、纯化,得到δ3型d-7-aca;
[0014]
s3、δ3型d-7-aca与bapa缩合后再与氯磺酰异氰酸酯反应得到δ3型bcn;
[0015]
s4、δ3型bcn与特戊酸碘甲酯发生酯化反应,经析晶、纯化得到δ3型保护卡品酯;
[0016]
s5、δ3型保护卡品酯在lewis酸存在下脱去boc保护基,然后析晶、纯化得到δ3型卡品酯。
[0017]
上述制备方法中涉及的主要反应方程式如下:
[0018][0019][0020]
优选的,步骤s1的具体方法为:将7-aca溶解于第一有机溶剂中,在氮气保护下,加入bsa,控温25~35℃,搅拌至溶清;滴加有机碱,于15~50℃反应50~70min,加水,搅拌5~15min后静置分层,水层冷却至2~5℃,调节ph至3.0~5.0,养晶0.5~1.5h,过滤,滤饼分别
用0~5℃水和0~5℃丙酮各洗涤若干次;将洗涤后的滤饼溶于甲醇,于35℃过滤,收集滤液,滤饼用甲醇洗涤若干次,合并滤液和洗涤液,旋蒸至干,得到δ3型7-aca。
[0021]
优选的,步骤s1中bsa与7-aca的质量比为(0.5~1.5):1;进一步优选为(0.5~1.2):1。
[0022]
优选的,步骤s1中第一有机溶剂与7-aca的体积质量比为(5~15)ml:1g。
[0023]
优选的,所述第一有机溶剂为二氯甲烷。
[0024]
优选的,步骤s1中有机碱与7-aca体积质量比为(0.3~1.0)ml:1g;进一步优选为(0.3~0.5)ml:1g。
[0025]
优选的,步骤s1中有机碱为三乙胺、二乙胺、二异丙基乙胺、吡啶中的至少一种。
[0026]
优选的,步骤s2中水解结束后控温0~5℃静置1h,加入稀盐酸结晶,过滤,滤饼依次用丙酮和水各洗涤若干次,收集滤饼即得δ3型d-7-aca。
[0027]
优选的,步骤s2中的碱溶液为0.4wt%~2.5wt%的氢氧化钠溶液或/和碳酸钠溶液;碱溶液的浓度进一步优选为0.5wt%~2.0wt%。
[0028]
优选的,步骤s3的具体方法为:将δ3型d-7-aca溶于第二有机溶剂,加入四甲基胍和二异丙胺,控温-20~-10℃,得到混合液i;将bapa溶于第二有机溶剂中,加入甲磺酰氯和二异丙胺,在-20~-10℃反应2h,得到混合液ii;将混合液i加入混合液ii中,于-20~-10℃反应1h;保持-20~-10℃的温度,加入含5wt%~8wt%氯化氢气体的乙酸乙酯,然后加入氯磺酰异氰酸酯,反应1h;反应结束后,加水,静置分层,再加入丙酮搅拌溶清后继续搅拌0.5h;反应结束后加入第二有机溶剂静置分层,有机层加水,析晶1.5h,过滤,滤饼用第二有机溶剂洗涤,离心,得δ3型bcn;第二有机溶剂优选二氯甲烷。
[0029]
优选的,步骤s3中bapa与δ3型d-7-aca的质量比为(1.4~1.6):1;进一步优选为(1.5~1.6):1。
[0030]
优选的,步骤s3中氯磺酰异氰酸酯与δ3型d-7-aca的质量比为(1.0~1.2):1。
[0031]
优选的,步骤s4的具体方法为:将δ3型bcn溶于第三有机溶剂和水中,调节ph至2.0~2.5,静置分层,加入特戊酸碘甲酯,反应3.5h,静置分层,有机层加水,在40~50℃下浓缩,然后加入甲基异丁基酮,在0℃以下搅拌析晶3h,过滤,滤饼用甲基异丁基酮洗涤,继续过滤至无液体流出,即得δ3型保护卡品酯;第三有机溶剂优选乙酸乙酯。
[0032]
优选的,步骤s4中特戊酸碘甲酯与δ3型bcn的质量比为(0.6~0.7):1;进一步优选为(0.6~0.65):1。
[0033]
优选的,步骤s5的具体方法为:将δ3型保护卡品酯溶于第四有机溶剂中,加入lewis酸,于-40℃~-30℃反应1h,然后加入乙醇和水,静置分层,有机层浓缩后加入甲基异丁基酮和水,再次静置分层,有机层加入浓盐酸,析出固体,过滤,滤饼用甲基异丁基酮洗涤后过滤,即得δ3型卡品酯;优选的,所述第四有机溶剂为二氯甲烷。
[0034]
优选的,所述lewis酸与δ3型保护卡品酯的质量比为(1.5~2.0):1;进一步优选为(1.5~1.8):1。
[0035]
优选的,所述lewis酸为ticl4。
[0036]
与现有技术相比,本发明的有益效果是:本发明以市场上常见的原料7-aca为起始物料,先对7-aca母核进行构型转化得到δ3型7-aca,排除正常异构体d-7-aca对后续反应的干扰,从源头直接减少其他类型各种杂质,使得各步反应的转化率大幅度提高;反应过程
中全程采用析晶纯化,无需额外的制备流动相进行柱层析纯化;从而使得到的最终产物δ3型卡品酯的纯度达到92.2%以上,摩尔收率达到75%以上。整个反应过程操作简便,原料和试剂易得,安全性高,技术人员具备基本的有机合成基础即可完成整个合成过程;且重现性好,适合规模化批量制备。本发明制备的δ3型卡品酯纯度较高,可将其作为杂质对照品使用,为盐酸头孢卡品酯生产过程的质量控制提供了有效的数据支持,进一步为药品安全使用提供了理论依据。
附图说明
[0037]
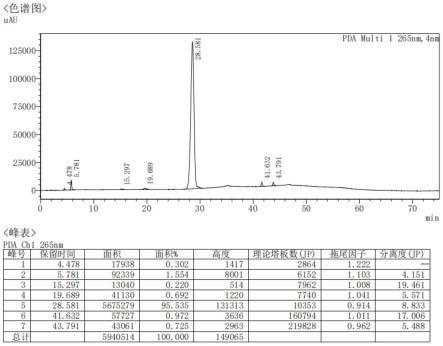
图1为本发明实施例1制备的δ3型卡品酯的hplc图谱;
[0038]
图2为本发明实施例2制备的δ3型卡品酯的hplc图谱;
[0039]
图3为本发明实施例3制备的δ3型卡品酯的hplc图谱。
具体实施方式
[0040]
以下结合实施例对本发明技术方案进行清楚、完整的描述,显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施例,都属于本发明所保护的范围。本领域技术人员依据以下实施方式所作的任何等效变换或替代,均属于本发明的保护范围之内。
[0041]
以下实施例1~3中δ3型卡品酯纯度检测方法参考《日本药典》jp18版中的盐酸头孢卡品酯的有关物质的检测方法,δ3型卡品酯的保留时间为28.8
±
1min。
[0042]
实施例1
[0043]
本实施例提供了一种δ3型卡品酯的制备方法,包括以下步骤:
[0044]
s1、在三颈烧瓶中加入30g 7-aca和450ml二氯甲烷,在氮气保护下,加入18ml n,o-双三甲硅基乙酰)(bsa),控温25℃~35℃,搅拌至溶清;滴加9ml三乙胺,在30℃反应60min,加水,搅拌8~9min后静置分层,收集水层;水层冷却到2~5℃,用10%盐酸调节ph=3.0,养晶0.5h后过滤,并依次用0~5℃水和0~5℃丙酮洗涤;滤饼用甲醇溶解,控温35℃,趁热过滤,滤饼用甲醇重复洗涤若干次,合并滤液和洗涤液,旋蒸至干,得28.55gδ3型7-aca,摩尔收率为95.2%;
[0045]
s2、将28.55gδ3型7-aca溶于100ml 0.5wt%氢氧化钠溶液中,控制温度0~5℃静置1h,加入120ml 1%稀盐酸结晶后,过滤、滤饼依次用丙酮和水洗涤,收集滤饼即得23.01gδ3型d-7-aca,摩尔收率为95.4%;
[0046]
s3、取23.01gδ3型d-7-aca溶于30ml二氯甲烷中,再加入12.15g四甲基胍和15ml二异丙胺,控制温度为-20℃,得到混合液i;取34.52g bapa((z)-2-(2-叔丁氧羰基氨基噻唑-4-基)-2-戊烯酸)溶于90ml二氯甲烷中,加入20.22g甲磺酰氯和15ml二异丙胺,控制温度为-20℃反应2h,得到混合液ii;将混合液i加入混合液ii中,在-20℃下继续反应1h,继续控制该温度加入90.5g含5wt%氯化氢气体的乙酸乙酯溶液,再加入23g氯磺酰异氰酸酯(csi),反应1h;然后加入200ml水静置分层,取上层水层再加入100ml丙酮搅拌溶清后,继续搅拌反应0.5h,反应结束后加入200ml二氯甲烷静置分层,取有机相加水,析晶1.5h,过滤,滤饼用二氯甲烷洗涤,然后离心至液体流出,即得61.39gδ3型bcn,摩尔收率为93.7%;
[0047]
s4、将61.39gδ3型bcn溶于80ml乙酸乙酯和20ml水的混合溶剂中,加入盐酸调节ph为2.0~2.5,静置分层,加37.90g特戊酸碘甲酯,反应3.5h,静置分层,取有机层加入100ml水,在40~50℃下浓缩,然后加入350ml甲基异丁基酮,在-2℃搅拌析晶3h,过滤,滤饼用甲基异丁基酮洗涤,继续过滤至无液体流出,即得59.87gδ3型保护卡品酯,摩尔收率为95.6%;
[0048]
s5、取59.87gδ3型保护卡品酯溶于200ml二氯甲烷中,加入89.8gticl4,在-35℃反应1h,加入150ml乙醇和100ml水,静置分层,取有机层浓缩后再加入180ml甲基异丁基酮和120ml水,再次静置分层,取有机层中加入浓盐酸10ml,析出固体,过滤,滤饼用甲基异丁基酮洗涤后继续过滤至液体流出,即得47.33gδ3型卡品酯,收率为93.0%,经hplc检测纯度为95.54%,hplc图谱如图1所示,δ3型卡品酯的保留时间为28.581min。以起始物料7-aca计算,δ3型卡品酯的总的摩尔收率为75.7%。
[0049]
实施例2
[0050]
本实施例提供了一种δ3型卡品酯的制备方法,包括以下步骤:
[0051]
s1、在三颈烧瓶中加入30g 7-aca和150ml二氯甲烷,在氮气保护下,加入42ml n,o-双三甲硅基乙酰)(bsa),控温25℃~35℃,搅拌至溶清;滴加8ml吡啶和8ml三乙胺,在27
±
2℃反应70min,加水,搅拌5~6min后静置分层,收集水层;水层冷却到2~5℃,用10%盐酸调节ph=4.5,养晶1h后过滤,并依次用0~5℃水和0~5℃丙酮洗涤;滤饼用甲醇溶解,控温35℃,趁热过滤,滤饼用甲醇重复洗涤若干次,合并滤液和洗涤液,旋蒸至干,得28.01gδ3型7-aca,摩尔收率为93.4%;
[0052]
s2、将28.01gδ3型7-aca溶于100ml 2wt%碳酸钠溶液中,控制温度0~5℃静置1h,加入120ml 2%稀盐酸结晶后,过滤、滤饼依次用丙酮和水洗涤,收集滤饼即得23.10gδ3型d-7-aca,摩尔收率为97.6%;
[0053]
s3、取23.10gδ3型d-7-aca溶于33ml二氯甲烷中,再加入12.51g四甲基胍和15ml二异丙胺,控制温度为-16℃,得到混合液i;取36.05g bapa溶于90ml二氯甲烷中,加入20.59g甲磺酰氯和15ml二异丙胺,控制温度为-16℃反应2h,得到混合液ii;将混合液i加入混合液ii中,在-16℃下继续反应1h,继续控制该温度加入93.71g含7wt%氯化氢气体的乙酸乙酯溶液,再加入28g氯磺酰异氰酸酯,反应1h;然后加入200ml水静置分层,再加入100ml丙酮搅拌溶清后,继续搅拌反应0.5h,反应结束后加入200ml二氯甲烷静置分层,取有机相加水,析晶1.5h,过滤,滤饼用二氯甲烷洗涤,然后离心至液体流出,即得62.92gδ3型bcn,摩尔收率为95.7%;
[0054]
s4、将62.92gδ3型bcn溶于80ml乙酸乙酯和20ml水的混合溶剂中,加入盐酸调节ph为2.0~2.5,静置分层,加40.05g特戊酸碘甲酯,反应3.5h,静置分层,取有机层加入100ml水,在40~50℃下浓缩,然后加入350ml甲基异丁基酮,在-4℃搅拌析晶3h,过滤,滤饼用甲基异丁基酮洗涤,继续过滤至无液体流出,即得60.89gδ3型保护卡品酯,摩尔收率为94.9%;
[0055]
s5、取60.89gδ3型保护卡品酯溶于200ml二氯甲烷中,加入110.0g ticl4,在-30℃反应1h,加入150ml乙醇和100ml水,静置分层,取有机层浓缩后再加入180ml甲基异丁基酮和120ml水,再次静置分层,取有机层中加入浓盐酸10ml,析出固体,过滤,滤饼用甲基异丁基酮洗涤后继续过滤至液体流出,即得48.99gδ3型卡品酯,收率为94.6%,经hplc检测
纯度为94.97%,hplc图谱如图2所示,δ3型卡品酯的保留时间为29.052min。以起始物料7-aca计算,δ3型卡品酯的总的摩尔收率为78.3%。
[0056]
实施例3
[0057]
本实施例提供了一种δ3型卡品酯的制备方法,包括以下步骤:
[0058]
s1、在三颈烧瓶中加入35g 7-aca和350ml二氯甲烷,在氮气保护下,加入63ml n,o-双三甲硅基乙酰)(bsa),控温25℃~35℃,搅拌至溶清;滴加33ml二乙胺,在33
±
2℃反应50min,加水,搅拌15min后静置分层,收集水层;水层冷却到2~5℃,用10%盐酸调节ph=5.0,养晶1.5h后过滤,并依次用0~5℃水和0~5℃丙酮洗涤;滤饼用甲醇溶解,控温35℃,趁热过滤,滤饼用甲醇重复洗涤若干次,合并滤液和洗涤液,旋蒸至干,得33.05gδ3型7-aca,摩尔收率为94.4%;
[0059]
s2、将33.05gδ3型7-aca溶于100ml 0.6wt%氢氧化钠溶液中,控制温度0~5℃静置1h,加入70ml 2%稀盐酸结晶后,过滤、滤饼依次用丙酮和水洗涤,收集滤饼即得26.13gδ3型d-7-aca,摩尔收率为93.6%;
[0060]
s3、取26.13gδ3型d-7-aca溶于40ml二氯甲烷中,再加入14.62g四甲基胍和16ml二异丙胺,控制温度为-10℃,得到混合液i;取36.68g bapa溶于90ml二氯甲烷中,加入25.07g甲磺酰氯和16ml二异丙胺,控制温度为-10℃反应2h,得到混合液ii;将混合液i加入混合液ii中,在-10℃下继续反应1h,继续控制该温度加入90.76g含8wt%氯化氢气体的乙酸乙酯溶液,再加入30g氯磺酰异氰酸酯,反应1h;然后加入220ml水静置分层,再加入120ml丙酮搅拌溶清后,继续搅拌反应0.5h,反应结束后加入220ml二氯甲烷静置分层,取有机相加水,析晶1.5h,过滤,滤饼用二氯甲烷洗涤,然后离心至液体流出,即得71.20gδ3型bcn,摩尔收率为95.7%;
[0061]
s4、将71.20gδ3型bcn溶于100ml乙酸乙酯和30ml水的混合溶剂中,加入盐酸调节ph为2.0~2.5,静置分层,加50.84g特戊酸碘甲酯,反应3.5h,静置分层,取有机层加入100ml水,在40~50℃下浓缩,然后加入400ml甲基异丁基酮,在0℃搅拌析晶3h,过滤,滤饼用甲基异丁基酮洗涤,继续过滤至无液体流出,即得69.17gδ3型保护卡品酯,摩尔收率为95.3%;
[0062]
s5、取69.17gδ3型保护卡品酯溶于230ml二氯甲烷中,加入138.3g ticl4,在-40℃反应1h,加入180ml乙醇和110ml水,静置分层,取有机层浓缩后再加入200ml甲基异丁基酮和150ml水,再次静置分层,取有机层中加入浓盐酸12ml,析出固体,过滤,滤饼用甲基异丁基酮洗涤后继续过滤至液体流出,即得54.70gδ3型卡品酯,收率为93.0%,经hplc检测纯度为92.17%,hplc图谱如图3所示,δ3型卡品酯的保留时间为28.834min。以起始物料7-aca计算,δ3型卡品酯的总的摩尔收率为87.5%。
[0063]
由以上实施例可知,本发明中制备δ3型卡品酯的方法由于先合成了高纯度高收率的δ3构型母核(δ3型7-aca),后续反应的每一步收率都很高,最终产物的摩尔收率达到75%以上,远高于现有技术中的收率。后续步骤中缩合、酯化和脱boc保护基的反应均可通过析晶纯化得到中间体和最终的成品,无需多次制备流动相和进行柱层析纯化,操作简单,产物纯度高,成本低,适合进行工业化生产。
[0064]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明的保护范围。对于任何熟悉本领域的技术人员来说,本发明可以有各种更改和变化。任何依据本发明申请保
护范围及说明书内容所作的简单的等效变化和修饰,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。