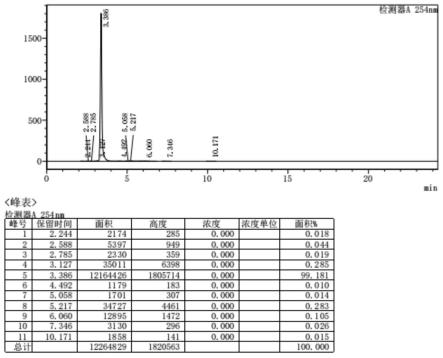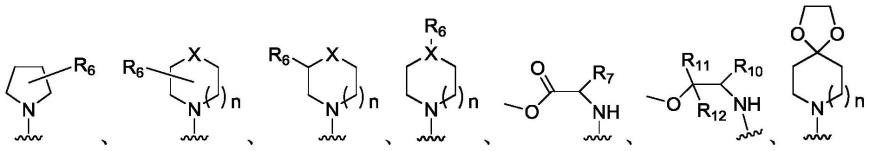
1.本发明属于化学合成技术领域,具体涉及芳基胺类化合物制备方法。
背景技术:
2.芳基胺类化合物广泛的存在于许多药物、天然产物、有机材料和催化剂中。鉴于苯胺类化合物广泛的药物和药理活性以及在有机合成中的应用,科学家们已经开发了许多种合成策略来制备苯胺衍生物。其中,报道最多的合成策略是过渡金属(pd和cu)介导的c-n交叉偶联反应(m.j.west,j.w.b.fyfe,j.c.vantourout,a.j.b.watson,chem.rev.2019,119,12491-12523)。根据与胺偶联物种的种类不同主要分为两种不同的方法:即以buchwald-hartwig(j.f.hartwig,nature 2008,455,314-322)和ullmann-goldberg(i.p.beletskaya,v.a.cheprakov,coord.chem.rev.2004,248,2337-2364)反应为代表的亲电试剂-亲核试剂交叉偶联和以chan-lam(m.j.west,j.w.b.fyfe,j.c.vantourout,a.j.b.watson,chem.rev.2019,119,12491-12523)反应为代表的亲核试剂-亲核试剂交叉偶联。然而,这些合成策略存在许多缺点阻碍反应的进行,例如底物胺和过渡金属之间会配位产生的非反应性络合物,n-h键的活化能高,单芳基化产物和胺之间的亲核竞争产生的多芳基化副产物,以及苛刻的反应条件(强酸或碱、高反应温度、化学计量的金属催化剂等)(d.m.roundhill,chem.rev.1992,92,1-27)。另外,具有位点选择性官能团的苯胺通常通过亲电取代制备,因此芳烃上的导向基团(电子效应或空间效应)是必不可少的(i.a.i.mkhalid,j.h.barnard,t.b.marder,j.m.murphy,j.f.hartwig,chem.rev.2010,110,890-931)。所以,化学家一直在寻求一种新的、高效的、位点选择性构筑c-n键的方法来合成苯胺及其衍生物。
3.由于光化学独特的反应模式和在可持续化学中的重要性,光化学为自由基官能化带来了极具吸引力的策略,其中光催化构筑c-n键合成苯胺类化合物受到了越来越多的关注。虽然,芳烃的光催化胺化来制备苯胺已经得到了很好的发展,但大多数反应都适用于富电子芳烃,并且表现出很强的对位选择性(a)n.a.romero,k.a.margrey,n.e.tay,d.a.nicewicz,science 2015,349,1326-1330;b)k.a.margrey,j.b.mcmanus,s.bonazzi,f.zecri and d.a.nicewicz,j.am.chem.soc.2017,139,11288-11299;c)k.a.margrey,a.levens,d.a.nicewicz,angew.chem.int.ed.2017,56,15644-15648;d)t.d.svejstrup,a.ruffoni,f.juli
á
,v.m.aubert,d.leonori,angew.chem.int.ed.2017,56,14948-14952;e)a.ruffoni,f.julia,t.d.svejstrup,a.j.mcmillan,j.j.douglas,d.leonori,nat.chem.2019,11,426-433;f)w.s.ham,j.hillenbrand,j.jacq,c.genicot,t.ritter,angew.chem.int.ed.2019,58,532-536;g)s.l.b.j.jelier,p.f.tripet,a.shemet,g.jeschke,a.togni,e.m.carreira,angew.chem.int.ed.2019,58,526-531)。由于苯胺在亲电取代反应中固有的邻位和对位偏向,间位取代苯胺的有效和选择性制备仍然是一个问题。更重要的是,在间位具有羰基取代基的苯胺作为生物活性剂的药效团已被广泛应用于药物化学(a)a.r.galeev,m.v.dmitriev,i.g.mokrushin,i.v.mashevskaya,
a.n.maslivets,m.rubin,org.biomol.chem.2019,17,10030-10044;b)i.nakamura,h.tashio,y.ishida,m.terada,org.lett.2020,22,3794-3798)。因此,化学家一直在探索间位取代苯胺的新合成方法。
4.因此,亟待一种绿色环保、产率高的间位取代芳基胺类化合物制备方法。
技术实现要素:
5.为解决上述间题,本发明提供以下技术方案。
6.一种化合物c的制备方法,其包括:
[0007][0008]
化合物a和化合物b在光敏剂、钴肟复合物、酸和碱存在并于惰性气体氛围条件下,在溶剂中,经光照照射发生反应,得到化合物c,
[0009]
其中,
[0010]
r3选自取代或未取代的c
1-c
15
烷基、取代或未取代的c
6-c
20
芳基;
[0011]
r4选自卤素原子;
[0012]
r5选自h、取代或未取代的c
1-c
15
烷基、取代或未取代的c
6-c
20
芳基;
[0013]
化合物b或化合物c中,r1和r2与相邻的n共同构成氨基醇或氨基酯;或者化合物b或化合物c中,r1和r2分别独立选自h、取代或未取代的c
1-c
15
烷基、取代或未取代的c
1-c
10
环烷基、取代或未取代的c
1-c
15
羟烷基、取代或未取代的c
6-c
20
芳基、取代或未取代的c
6-c
20
羟基芳基;或者化合物b或化合物c中,r1和r2与相邻的n共同构成以下结构:
[0014][0014]
其中,n为1或2,x为c、o、n或s;r6选自h、羟基、c
1-c
15
羟烷基、c
3-c
10
环烷基、c
1-c
15
烷氧基、-ch(ph)2、-c(ph)2oh、-(ch2)mnhc(=o)r8、苯基、吗啉基、4-吡啶基、嘧啶基、-coor9、-c(=o)r9、-cooch2ph、甲磺酰基、-cf3,-conh2,其中m为1、2、3、4或5;r7、r8或r9分别独立选自取代或未取代的c
1-c
15
烷基、取代或未取代的c
6-c
20
芳基;r10选自取代或未取代的c
1-c
15
烷基、取代或未取代的c
6-c
20
芳基、取代或未取代的c
1-c
15
烷氧基、-coor
13
,r
13
选自c
1-c
15
烷基;r
11
和r
12
分别独立选自h、取代或未取代的c
6-c
20
芳基、取代或未取代的c
3-c
10
环烷基。
[0015]
在一些实施例中,所述光照为蓝光。
[0016]
在一些实施例中,所述蓝光的波长为400nm~480nm。在一些实施例中,所述蓝光的波长为440nm。
[0017]
在一些实施例中,化合物b或化合物c中,r1和r2与相邻的n共同构成以下结构:
[0018][0019][0020]
在一些实施例中,所述取代为被取代基中一个或多个氢原子各自独立地被羟基、c
1-c
15
烷基、c
1-c
15
羟烷基或代替。
[0021]
在一些实施例中,所述光敏剂选自铱类光敏剂、钌类光敏剂或有机光敏剂中的至少一种。
[0022]
在一些实施例中,所述铱类光敏剂包括[lr(ppy)2(dtbbpy)]pf6、[ir(df(cf3)(ppy)2(dtbbpy)]pf6、[ir(p-f(cf3)ppy)2(bpy)]pf6、[ir(p-cf
3-ppy)2(bpy)]pf6、[ir(dtbppy)2(dtbbpy)]pf6、[ir(m-cf3(cf3)ppy)2bpy]pf6、[ir(dtbppy)2(bpy)]pf6、[ir(ppy)2(bpy)]pf6、fac-ir(ppy)3、[ir(dmppy)2(dtbbpy)]pf6、[ir(ppycf3)2bpy]pf6、[ir(df(me)ppy2)(dtbbpy)]pf6、[ir(df(cf3)ppy)2bpy]pf6中的至少一种。在一些优选的实施例中,所述铱类光敏剂包括[ir(ppy)2(dtbbpy)]pf6、[ir(p-f(cf3)ppy)2(bpy)]pf6、[ir(dmppy)2(dtbbpy)]pf6、[ir(ppy)2(bpy)]pf6、[ir(dtbppy)2(dtbbpy)]pf6或[ir(ppycf3)2bpy]pf6中的至少一种。在一些更优选的实施例中,所述铱类光敏剂包括[ir(ppy)2(dtbbpy)]pf6,
[0023][0024]
在一些实施例中,所述钌类光敏剂包括ru(phen)3(pf6)2、[ru(dtbbpy)3](pf6)2或[ru(bpz)3](pf6)中的至少一种。在一些优选的实施例中,所述钌类光敏剂包括ru(phen)3(pf6)2,
[0025][0026]
在一些实施例中,所述有机光敏剂包括eosiny或4-czipn中的至少一种。在一些实施例中,所述有机光敏剂包括eosiny,
[0027][0028]
在一些实施例中,所述钴肟复合物包括co(dmgh)(dmgh2)pycl、co(dmgh)(dmgh2)cl2、co(dmgbf2)2(h2o)2、co(dmgh)2(4-cnpy)cl、co(dmgh)2cl(dmap)中的至少一种,
[0029][0030]
在一些优选的实施例中,所述钴肟复合物包括co(dmgh)(dmgh2)pycl、co(dmgh)(dmgh2)cl2、co(dmgbf2)2(h2o)2、co(dmgh)2(4-cnpy)ci、co(dmgh)2cl(dmap)中的至少一种。在一些更优选的实施例中,所述钴肟复合物包括co(dmgh)(dmgh2)cl2。
[0031]
在一些实施例中,所述酸包括三氟乙酸(tfa)、醋酸(ch3cooh)、对甲基苯磺酸(tsoh)、甲基磺酸(msoh)、三氟甲磺酸钪(sc(otf)3)或三氟甲磺酸钇(y(otf)3)中的至少一种。在一些优选的实施例中,所述酸为醋酸或三氟甲磺酸钪中的至少一种。在一些更优选的实施例中,所述酸为三氟甲磺酸钪。酸的作用是活化羰基,促进烯胺的生成。
[0032]
在一些实施例中,所述碱包括醋酸钠(naoac)或三乙烯二胺(dabco)中的至少一种,优选为三乙烯二胺。碱的作用是作为质子源提供氢原子。
[0033]
在一些实施例中,所述惰性气体包括氮气、氦气、氖气、氩气中的至少一种。
[0034]
在一些实施例中,所述溶剂包括水、乙酸乙酯、乙腈中的至少一种。在一些优选的实施例中,所述溶剂为乙腈。
[0035]
在一些实施例中,所述制备方法还包括在反应前加入分子筛。
[0036]
在一些实施例中,所述c
1-c
15
烷基包括c1烷基、c2烷基、c3烷基、c4烷基、c5烷基、c6烷基、c7烷基、c8烷基、c9烷基、c
10
烷基、c
11
烷基、c
12
烷基、c
13
烷基、c
14
烷基或c
15
烷基。在一些实施例中,所述c
1-c
15
烷基包括甲基、乙基、1-丙基、2-丙基、1-丁基、2-甲基-1-丙基、2-丁基、
2-甲基-2-丙基、1-戊基、2-戊基、3-戊基、2-甲基-2-丁基、3-甲-2-丁基、3-甲基-1-丁基、2-甲基-1-丁基、1-己基、2-己基、3-己基、2-甲基-2-戊基、3-甲基-2-戊基、4-甲基-2-戊基、3-甲基-3-戊基、2-甲基-3-戊基、2,3-二甲基-2-丁基、3,3-二甲基-2-丁基、辛基或
[0037]
在一些实施例中,所述c
6-c
20
芳基包括c6芳基、c7芳基、c8芳基、c9芳基、c
10
芳基、c
11
芳基、c
12
芳基、c
13
芳基、c
14
芳基、c
15
芳基、c16芳基、c
17
芳基、c
18
芳基、c
19
芳基或c
20
芳基。在一些实施例中,所述c
6-c
20
芳基包括芳基包括苯基或苄基。
[0038]
在一些实施例中,所述c
1-c
15
羟烷基包括含一个羟基取代基的c
1-c
15
烷基。在一些实施例中,所述c
1-c
15
羟烷基包括含一个羟基取代基的c1烷基、含一个羟基取代基的c2烷基、含一个羟基取代基的c3烷基、含一个羟基取代基的c4烷基、含一个羟基取代基的c5烷基、含一个羟基取代基的c6烷基、含一个羟基取代基的c7烷基、含一个羟基取代基的c8烷基、含一个羟基取代基的c9烷基、含一个羟基取代基的c
10
烷基、含一个羟基取代基的c
11
烷基、含一个羟基取代基的c
12
烷基、含一个羟基取代基的c
13
烷基、含一个羟基取代基的c
14
烷基或含一个羟基取代基的c
15
烷基。在一些实施例中,所述c
1-c
15
羟烷基包括羟甲基、羟乙基或
[0039]
在一些实施例中,所述c
3-c
10
环烷基包括c3环烷基、c4环烷基、c5环烷基、c6环烷基、c7环烷基、c8环烷基、c9环烷基或c
10
环烷基。在一些实施例中,所述c
3-c
10
环烷基包括环丙基、环丁基、环戊基、环己基、环庚基或环辛基。
[0040]
在一些实施例中,所述c
1-c
15
烷氧基包括-o-c
1-c
15
烷基。在一些实施例中,所述c
1-c
15
烷氧基包括-o-c1烷基、-o-c2烷基、-o-c3烷基、-o-c4烷基、-o-c5烷基、-o-c6烷基、-o-c7烷基、-o-c8烷基、-o-c9烷基、-o-c
10
烷基、-o-c
11
烷基、-o-c
12
烷基、-o-c
13
烷基、-o-c
14
烷基或-o-c
15
烷基。在一些实施例中,所述c
1-c
15
烷氧基包括甲氧基、乙氧基或丙氧基。
[0041]
在一些实施例中,所述c
6-c
20
羟基芳基包括含一个羟基取代基的c
6-c
20
芳基。在一些实施例中,所述c
6-c
20
羟基芳基包括含一个羟基取代基的c6芳基、含一个羟基取代基的c7芳基、含一个羟基取代基的c8芳基、含一个羟基取代基的c9芳基、含一个羟基取代基的c
10
芳基、含一个羟基取代基的c
11
芳基、含一个羟基取代基的c
12
芳基、含一个羟基取代基的c
13
芳
基、含一个羟基取代基的c
14
芳基、含一个羟基取代基的c
15
芳基、含一个羟基取代基的c
16
芳基、含一个羟基取代基的c
17
芳基、含一个羟基取代基的c
18
芳基、含一个羟基取代基的c
19
芳基或含一个羟基取代基的c
20
芳基。在一些实施例中,所述c
6-c
20
羟基芳基包括羟基芳基包括
[0042]
在一些实施例中,所述卤素原子为碘原子。
[0043]
在一些实施例中,所述化合物b与化合物a的投料摩尔比为1∶1-5∶1。在一些实施例中,所述化合物b与化合物a的投料摩尔比为1∶1、2∶1、3∶1、4∶1或5∶1。
[0044]
在一些实施例中,所述酸与化合物a的投料摩尔比为0.05∶1.00-0.50∶1.00。在一些实施例中,所述酸与化合物a的投料摩尔比为0.05∶1.00、0.10∶1.00、0.20∶1.00、0.30∶1.00、0.40∶1.00或0.50∶1.00。在一些优选的实施例中,所述酸与化合物a的投料摩尔比为0.30∶1.00-0.50∶1.00。在一些更优选的实施例中,所述酸与化合物a的投料摩尔比为0.40∶1.00。
[0045]
在一些实施例中,所述碱与化合物a的投料摩尔比为0.5∶1.0-10.0∶1.0。在一些实施例中,所述碱与化合物a的投料摩尔比为0.5∶1.0、1.0∶1.0、2.0∶1.0、3.0∶1.0、4.0∶1.0、5.0∶1.0、6.0∶1.0、7.0∶1.0、8.0∶1.0、9.0∶1.0或10.0∶1.0。在一些优选的实施例中,所述碱与化合物a的投料摩尔比为5.0∶1.0~9.0∶1.0。在一些更有选的实施例中,所述碱与化合物a的投料摩尔比为6.0∶1.0~7.0∶1.0。
[0046]
在一些实施例中,所述光敏剂与化合物a的投料摩尔比为0.005∶1.000-0.100∶1.000。在一些实施例中,所述光敏剂与化合物a的投料摩尔比为0.005∶1.000、0.010∶1.000、0.020∶1.000、0.030∶1.000、0.040∶1.000、0.050∶1.000、0.060∶1.000、0.070∶1.000、0.080∶1.000、0.090∶1.000或0.100∶1.000。在一些实施例中,所述光敏剂与化合物a的投料摩尔比为0.010∶1.000-0.100∶1.000。在一些实施例中,所述光敏剂与化合物a的投料摩尔比为0.020∶1.000-0.100∶1.000。
[0047]
在一些实施例中,所述钴肟复合物与化合物a的投料摩尔比为0.005∶1.000-0.100∶1.000。在一些实施例中,所述钴肟复合物与化合物a的投料摩尔比为0.005∶1.000、0.010∶1.000、0.020∶1.000、0.030∶1.000、0.040∶1.000、0.050∶1.000、0.060∶1.000、0.070∶1.000、0.080∶1.000、0.090∶1.000、0.100∶1.000、0.200∶1.000、0.300∶1.000、0.400∶1.000、0.500∶1.000、0.600∶1.000、0.700∶1.000、0.800∶1.000、0.900∶1.000或1.000∶1.000。在一些实施例中,所述钴肟复合物与化合物a的投料摩尔比为0.030∶1.000-0.100∶1.000。在一些实施例中,所述钴肟复合物与化合物a的投料摩尔比为0.040∶1.000-0.100∶1.000。
[0048]
在一些实施例中,所述反应的反应温度为10℃-40℃。在一些实施例中,所述反应的反应温度为15℃-35℃。在一些实施例中,所述反应的反应温度为20℃-30℃。在一些实施例中,所述反应的反应温度为22℃-28℃。在一些实施例中,所述反应的反应温度为24℃-26℃。
[0049]
在一些实施例中,所述化合物c选自以下结构∶化合物1~化合物65,
[0050]
[0051]
[0052]
[0053]
[0054][0055]
在一些实施例中,所述化合物b选自以下结构:化合物1-b~化合物51-b,
[0056]
[0057]
[0058][0059]
在一些实施例中,所述化合物a选自以下结构:化合物1a、化合物51a~化合物65a,
[0060]
[0061][0062]
在一些实施例中,所述化合物c为化合物1~化合物50中的任一项,所述化合物a为化合物1a,所述化合物b分别对应为化合物1-b~化合物50-b。
[0063]
在一些实施例中,所述化合物c为化合物51~化合物65中的任一项,所述化合物b为化合物51-b,所述化合物a分别对应为化合物51a~化合物65a。
[0064]
有益效果
[0065]
相比现有技术,本发明的某一个实施例具有以下至少一种有益效果:
[0066]
(1)采用本发明所提供的光敏剂和钴肟复合物,尤其是采用本发明优选的光敏剂和钴肟复合物,有利于将间位羰基取代的苯胺类化合物的合成,同时克服了现有技术反应条件苛刻、底物胺和过渡金属之间会配位产生的非反应性络合物、n-h键的反应活化能高、单芳基化产物和胺之间的亲核竞争产生的多芳基化副产物等缺点。
[0067]
(2)采用本发明所提供的光敏剂和钴肟复合物,尤其是采用本发明优选的光敏剂和钴肟复合物,有利于大大促进和加速反应的发生并提高产物的收率。
[0068]
(3)本发明所提供的反应中,光敏剂可以为铱类光敏剂、钌类光敏剂或有机光敏剂;其中铱类光敏剂优选包括[ir(ppy)2(dtbbpy)]pf6、[ir(p-f(cf3)ppy)2(bpy)]pf6、[ir(dmppy)2(dtbbpy)]pf6、[ir(ppy)2(bpy)]pf6、[ir(dtbppy)2(dtbbpy)]pf6或[ir(ppycf3)2bpy]pf6中的至少一种;所述钌类光敏剂优选包括ru(phen)3(pf6)2;有机光敏剂优选包括eosiny;在所有光敏剂中,最优选包括ru(phen)3(pf6)2,有利于提高产物产率。
[0069]
(4)本发明所提供的反应中,钴肟复合物优选包括co(dmgh)(dmgh2)pycl、co(dmgh)2(4-cnpy)cl、co(dmgh)(dmgh2)cl2、co(dmgh)2cl(dmap),更优选包括co(dmgh)(dmgh2)cl2,有利于提高产物产率。
[0070]
(5)采用本发明所提供的酸,有利于提高产物的收率,其中所述酸优选为醋酸或三氟甲磺酸钪中的至少一种;更优选为三氟甲磺酸钪。
[0071]
(6)所述酸与化合物a的投料摩尔比优选为0.3∶1-0.5∶1,有利于提高产物的收率。所述酸与化合物a的投料摩尔比更优选为0.4∶1,更有利于提高产物的收率。
[0072]
(7)采用本发明所提供的碱,尤其是优选的dabco,有利于提高产物的收率。
[0073]
(8)所述碱与化合物a的投料摩尔比优选为为5∶1~9∶1,有利于提高产物的收率。所述碱与化合物a的投料摩尔比更优选为6∶1~7∶1,更有利于提高产物的收率。
[0074]
(9)采用本发明所提供的溶剂,有利于提高产物的收率,尤其是优选的乙腈,更有利于提高产物的收率。
[0075]
(10)光敏剂和光照为本发明反应的必要条件,缺失任何一个条件,反应均无法进行;另外,若要获得高产率的产物,除光敏剂和光照外,钴肟复合物、酸和碱也缺一不可;光敏剂、钴肟复合物、酸、碱和光照彼此相互协同,共同促进产物产率的提高。
[0076]
(11)通过加入分子筛,有利于进一步提高产物的收率。
[0077]
术语说明
[0078]
本发明中,“室温”表示环境温度,可以为10℃-40℃,可以为15℃-35℃,或者可以为20℃-30℃;在一些实施例中,为22℃-28℃;在一些实施例中,为24℃-26℃;在一些实施例中,为25℃。
[0079]
在本发明的上文中,无论是否使用“大约”或“约”等字眼,所有在此公开了的数字均为近似值。基于公开的数字,每一个数字的数值有可能会出现
±
10%以下的差异或者本领域人员认为的合理的差异,如
±
1%、
±
2%、
±
3%、
±
4%或
±
5%的差异。
[0080]
术语“卤素原子”表示氟原子、氯原子、溴原子或碘原子。
[0081]
术语“任选”、“任选的”或“任选地”是指随后描述的事件或情形可以但不一定出现。例如,“任选地,所述还原胺化反应的反应压强为0.5mpa~3mpa”表示“所述还原胺化反应的反应压强为0.5mpa~3mpa”的情形可以存在或者可以不存在。
[0082]
术语“重量百分比”或“以重量计的百分比”或“wt%”定义为组合物中单个组分的重量除以组合物所有组分的总重量然后乘以100%。
[0083]
基团“ph”表示苯基。
[0084]
基团“t-bu”表示叔丁基。
[0085]
术语“和/或”应理解为意指可选项中的任一项或可选项中的任意两项或多项的组合。
[0086]“烷基”是包含正碳原子、仲碳原子、叔碳原子或环碳原子的烃。例如,烷基可以具有1至15个碳原子(即,c1烷基、c2烷基、c3烷基、c4烷基、c5烷基、c6烷基、c7烷基、c8烷基、c9烷基、c
10
烷基、c
11
烷基、c
12
烷基、c
13
烷基、c
14
烷基或c
15
烷基)、1至8个碳原子(即,c
1-c8烷基)或1至6个碳原子(即,c
1-c6烷基)。合适的烷基的实施例包括但不限于,甲基(me、-ch3)、乙基(et、-ch2ch3)、1-丙基(i-pr、i-丙基、-ch2ch2ch3)、2-丙基(i-pr、i-丙基、-ch(ch3)2)、1-丁基(n-bu、n-丁基、-ch2ch2ch2ch3)、2-甲基-1-丙基(i-bu、i-丁基、-ch2ch(ch3)2)、2-丁基(s-bu、s-丁基、-ch(ch3)ch2ch3)、2-甲基-2-丙基(t-bu、t-丁基、-c(ch3)3)、1-戊基(n-戊基、-ch2ch2ch2ch2ch3)、2-戊基(-ch(ch3)ch2ch2ch3)、3-戊基(-ch(ch2ch3)2)、2-甲基-2-丁基(-c(ch3)2ch2ch3)、3-甲-2-丁基(-ch(ch3)ch(ch3)2)、3-甲基-1-丁基(-ch2ch2ch(ch3)2)、2-甲基-1-丁基(-ch2ch(ch3)ch2ch3)、1-己基(-ch2ch2ch2ch2ch2ch3)、2-己基(-ch(ch3)ch2ch2ch2ch3)、3-己基(-ch(ch2ch3)(ch2ch2ch3))、2-甲基-2-戊基(-c(ch3)2ch2ch2ch3)、3-甲基-2-戊基(-ch(ch3)ch(ch3)ch2ch3)、4-甲基-2-戊基(-ch(ch3)ch2ch(ch3)2)、3-甲基-3-戊基(-c(ch3)(ch2ch3)2)、2-甲基-3-戊基(-ch(ch2ch3)ch(ch3)2)、2,3-二甲基-2-丁基(-c(ch3)2ch(ch3)2)、3,3-二甲基-2-丁基(-ch(ch3)c(ch3)3和辛基(-(ch2)7ch3)。
[0087]“芳基”意指通过从母体芳环系统的单个碳原子除去一个氢原子衍生的芳族烃基。例如,芳基可以具有6至20个碳原子(例如c6芳基、c7芳基、c8芳基、c9芳基、c
10
芳基、c
11
芳基、c
12
芳基、c
13
芳基、c
14
芳基、c
12
芳基、c
16
芳基、c
17
芳基、c
18
芳基、c
19
芳基或c
20
芳基)、6至14个碳原子或6至10个碳原子。典型的芳基包括但不限于,从苯(例如,苯基)、取代的苯、萘、蒽、联苯等衍生的基团以及类似基团。
[0088]
术语“环烷基”表示含有3-12个碳原子的,单价或多价的饱和单环,双环或三环体系。在一实施方案中,环烷基包含3-12个碳原子(例如3、4、5、6、7、8、9、10、11或12个碳原子);在另一实施方案中,环烷基包含3-8个碳原子;在又一实施方案中,环烷基包含3-6个碳原子,例如环丙基、环丁基、环戊基或环己基。所述环烷基基团可以独立地未被取代或被一
个或多个本发明所描述的取代基所取代。
[0089]
涉及烷基、芳基、芳基烷基、杂环基、杂芳基、碳环基等的术语“取代的”例如“取代的c
1-c
15
烷基”、“取代的c
6-c
20
芳基”、“取代的芳基烷基”、“取代的c
1-c
20
杂环”和“取代的碳环基”分别意指其中一个或多个氢原子各自独立地被非氢取代基(如羟基、c
1-c
15
烷基、c
1-c
1s
羟烷基或)代替的c
1-c
15
烷基、c
6-c
20
芳基、芳基烷基、c
1-c
20
杂环、碳环基。除非另外表明,否则当术语“取代的”与具有能够取代的两个或更多个部分的基团例如芳基烷基结合使用时,取代基可以连接至芳基部分、烷基部分或两者。
[0090]
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
具体实施方式
[0091]
为了使本领域的技术人员更好地理解本发明的技术方案,下面进一步披露一些非限制实施例以对本发明作进一步的详细说明。
[0092]
本发明所使用的试剂均可以从市场上购得或者可以通过本发明所描述的方法制备而得。
[0093]
本发明所用试剂的简称或化学式所对应的中文含义:
[0094]
简称或化学式中文含义pth10-苯基吩噻嗪ch3cooh乙酸tfa三氟乙酸tsoh对甲苯磺酸tbsotf叔丁基二甲硅基三氟甲磺酸酯sc(otf)3三氟甲磺酸钪mecn乙腈dcm二氯甲烷chcl3三氯甲烷dce1,2-二氯乙烷acetone丙酮ea乙酸乙酯dmpun,n-二甲基丙烯基脲etoh乙醇toulene甲苯dmfn,n-二甲基甲酰胺
et2o乙醚dbu1,8-二氮杂二环十一碳-7-烯quinuclidine奎宁环tmedan,n,n
′
,n
′‑
四甲基乙二胺dbn1,5-二氮杂双环[4.3.0]-5-壬烯et3n三乙胺dabcon,n-二甲基乙醇胺
[0095]
本发明所用试剂的简称或化学式所对应的结构或cas号:
[0096][0097][0098]
本发明中,“equiv”表示当量,“mmol”表示毫摩尔;“μl”表示微升;“ml”表示毫升;“mg”表示毫克。
[0099]
实施例1:光敏剂的筛选
[0100][0101]
将如表1所述光敏剂(0.002mmol),co(dmgh)(dmgh2)pycl(1.4mg,0.004mmol),dabco(22.4mg,0.2mmol,2equiv)和乙酸(1.2μl,0.02mmol)混合,于氮气氛围条件下加入1.0ml乙腈,化合物1a(28.2mg,0.1mmol,1.0equiv)和化合物1-b(22mg,0.25mmol,2.5equiv),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率,结果如表1所述。柱层析分离纯化,得到化合物1(无色油状液体)。取适量所得化合物1进行氢谱和碳谱检测,结果如下:
[0102]1h nmr(400mhz,chloroform-d)δ7.58(dd,j=2.7,1.5hz,1h),7.54(dt,j=7.6,1.2hz,1h),7.33(t,j=7.9hz,1h),7.09(ddd,j=8.4,2.7,0.9hz,1h),3.92-3.83(m,7h),3.23-3.16(m,4h)。
[0103]
13
c nmr(101mhz,chloroform-d)δ167.33,151.22,131.03,129.18,121.04,120.04,116.40,66.79,52.13,49.14。
[0104]
表1:光敏剂的筛选
[0105]
实验光敏剂化合物1的转化率产物产率1[ir(ppy)2(dtbbpy)]pf6>90%22%24-czlpn>90%13%3[ir(df(cf3)(ppy)2(dtbbpy)]pf6>90%11%4[ir(p-f(cf3)ppy)2(bpy)]pf6>90%20%5[ir(p-cf
3-ppy)2(bpy)]pf6>90%16%6[ir(dtbppy)2(dtbbpy)]pf6>90%19%7[ir(m-cf3(cf3)ppy)2bpy]pf6>90%17%8[ir(dtbppy)2(bpy)]pf6>90%16%9[ir(ppy)2(bpy)]pf6>90%19%10fac-ir(ppy)3>90%14%11[ir(dmppy)2(dtbbpy)]pf6>90%20%12[ir(ppycf3)2bpy]pf6>90%20%13[ir(df(me)ppy2)(dtbbpy)]pf6>90%10%14[ir(df(cf3)ppy)2bpy]pf6>90%18%15[ru(dtbbpy)3](pf6)2>90%13%16[ru(bpz)3](pf6)>90%9%17ru(phen)3(pf6)2>90%33%
189h-thioxanthen-9-one>90%0%19pth>90%2%20eosiny>90%22%
[0106]
结论:在钴肟复合物、酸和碱存在并于惰性气体氛围条件下,通过加入光敏剂能促使化合物1a和化合物1-b在光照条件下反应得到化合物1;光敏剂可以为铱类光敏剂、钌类光敏剂或有机光敏剂;其中铱类光敏剂优选包括[ir(ppy)2(dtbbpy)]pf6、[ir(p-f(cf3)ppy)2(bpy)]pf6、[ir(dmppy)2(dtbbpy)]pf6、[ir(ppy)2(bpy)]pf6、[ir(dtbppy)2(dtbbpy)]pf6或[ir(ppycf3)2bpy]pf6中的至少一种,更优选包括[ir(ppy)2(dtbbpy)]pf6;钌类光敏剂优选包括ru(phen)3(pf6)2;有机光敏剂优选包括eosiny;在所有光敏剂中,最优选包括ru(phen)3(pf6)2,有利于提高产物产率。
[0107]
实施例2:钴肟复合物的筛选
[0108][0109]
将ru(phen)3(pf6)2(0.002mmol),如表2所述钴肟复合物(0.004mmol),dabco(表2中实验2-5中dabco的投料量为56mg,0.5mmol,5equiv;表2中实验1中dabco的投料量为0.2mmol)和乙酸(1.2μl,0.02mmol)混合,于氮气氛围条件下加入1.0ml乙腈,化合物1a(28.2mg,0.1mmol,1.0equiv)和化合物1-b(22mg,0.25mmol,2.5equiv),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率,结果如表2所述。柱层析分离纯化,得到化合物1(无色油状液体)。取适量所得化合物1进行氢谱和碳谱检测,结果同实施例1。
[0110]
表2:钴肟复合物的筛选
[0111]
实验钴肟复合物化合物1的转化率产率1co(dmgh)(dmgh2)pycl>90%36%2co(dmgh)(dmgh2)pycl>90%53%2co(dmgbf2)2(h2o)2>90%20%3co(dmgh)2(4-cnpy)cl>90%46%4co(dmgh)(dmgh2)cl2>90%60%5co(dmgh)2cl(dmap)>90%45%
[0112]
注:实验1中dabco的投料量为0.2mmol。
[0113]
结论:在光敏剂、酸和碱存在并于惰性气体氛围条件下,通过加入钴肟复合物能促使化合物1a和化合物1-b在光照条件下反应得到化合物1;其中钴肟复合物优选包括co(dmgh)(dmgh2)pycl、co(dmgh)2(4-cnpy)cl、co(dmgh)(dmgh2)cl2、co(dmgh)2cl(dmap),更优
选包括co(dmgh)(dmgh2)cl2,有利于提高产物产率。
[0114]
实施例3:酸的筛选
[0115][0116]
将光敏剂ru(phen)3(pf6)2(0.002mmol),钴肟复合物co(dmgh)(dmgh2)cl2(0.004mmol),dabco(56mg,0.5mmol,5equiv)和如表3所示酸(0.02mmol)混合,于氮气氛围条件下加入1.0ml乙腈,化合物1a(28.2mg,0.1mmol,1.0equiv)和化合物1-b(22mg,0.25mmol,2.5equiv),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率,结果如表3所述。柱层析分离纯化,得到化合物1(无色油状液体)。取适量所得化合物1进行氢谱和碳谱检测,结果同实施例1。
[0117]
表3:酸的筛选
[0118]
实验酸化合物1的转化率产率1ch3cooh(0.02mmol)>90%60%2tfa(0.02mmol)>90%38%3sc(otf)3(0.02mmol)>90%68%4tsoh(0.02mmol)>90%51%5tbsotf(0.02mmol)>90%38%6sc(otf)3(0.01mmol)>90%62%7sc(otf)3(0.03mmol)>90%72%8sc(otf)3(0.04mmol)>90%77%9sc(otf)3(0.05mmol)>90%74%10sc(otf)3(0.06mmol)>90%64%11sc(otf)3(0.08mmol)>90%53%
[0119]
结论:在化合物1的制备方法中,所述酸优选为ch3cooh或sc(otf)3,更优选为sc(otf)3。所述酸与化合物1a的投料摩尔比优选为0.30∶1.00-0.50∶1.00,更优选为0.40∶1.00。
[0120]
实施例4:溶剂的筛选
[0121][0122]
将ru(phen)3(pf6)2(0.002mmol),co(dmgh)(dmgh2)pycl(1.4mg,0.004mmol),dabco(56mg,0.5mmol,5equiv)和乙酸(1.2μl,0.02mmol)混合,于氮气氛围条件下分别加入1.0ml如表4所示溶剂,化合物1a(28.2mg,0.1mmol,1.0equiv)和化合物1-b(22mg,0.25mmol,2.5equiv),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率,结果如表4所述。柱层析分离纯化,得到化合物1(无色油状液体)。取适量所得化合物1进行氢谱和碳谱检测,结果同实施例1。
[0123]
表4:溶剂的筛选
[0124]
实验溶剂化合物1的转化率产率1mecn>90%77%2dcm>90%27%3chcl3>90%4%4dce>90%39%5acetone>90%32%6ea>90%8%7dmpu>90%痕量8etoh>90%6%9toulene>90%9%10dmf>90%40%11et2o>90%6%
[0125]
结论:所述溶剂优选为乙腈,有利于提高产物产率。
[0126]
实施例5:碱的筛选
[0127][0128]
将ru(phen)3(pf6)2(0.002mmol),co(dmgh)(dmgh2)pycl(1.4mg,0.004mmol),如表5所示碱(投料量如表5所示)和如表5所示酸(投料量如表5所示)混和(表5中实验14还加入
50mg分子筛,其他实验组不加入分子筛),于氮气氛围条件下分别加入1.0ml乙腈,化合物1a(28.2mg,0.1mmol,1.0equiv)和化合物1-b(22mg,0.25mmol,2.5equiv),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率,结果如表5所述。柱层析分离纯化,得到化合物1(无色油状液体)。取适量所得化合物1进行氢谱和碳谱检测,结果同实施例1。
[0129]
表5:碱的筛选
[0130][0131][0132]
结论:在化合物1的制备过程中,所述碱优选为quinuclidine或dabco,更优选为dabco,有利于提高产物产率。所述碱与化合物1a的投料摩尔比优选为5.0∶1.0~9.0∶1.0,更优选为6.0∶1.0~7.0∶1.0,有利于提高产物产率。
[0133]
另外,分子筛的加入(表5的实验14)有利于提高产物产率。
[0134]
实施例6:控制实验
[0135][0136]
将如表6所示的光敏剂(0.002mmol,0.02equiv.),如表6所示的钴肟复合物(0.004mmol),如表6所示的碱(0.7mmol,7equiv),如表6所示的酸(0.04mmol)和50mg分子筛混和,于氮气氛围条件下分别加入1.0ml乙腈,化合物1a(28.2mg,0.1mmol,1.0equiv)和化合物1-b(22mg,0.25mmol,2.5equiv),在室温条件下,表6中实验1~4于30w蓝光灯照射20小时,表6中实验5在暗处放置20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率,结果如表6所述。柱层析分离纯化,得到化合物1(无色油状液体)。取适量所得化合物1进行氢谱和碳谱检测,结果同实施例1。
[0137]
表6:控制实验各试剂
[0138][0139]
结论:光敏剂和光照为反应的必要条件,缺失任何一个条件,反应均无法进行;另外,若要获得高产率的产物,除光敏剂和光照外,钴肟复合物、酸和碱也缺一不可;光敏剂、钴肟复合物、酸、碱和光照彼此相互协同,共同促进产物产率的提高。
[0140]
实施例7:化合物1的制备
[0141]
[0142]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.)和50mg分子筛混和,在氮气氛围的条件下加入1.0ml乙腈,化合物1a(28.2mg,0.1mmol,1equiv.)和化合物1-b(22mg,0.25mmol,2.5equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为85%。柱层析分离纯化,得到无色油状液体化合物1,产率为82%。取适量所得化合物1进行氢谱和碳谱检测,结果同实施例1。
[0143]
实施例8:化合物2的制备
[0144][0145]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.)和50mg分子筛混和,在氮气氛围条件下加入1.0ml乙腈,28.2mg化合物55-a(0.1mmol,1equiv.)和化合物2-b(32mg,0.25mmol,2.5equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为80%。柱层析分离纯化,得到无色油状液体化合物2,产率为71%。取适量所得化合物2进行氢谱和碳谱检测,结果如下:
[0146]1h nmr(400mhz,chloroform-d)δ7.60(t,j=2.1hz,1h),7.48(d,j=7.6hz,1h),7.29(t,j=7.9hz,1h),7.13(d,j=7.5hz,1h),3.89(s,3h),3.72(q,j=5.7,5.2hz,4h),2.74(td,j=12.2,2.6hz,2h),1.82(dt,j=13.0,2.7hz,2h),1.56(q,j=6.1hz,3h),1.41(ddd,j=15.1,11.8,3.6hz,2h)。
[0147]
13
c nmr(101mhz,chloroform-d)δ167.6,151.6,130.9,129.1,121.0,120.3,117.1,60.4,52.1,49.8,39.3,32.2,32.0。
[0148]
实施例9:化合物3的制备
[0149][0150]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.)和50mg分子筛混和,在氮气氛围条件下加入1.0ml乙腈,化合物1a(28.2mg,0.1mmol,1equiv.)和化合物3-b(36mg,0.25mmol,2.5equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为88%。柱层析分离纯化,得到无色油状液体化合物3,产率为80%。取适量所得化合物3进行氢谱和碳谱检测,结果如下:
[0151]1h nmr(600mhz,chloroform-d)δ7.56(d,j=4.4hz,1h),7.43(t,j=6.3hz,1h),7.23(dq,j=14.6,5.1,4.3hz,1h),7.07(d,j=7.5hz,1h),3.94(d,j=5.1hz,4h),3.84(d,j=5.1hz,3h),3.33(q,j=5.3hz,4h),1.79(q,j=5.2hz,4h)。
[0152]
13
c nmr(151mhz,chloroform-d)δ167.44,150.84,130.97,129.13,120.79,120.32,117.24,107.04,64.38,52.08,47.51,34.40。
[0153]
实施例10:化合物4的制备
[0154][0155]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈,28.2mg化合物55-a(0.1mmol,1equiv.)和化合物4-b(25mg,0.25mmol,2.5equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为89%。柱层析分离纯化,得到无色油状液体化合物4,产率为80%。取适量所得化合物4进行氢谱和碳谱检测,结果如下:
[0156]1h nmr(400mhz,chloroform-d)δ7.62-7.58(m,1h),7.49(dt,j=7.6,1.2hz,1h),
7.29(t,j=7.9hz,1h),7.12(dd,j=8.5,2.6hz,1h),3.89(s,3h),3.88-3.81(m,1h),3.65-3.55(m,2h),2.97(ddd,j=12.8,9.8,3.1hz,2h),2.06-1.96(m,2h),1.69(dtd,j=13.0,9.3,3.8hz,2h)。
[0157]
13
c nmr(101mhz,chloroform-d)δ167.50,151.13,130.93,129.12,120.88,120.46,117.14,67.64,52.11,47.07,33.96。
[0158]
实施例11:化合物5的制备
[0159][0160]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈,化合物1a(28.2mg,0.1mmol,1equiv.)和化合物5-b(29mg,0.25mmol,2.5equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为89%。柱层析分离纯化,得到无色油状液体化合物5,产率为80%。取适量所得化合物5进行氢谱和碳谱检测,结果如下:
[0161]1h nmr(400mhz,chloroform-d)δ7.60(dd,j=2.7,1.5hz,1h),7.48(dt,j=7.6,1.3hz,1h),7.29(1,j=7.9hz,1h),7.11(dd,j=8.4,2.7hz,1h),3.89(s,3h),3.59-3.50(m,2h),3.37(s,4h),2.98(ddd,j=12.6,9.3,3.3hz,2h),2.08-1.92(m,2h),1.70(dtd,j=12.8,8.8,3.8hz,2h)。
[0162]
13
c nmr(101mhz,chloroform-d)δ167.47,151.27,130.90,129.07,120.80,120.33,117.09,75.86,55.62,52.07,46.96,30.44。
[0163]
实施例12:化合物6的制备
[0164][0165]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物6-b(67mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入
1.0ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为70%。柱层析分离纯化,得到无色油状液体化合物6,产率为68%。取适量所得化合物6进行氢谱和碳谱检测,结果如下:
[0166]1h nmr(400mhz,chloroform-d)δ7.60(dd,j=2.7,1.6hz,1h),7.51(tt,j=6.4,1.4hz,5h),7.32(dt,j=8.2,7.0hz,5h),7.25-7.19(m,2h),7.11(dd,j=8.2,2.6hz,1h),3.89(s,3h),3.83-3.72(m,2h),2.79(td,j=11.7,4.0hz,2h),2.66-2.54(m,1h),1.65-1.62(m,2h),0.88(qd,j=7.2,6.0,3.3hz,2h)。
[0167]
13
c nmr(101mhz,chloroform-d)δ167.50,151.37,145.74,130.91,129.08,128.30,126.72,125.84,121.00,120.51,117.09,79.60,52.10,50.04,44.02,26.27。
[0168]
实施例13:化合物7的制备
[0169][0170]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈,化合物1a(28.2mg,0.1mmol,1equiv.)和化合物7-b(50mg,0.25mmol,2.5equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为84%。柱层析分离纯化,得到无色油状液体化合物7,产率为76%。取适量所得化合物7进行氢谱和碳谱检测,结果如下:
[0171]1h nmr(400mhz,chloroform-d)δ7.60(t,j=2.0hz,1h),7.49(d,j=7.6hz,1h),7.29(t,j=7.9hz,1h),7.14(t,j=5.4hz,1h),4.67(s,1h),3.89(s,3h),3.73(dt,j=12.3,2.7hz,2h),3.06(t,j=6.4hz,2h),2.74(td,j=12.4,2.6hz,2h),1.84-1.75(m,2h),1.62(d,j=13.1hz,1h),1.44(s,9h),1.42-1.32(m,2h)。
[0172]
13
c nmr(101mhz,chloroform-d)δ167.46,156.10,151.45,130.92,129.08,121.07,120.53,117.25,79.26,52.09,49.55,46.04,36.40,29.50,28.43。
[0173]
实施例14:化合物8的制备
[0174][0175]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈,化合物1a(28.2mg,0.1mmol,1equiv.)和化合物8-b(40mg,0.25mmol,2.5equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为75%。柱层析分离纯化,得到无色油状液体化合物8,产率为68%。取适量所得化合物8进行氢谱和碳谱检测,结果如下:
[0176]1h nmr(600mhz,chloroform-d)δ7.58(s,1h),7.44(d,j=7.6hz,1h),7.25(t,j=7.6hz,3h),7.18(d,j=7.6hz,2h),7.15(t,j=7.3hz,1h),7.12-7.06(m,1h),3.83(s,3h),3.79(dt,j=13.1,3.0hz,2h),2.83-2.75(m,2h),2.60(tt,j=11.9,3.9hz,1h),1.93-1.87(m,2h),1.87-1.77(m,2h)。
[0177]
13
c nmr(151mhz,chloroform-d)δ167.52,151.71,145.89,130.97,129.11,128.55,126.86,126.38,120.98,120.41,117.21,52.11,50.28,42.40,33.15。
[0178]
实施例15:化合物9的制备
[0179][0180]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈,化合物1a(28.2mg,0.1mmol,1equiv.)和化合物9-b(43mg,0.25mmol,2.5equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸
乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为82%。柱层析分离纯化,得到无色油状液体化合物9,产率为77%。取适量所得化合物9进行氢谱和碳谱检测,结果如下:
[0181]1h nmr(400mhz,chloroform-d)δ7.59(dd,j=2.7,1.5hz,1h),7.48(dt,j=7.6,1.2hz,1h),7.29(t,j=7.9hz,1h),7.10(ddd,j=8.3,2.7,1.0hz,1h),3.89(s,3h),3.82-3.70(m,6h),2.76(td,j=12.4,2.5hz,2h),2.59(dd,j=5.9,3.7hz,4h),2.36(ddt,j=11.3,7.4,3.7hz,1h),1.96(dt,j=12.8,2.9hz,2h),1.65(qd,j=12.1,4.0hz,2h)。
[0182]
13
c nmr(101mhz,chloroform-d)δ167.47,151.19,130.91,129.07,120.76,120.36,117.01,67.17,61.98,52.09,49.79,48.83,27.91。
[0183]
实施例16:化合物10的制备
[0184][0185]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈,化合物1a(28.2mg,0.1mmol,1equiv.)和化合物10-b(39mg,0.25mmol,2.5equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为80%。柱层析分离纯化,得到无色油状液体化合物10,产率为72%。取适量所得化合物10进行氢谱和碳谱检测,结果如下:
[0186]1h nmr(400mhz,chloroform-d)δ7.59(dd,j=2.7,1.5hz,1h),7.49(dt,j=7.7,1.2hz,1h),7.29(t,j=7.9hz,1h),7.15-7.07(m,1h),4.15(q,j=7.1hz,2h),3.89(s,3h),3.73-3.62(m,2h),2.83(td,j=11.9,2.8hz,2h),2.44(tt,j=11.1,4.0hz,1h),2.08-1.98(m,2h),1.93-1.80(m,2h),1.25(d,j=7.2hz,3h)。
[0187]
13
c nmr(101mhz,chloroform-d)δ174.73,167.42,151.39,130.93,129.09,121.02,120.57,117.19,60.50,52.09,49.02,40.91,27.92,14.24。
[0188]
实施例17:化合物11的制备
[0189][0190]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈,化合物1a(28.2mg,0.1mmol,1equiv.)和化合物11-b(39mg,0.25mmol,2.5equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为63%。柱层析分离纯化,得到无色油状液体化合物11,产率为59%。取适量所得化合物11进行氢谱和碳谱检测,结果如下:
[0191]1h nmr(400mhz,chloroform-d)δ7.64-7.59(m,1h),7.50(d,j=7.6hz,1h),7.30(t,j=7.9hz,1h),7.18-7.10(m,1h),4.17(q,j=7.1hz,2h),3.89(s,3h),3.73(ddt,j=12.5,3.5,1.5hz,1h),3.51(dt,j=12.5,4.0hz,1h),3.09(dd,j=12.4,9.7hz,1h),2.92-2.83(m,1h),2.67(s,1h),2.03(dd,j=9.7,5.5hz,1h),1.88-1.79(m,1h),1.75-1.64(m,2h),1.28(t,j=7.1hz,3h)。
[0192]
13
c nmr(101mhz,chloroform-d)δ173.70,167.41,151.44,130.97,129.11,121.24,120.65,117.51,60.59,52.09,49.71,41.32,26.82,24.05,14.24。
[0193]
实施例18:化合物17的制备
[0194][0195]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈,化合物1a(28.2mg,0.1mmol,1equiv.)和化合物17-b(41mg,0.25mmol,2.5equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干
燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为63%。柱层析分离纯化,得到白色固体化合物17,产率为59%。取适量所得化合物17进行氢谱和碳谱检测,结果如下:
[0196]1h nmr(400mhz,chloroform-d)δ8.36(d,j=4.7hz,2h),7.70-7.63(m,1h),7.57(dt,j=7.6,1.2hz,1h),7.36(t,j=7.9hz,1h),7.18(dd,j=8.3,2.6hz,1h),6.55(t,j=4.7hz,1h),4.06-3.98(m,4h),3.93(s,3h),3.38-3.28(m,4h)。
[0197]
13
c nmr(101mhz,chloroform-d)δ167.36,161.57,157.79,151.28,131.03,129.20,121.07,120.77,117.12,110.23,52.15,49.06,43.60。
[0198]
实施例19:化合物18的制备
[0199][0200]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈,化合物1a(28.2mg,0.1mmol,1equiv.)和化合物18-b(46.5mg,0.25mmol,2.5equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为63%。柱层析分离纯化,得到无色油状液体化合物18,产率为59%。取适量所得化合物18进行氢谱和碳谱检测,结果如下:
[0201]1h nmr(400mhz,chloroform-d)δ7.59(dd,j=2.7,1.5hz,1h),7.54(dt,j=7.6,1.2hz,1h),7.32(t,j=7.9hz,1h),7.11(dd,j=8.3,2.7hz,1h),3.89(s,3h),3.63-3.53(m,4h),3.17(t,j=5.2hz,4h),1.48(s,9h)。
[0202]
13
c nmr(101mhz,chloroform-d)δ167.27,154.69,151.17,131.05,129.20,121.28,120.95,117.27,80.01,52.13,49.21,43.46,28.43。
[0203]
实施例20:化合物25的制备
[0204][0205]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,
0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物25-b(48mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入1.0ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为54%。柱层析分离纯化,得到无色油状液体化合物25,产率为51%。取适量所得化合物25进行氢谱和碳谱检测,结果如下:
[0206]1h nmr(600mhz,chloroform-d)δ7.49(dt,j=7.7,1.2hz,1h),7.43(dd,j=2.6,1.5hz,1h),7.30(dd,j=9.0,6.8hz,1h),7.24(t,j=7.9hz,1h),6.95(ddd,j=8.3,2.7,1.0hz,1h),6.47(d,j=8.9hz,1h),6.10(d,j=6.7hz,1h),4.14(d,j=15.5hz,1h),4.02-3.95(m,1h),3.87(s,3h),3.78-3.72(m,1h),3.71-3.63(m,1h),3.19-3.14(m,1h),3.04(ddd,j=20.9,11.7,1.9hz,2h),2.63(d,j=6.2hz,1h),2.03(d,j=13.0hz,1h),1.94(dt,j=12.8,3.2hz,1h)。
[0207]
13
c nmr(151mhz,chloroform-d)δ167.22,163.50,151.23,150.36,138.97,130.89,129.01,121.62,121.42,117.37,117.11,105.26,57.07,56.59,52.12,49.87,35.20,27.85,25.48。
[0208]
实施例21:化合物32的制备
[0209][0210]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物32-b(71mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为49%。柱层析分离纯化,得到无色油状液体化合物32,产率为45%。取适量所得化合物32进行氢谱和碳谱检测,结果如下:
[0211]1h nmr(600mhz,chloroform-d)δ7.34(d,j=7.6hz,1h),7.28(s,1h),7.21-7.18(m,2h),7.01(dd,j=8.1,2.1hz,1h),6.91-6.89(m,1h),6.79(d,j=7.8hz,1h),3.90(s,3h),3.09(d,j=12.5hz,1h),2.96(d,j=12.5hz,1h),2.92(ddd,j=17.2,6.7,1.9hz,1h),
2.87-2.79(m,2h),2.34-2.28(m,1h),1.85-1.75(m,3h),1.69(dp,j=14.1,3.6hz,1h),1.63-1.60(m,1h),1.50(d,j=13.0hz,1h),1.43(tt,j=13.2,4.5hz,2h),1.25(s,3h),1.24(s,3h),1.23(s,3h),1.04(s,3h)。
[0212]
13
c nmr(151mhz,chloroform-d)δ167.58,148.87,147.24,145.72,134.66,131.05,129.14,126.89,124.24,123.95,118.14,117.35,113.07,52.04,45.38,38.43,37.59,37.51,36.35,33.47,30.07,25.29,24.01,24.00,19.26,18.95,18.73。
[0213]
实施例22:化合物33的制备
[0214][0215]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物33-b(26mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入1.0ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为57%。柱层析分离纯化,得到无色油状液体化合物33,产率为55%。取适量所得化合物33进行氢谱和碳谱检测,结果如下:
[0216]1h nmr(400mhz,chloroform-d)δ7.35(dt,j=7.6,1.3hz,1h),7.33(dd,j=2.6,1.5hz,1h),7.20(t,j=7.8hz,1h),6.84(ddd,j=8.1,2.6,1.0hz,1h),3.88(s,3h),3.77(dd,j=11.0,4.2hz,1h),3.58(dd,j=11.0,6.5hz,1h),3.34(td,j=6.5,4.2hz,1h),1.97-1.83(m,j=6.8hz,1h),0.97(t,j=6.5hz,6h)。
[0217]
13
c nmr(101mhz,chloroform-d)δ167.49,148.44,131.15,129.35,118.81,118.08,114.25,62.61,60.89,52.08,30.12,19.23,19.06。
[0218]
实施例23:化合物34的制备
[0219][0220]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,
0.4equiv.),化合物34-b(29mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为75%。柱层析分离纯化,得到无色油状液体化合物34,产率为71%。取适量所得化合物34进行氢谱和碳谱检测,结果如下:
[0221]1h nmr(400mhz,chloroform-d)δ7.35(dt,j=7.6,1.4hz,1h),7.32(dt,j=2.9,1.5hz,1h),7.20(tt,j=7.8,1.5hz,1h),6.84(ddd,j=8.2,2.7,1.4hz,1h),3.88(d,j=1.3hz,3h),3.78(ddd,j=10.9,4.0,1.3hz,1h),3.58(ddt,j=9.9,6.8,1.5hz,1h),3.42(tt,j=6.6,2.7hz,1h),1.73-1.51(m,2h),1.28-1.12(m,1h),0.97-0.86(m,6h)。
[0222]
13
c nmr(101mhz,chloroform-d)δ167.49,148.27,131.15,129.36,118.84,118.07,114.32,62.28,59.49,52.08,36.58,25.92,15.25,11.65。
[0223]
实施例24:化合物35的制备
[0224][0225]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物35-b(22mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为56%。柱层析分离纯化,得到无色油状液体化合物35,产率为52%。取适量所得化合物35进行氢谱和碳谱检测,结果如下:
[0226]1h nmr(400mhz,chloroform-d)δ7.37(dt,j=7.7,1.3hz,1h),7.32(dd,j=2.6,1.6hz,1h),7.22(t,j=7.8hz,1h),6.84(ddd,j=8.1,2.6,1.1hz,1h),3.88(s,3h),3.76(dd,j=11.0,4.2hz,1h),3.58(dd,j=11.0,5.6hz,1h),3.46(qd,j=6.3,4.2hz,1h),1.60(dtt,j=26.5,14.0,6.7hz,2h),0.97(t,j=7.5hz,3h)。
[0227]
13
c nmr(101mhz,chloroform-d)δ167.47,147.74,131.15,129.34,119.02,118.27,114.31,63.88,56.78,52.09,24.75,10.58。
[0228]
实施例25:化合物36的制备
[0229][0230]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物36-b(19mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为55%。柱层析分离纯化,得到无色油状液体化合物36,产率为50%。取适量所得化合物36进行氢谱和碳谱检测,结果如下:
[0231]1h nmr(400mhz,chloroform-d)δ7.39(dt,j=7.6,1.3hz,1h),7.33(t,j=2.0hz,1h),7.22(t,j=7.9hz,1h),6.85(ddd,j=8.1,2.5,1.0hz,1h),3.88(s,3h),3.70(dtd,j=12.7,10.6,5.3hz,2h),3.56(dd,j=10.4,5.3hz,1h),1.21(d,j=6.3hz,3h)。
[0232]
13
c nmr(101mhz,chloroform-d)δ167.46,147.21,131.16,129.33,119.26,118.54,114.48,65.94,52.10,50.86,17.29。
[0233]
实施例26:化合物37的制备
[0234][0235]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物39-b(34mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为76%。柱层析分离纯化,得到无色油状液体化合物39,产率为70%。取适量所得化合物39进行氢谱和碳谱检测,结果如下:
[0236]1h nmr(400mhz,chloroform-d)δ7.41-7.33(m,5h),7.33-7.26(m,2h),7.15(t,j=7.9hz,1h),6.72(ddd,j=8.1,2.5,1.0hz,1h),4.56(dd,j=6.9,4.1hz,1h),3.97(dd,j
=11.2,4.1hz,1h),3.86(s,3h),3.79(dd,j=11.2,6.9hz,1h)。
[0237]
13
c nmr(101mhz,chloroform-d)δ167.51,147.32,139.71,130.89,129.17,128.89,127.75,126.75,118.91,117.94,114.84,67.22,59.78,52.05。
[0238]
实施例27:化合物40的制备
[0239][0240]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物40-b(30mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为76%。柱层析分离纯化,得到无色油状液体化合物40,产率为70%。取适量所得化合物40进行氢谱和碳谱检测,结果如下:
[0241]1h nmr(400mhz,chloroform-d)δ7.43(dt,j=7.7,1.2hz,1h),7.31(t,j=2.0hz,1h),7.23(t,j=7.9hz,1h),6.88-6.82(m,1h),4.25(t,j=4.0hz,1h),3.98(d,j=4.0hz,2h),3.88(s,3h),3.78(s,3h)。
[0242]
13
c nmr(101mhz,chloroform-d)δ172.40,167.30,146.58,131.20,129.40,120.01,118.46,114.20,62.82,58.32,52.69,52.14。
[0243]
实施例28:化合物42的制备
[0244][0245]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物42-b(37mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取
两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为47%。柱层析分离纯化,得到无色油状液体化合物42,产率为44%。取适量所得化合物42进行氢谱和碳谱检测,结果如下:
[0246]1h nmr(400mhz,chloroform-d)δ7.45(dd,j=2.5,1.5hz,1h),7.43(dt,j=7.7,1.2hz,1h),7.26-7.15(m,5h),6.96(ddd,j=8.1,2.6,1.0hz,1h),4.88(d,j=4.7hz,1h),4.68(td,j=4.9,1.8hz,1h),3.84(s,3h),3.15(dd,j=16.7,5.1hz,1h),2.99(dd,j=16.7,1.8hz,1h)。
[0247]
13
c nmr(101mhz,chloroform-d)δ167.29,147.35,141.14,140.41,131.38,129.52,128.52,127.21,125.63,124.27,119.98,118.47,114.40,72.61,62.85,52.16,39.60。
[0248]
实施例29:化合物43的制备
[0249][0250]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物43-b(53mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为70%。柱层析分离纯化,得到无色油状液体化合物43,产率为64%。取适量所得化合物43进行氢谱和碳谱检测,结果如下:
[0251]1h nmr(600mhz,chloroform-d)δ7.30(dt,j=7.6,1.2hz,1h),7.29-7.26(m,3h),7.25-7.21(m,4h),7.14-7.07(m,5h),6.66(dd,j=8.1,2.5hz,1h),5.09(d,j=4.7hz,1h),4.71(d,j=4.7hz,1h),3.83(s,3h)。
[0252]
13
c nmr(151mhz,chloroform-d)δ167.41,146.76,139.90,137.94,130.86,129.13,128.31,128.28,128.10,127.98,127.74,126.53,118.98,117.98,115.03,77.10,63.58,52.03。
[0253]
实施例30:化合物44的制备
[0254][0255]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物44-b(39mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入3.5ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为62%。柱层析分离纯化,得到无色油状液体化合物44,产率为57%。取适量所得化合物44进行氢谱和碳谱检测,结果如下:
[0256]1h nmr(400mhz,chloroform-d)δ7.32-7.30(m,1h),7.28(dt,j=7.7,1.3hz,1h),7.17(t,j=7.8hz,1h),6.81(ddd,j=8.1,2.6,1.0hz,1h),4.24(d,j=10.0hz,1h),3.88(s,3h),3.32(dd,j=9.5,2.8hz,1h),2.16(pd,j=6.8,2.7hz,1h),1.86-1.71(m,3h),1.65-1.54(m,4h),1.01(d,j=6.8hz,3h),0.98(d,j=6.9hz,3h)。
[0257]
13
c nmr(101mhz,chloroform-d)δ167.66,149.86,131.08,129.27,117.72,117.34,113.49,86.53,64.46,52.02,39.91,37.95,30.56,23.58,23.22,22.89,17.60。
[0258]
实施例31:化合物45的制备
[0259][0260]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物45-b(48mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入1.0ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为67%。柱层析分离纯化,得到无色油状液体化合物45,产率为63%。取适量所得化合物45进行氢谱和碳谱检测,结果如下:
[0261]1h nmr(400mhz,chloroform-d)δ7.43-7.39(m,2h),7.38-7.32(m,2h),7.29(d,j
=7.3hz,2h),7.26(t,j=2.0hz,1h),7.12(t,j=7.9hz,1h),6.72-6.66(m,1h),5.12(s,1h),4.37(s,1h),3.87(s,3h),2.03-1.77(m,5h),1.31(qd,j=9.0,8.1,6.1hz,1h),1.21(q,j=7.4,6.5hz,1h),0.95-0.86(m,2h)。
[0262]
13
c nmr(101mhz,chloroform-d)δ167.51,147.13,140.03,130.84,129.06,128.51,127.82,127.61,118.10,117.42,114.23,84.60,65.18,51.97,39.19,37.97,23.70,23.50。
[0263]
实施例32:化合物48的制备
[0264][0265]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物48-b(41mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入1.0ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为63%。柱层析分离纯化,得到无色油状液体化合物48,产率为58%。取适量所得化合物48进行氢谱和碳谱检测,结果如下:
[0266]1h nmr(400mhz,chloroform-d)δ7.51-7.46(m,2h),7.39-7.29(m,4h),7.27(dd,j=2.6,1.6hz,1h),7.17(t,j=7.9hz,1h),6.71(ddd,j=8.1,2.6,1.0hz,1h),5.13(s,1h),3.86(s,3h),3.74(s,3h)。
[0267]
13
c nmr(101mhz,chloroform-d)δ172.06,167.26,145.86,137.13,131.08,129.24,128.98,128.49,127.25,119.28,117.68,114.30,60.53,52.92,52.04。
[0268]
实施例33:化合物49的制备
[0269][0270]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物49-b(45mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入1.0ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小
时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为58%。柱层析分离纯化,得到无色油状液体化合物49,产率为50%。取适量所得化合物49进行氢谱和碳谱检测,结果如下:
[0271]1h nmr(400mhz,chloroform-d)δ7.42(dt,j=7.6,1.2hz,1h),7.34-7.27(m,4h),7.23(t,j=7.9hz,1h),7.19-7.15(m,2h),6.78(ddd,j=8.1,2.6,1.0hz,1h),4.44(t,j=6.1hz,1h),3.90(s,3h),3.70(s,3h),3.16(qd,j=13.6,6.1hz,2h)。
[0272]
13
c nmr(101mhz,chloroform-d)δ173.26,167.29,146.37,136.07,131.20,129.36,129.27,128.61,127.13,119.59,118.02,114.11,57.48,52.20,52.08,38.50。
[0273]
实施例34:化合物50的制备
[0274][0275]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物50-b(36mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入1.0ml乙腈和化合物1a(28.2mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为53%。柱层析分离纯化,得到无色油状液体化合物50,产率为48%。取适量所得化合物50进行氢谱和碳谱检测,结果如下:
[0276]1h nmr(400mhz,chloroform-d)δ7.40(dq,j=7.6,1.3hz,1h),7.29(dd,j=2.7,1.5hz,1h),7.22(t,j=7.9hz,1h),6.79(ddt,j=8.2,2.5,1.2hz,1h),4.14(t,j=7.1hz,1h),3.88(s,3h),3.71(s,3h),1.80(dt,j=11.8,6.7hz,1h),1.73-1.59(m,2h),0.99(d,j=6.6hz,3h),0.94(d,j=6.5hz,3h)。
[0277]
13
c nmr(101mhz,chloroform-d)δ174.85,167.30,146.96,131.18,129.34,119.57,117.83,114.16,55.07,52.14,52.06,42.23,24.90,22.76,22.19。
[0278]
实施例35:化合物51的制备
[0279][0280]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物51-b(36mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入1.0ml乙腈和化合物51-a(29.6mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为80%。柱层析分离纯化,得到无色油状液体化合物51,产率为75%。取适量所得化合物51进行氢谱和碳谱检测,结果如下
[0281]1h nmr(400mhz,chloroform-d)δ7.42-7.25(m,6h),7.15-7.08(m,2h),4.65(dd,j=6.7,4.2hz,1h),4.01(dd,j=11.2,4.1hz,1h),3.85(dd,j=11.2,6.7hz,1h),3.80(s,3h),2.32(s,3h)。
[0282]
13
c nmr(101mhz,chloroform-d)δ167.6,145.1,139.8,130.0,128.9,128.8,128.1,127.8,126.7,119.0,112.0,67.2,59.5,51.9,17.9。
[0283]
实施例36:化合物52的制备
[0284][0285]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物52-b(36mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入1.0ml乙腈和化合物52-a(35mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为68%。柱层析分离纯化,得到无色油状液体化合物52,产率为62%。取适量所得化合物52进行氢谱和碳谱检测,结果如下1h nmr(400mhz,chloroform-d)δ7.40-7.32(m,5h),7.29-7.26(m,1h),7.19(d,j=8.0hz,1h),7.09(d,j=1.7hz,1h),4.63(dd,j=6.6,4.1hz,1h),4.01(dd,j=11.1,
4.1hz,1h),3.86(dd,j=11.2,6.6hz,1h),3.78(s,3h),3.19-3.09(m,1h),2.21-2.09(m,2h),1.87-1.80(m,2h),1.79-1.66(m,4h).
13
c nmr(101mhz,chloroform-d)δ167.5,144.4,139.7,136.1,128.9,128.3,127.8,126.8,125.4,119.1,112.767.3,59.8,51.8,39.9,32.10,32.09,25.35,25.33.
[0286]
实施例37:化合物55的制备
[0287][0288]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物55-b(36mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入1.0ml乙腈和化合物55-a(37mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为81%。柱层析分离纯化,得到白色固体化合物55,产率为73%。取适量所得化合物55进行氢谱、碳谱和质谱检测,结果如下:
[0289]1h nmr(400mhz,chloroform-d)δ7.42-7.18(m,10h),7.13(d,j=1.6hz,1h),6.99(dd,j=7.4,2.2hz,2h),4.58(dd,j=6.3,4.2hz,1h),4.12-4.00(m,2h),3.82(m,4h),3.64(dd,j=11.2,6.3hz,1h)。
[0290]
13
c nmr(101mhz,chloroform-d)δ167.5,144.9,139.5,138.6,130.7,130.4,129.6,128.9,128.8,128.7,127.6,126.9,126.5,118.9,112.7,67.2,59.0,51.9,38.7。
[0291]
实施例38:化合物59的制备
[0292][0293]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物59-b(36mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入1.0ml乙腈和化合物59-a(32mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取
两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为75%。柱层析分离纯化,得到无色油状液体化合物59,产率为69%。取适量所得化合物59进行氢谱和碳谱检测,结果如下
[0294]1h nmr(400mhz,chloroform-d)δ7.41-7.33(m,5h),7.29(dq,j=6.1,3.9,3.0hz,2h),7.16(t,j=7.9hz,1h),6.73(dd,j=8.2,2.5hz,1h),5.85(ddt,j=17.0,10.3,6.7hz,1h),5.25-5.08(m,2h),4.77(s,1h),4.56(dd,j=6.9,4.1hz,1h),4.32(td,j=6.7,2.4hz,2h),3.98(dd,j=11.4,4.0hz,1h),3.79(dd,j=11.2,6.9hz,1h),2.49(qt,j=6.8,1.4hz,2h),2.04(s,1h)。
[0295]
13
c nmr(101mhz,chloroform-d)δ166.9,147.3,139.7,134.1,131.1,129.1,128.9,127.8,126.8,118.9,118.0,117.3,114.7,67.3,63.9,59.8,33.1。
[0296]
实施例39:化合物61的制备
[0297][0298]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物61-b(36mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入1.0ml乙腈和化合物61-a(51mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为69%。柱层析分离纯化,得到自色固体化合物61,产率为62%。取适量所得化合物61进行氢谱和碳谱检测,结果如下
[0299]1h nmr(400mhz,chloroform-d)δ7.40-7.34(m,5h),7.34-7.29(m,2h),7.15(t,j=7.9hz,1h),6.74(ddd,j=8.1,2.6,0.9hz,1h),4.67-4.61(m,2h),4.55(dd,j=6.8,4.1hz,1h),4.42(d,j=2.6hz,1h),4.30-4.22(m,2h),3.97(ddd,j=13.0,5.5,3.0hz,2h),3.84-3.77(m,2h),1.55(s,3h),1.47(s,3h),1.36(s,3h),1.34(s,3h)。
[0300]
13
c nmr(101mhz,chloroform-d)δ166.2,147.3,139.6,130.7,129.1,128.9,127.8,126.7,119.1,118.2,115.1,109.2,108.9,101.7,70.8,70.4,70.1,67.2,65.1,61.4,59.8,26.5,25.9,25.6,24.1。
[0301]
实施例40:化合物63的制备
[0302][0303]
将ru(phen)3(pf6)2(0.002mmol,0.02equiv.),co(dmgh)(dmgh2)cl2(0.004mmol,0.04equiv.),dabco(78.4mg,0.7mmol,7equiv.),三氟甲磺酸钪(19.7mg,0.04mmol,0.4equiv.),化合物63-b(36mg,0.25mmol)和50mg分子筛混和,在氮气氛围条件下加入1.0ml乙腈和化合物63-a(64mg,0.1mmol,1equiv.),在室温条件下于30w蓝光灯照射20小时,反应结束后,反应液用5ml水和10ml乙酸乙酯稀释。待应液分层后,水相用乙酸乙酯萃取两次(5ml
×
2)。然后合并有机相并用饱和氯化钠水溶液清洗,再用无水硫酸镁干燥,然后将有机相浓缩,加入二溴甲烷作为核磁内标,经核磁氢谱确定反应产率为73%。柱层析分离纯化,得到无色油状液体化合物63,产率为65%。取适量所得化合物63进行氢谱和碳谱检测,结果如下:
[0304]1h nmr(400mhz,chloroform-d)δ7.41-7.33(m,5h),7.32-7.27(m,2h),7.14(t,j=7.9hz,1h),6.70(dd,j=8.0,2.5hz,1h),4.97-4.82(m,1h),4.56(dd,j=7.0,4.1hz,1h),3.97(dd,j=11.1,4.1hz,1h),3.79(dd,j=11.2,7.0hz,1h),2.04-1.97(m,1h),1.94-1.84(m,2h),1.82-1.76(m,1h),1.72-1.66(m,2h),1.61-1.51(m,4h),1.41-1.00(m,21h),0.94(d,j=6.5hz,3h),0.90(d,j=1.9hz,3h),0.89(d,j=2.2hz,6h),0.69(s,3h)。
[0305]
13
c nmr(101mhz,chloroform-d)δ166.5,147.2,139.8,131.7,129.0,128.9,127.7,126.8,118.9,117.7,115.0,74.2,67.2,59.9,56.5,56.3,54.3,44.7,42.6,40.0,39.6,36.8,36.2,35.8,35.5,34.1,32.0,28.7,28.3,28.0,27.5,24.3,23.9,22.9,22.6,21.3,18.7,12.3,12.1。
[0306]
上述实施例仅用以例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟习此项技艺的人士均可在不违背本发明的精神及范畴下,对上述实施例进行修改。因此本发明的权利保护范围,应如权利要求书所列。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。