一种1-[3-(2-氨基-4-乙基-1h-咪唑-5-基)丙基]胍的制备方法
技术领域
[0001]
本发明涉及天然产物合成技术领域,具体涉及一种1-[3-(2-氨基-4-乙基
ꢀ‑
1h-咪唑-5-基)丙基]胍的制备方法。
背景技术:
[0002]
河豚毒素(tetrodotoxin,ttx)和石房蛤毒素(saxitoxin,stx)均是剧毒的非蛋白类生物毒素。ttx是一种氨基全氢化喹唑啉化合物,主要存在于河豚鱼体内,是世界上最致命的生物毒素之一,其毒性比氰化钠强1250倍,0.5mg 即可致死。stx属于海洋麻痹性贝类毒素,其小鼠ld50为10μg/kg,源于海洋或淡水中的某些有毒藻类。河豚毒素和石房蛤毒素都属于钠离子通道抑制剂,其结构中的胍基可特异性地结合于神经细胞膜的钠离子通道上,它可以选择性地抑制钠离子通过神经细胞膜而不影响钾离子的通过,进而阻断神经兴奋的传导。河豚毒素具有多种药用功能:河豚毒素联合多卡因治疗严重心律失常,死亡率大大下降;河豚毒素与茚虫威联合应用有协同镇痛抗炎作用,在晚期给药作用远远超过吗啡,且不产生依赖性;河豚毒素还具有很强的局部麻醉作用,其效力比目前常用的局部麻醉药强千倍以上。河豚毒素虽然有剧毒,但是其高活性和高特异性使其具有潜在的巨大的医药开发价值。
[0003]
河豚毒素的化学名称为(4r,4ar,5r,6s,7s,8s,8ar,10s, 12s)-2-azaniumylidene-4,6,8,12-tetrahydroxy-6-(hydroxymethyl)-2,3,4, 4a,5,6,7,8-octahydro-1h-8a,10-methano-5,7-(epoxymethanooxy)quinazoli n-10-olate,结构式如下:
[0004][0005]
石房蛤毒素的化学名称为:(3as,4r,10as)-4-(carbamoyloxymethyl)-10, 10-dihydrox-y-hexahydropyrrolo[1,2-d]purine-2,6(1h,8h)-diiminium,结构式如下:
[0006][0007]
目前文献报道的生物合成途径中都涉及到的非常关键的中间体是1-[3-(2
‑ꢀ
氨基-4-乙基-1h-咪唑-5-基)丙基]胍,其结构式如下:
[0008]
技术实现要素:
[0009]
为了简化合成路线、提高反应选择性和产品收率且避免使用昂贵的原料,本发明提供一种新的1-[3-(2-氨基-4-乙基-1h-咪唑-5-基)丙基]胍的制备方法。
[0010]
本发明为实现上述发明目的,所采用的技术方案是:一种1-[3-(2-氨基-4
‑ꢀ
乙基-1h-咪唑-5-基)丙基]胍的制备方法,合成路线如下:
[0011][0012]
制备方法具体包括以下步骤:
[0013]
(1)在反应溶剂a中,在碱a作用下,起始原料精氨酸甲酯二盐酸盐与二碳酸二叔丁酯在保护气下反应得到式i所示化合物:三boc保护精氨酸甲酯;
[0014]
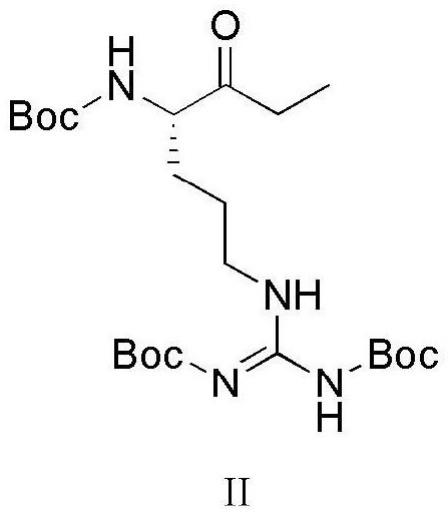
(2)将式i所示化合物:三boc保护精氨酸甲酯溶于有机溶剂b中,在低温的条件下加入乙基溴化镁溶液,在保护气下反应得到式ii所示化合物:三boc 保护(s)-1-(4-氨基-5-氧代庚基)胍;
[0015]
(3)将式ii所示化合物:三boc保护的(s)-1-(4-氨基-5-氧代庚基)胍在有机溶剂c中,经酸a脱boc得到式iii所示化合物:(s)-1-(4-氨基-5-氧代庚基)胍。
[0016]
(4)将式iii所示化合物:(s)-1-(4-氨基-5-氧代庚基)胍在有机溶剂d 中,在碱b的作用下且在保护气下,与氰胺关环得到式iv所示化合物:1-[3-(2
‑ꢀ
氨基-4-乙基-1h-咪唑-5-基)丙基]胍。
[0017]
其中,在所述步骤(1)中,原料精氨酸甲酯二盐酸较为廉价易得。
[0018]
更进一步地,在所述步骤(1)中,所用碱a可以是氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸氢钠、磷酸钠、三乙胺、n,n-二异丙基乙胺、n-甲基吗啉、吡啶或哌啶。
[0019]
更进一步地,在所述步骤(1)中,所述溶剂a选自甲醇、乙醇、异丙醇、叔丁醇、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、四氢呋喃、1,4
‑ꢀ
二氧六环或乙腈。
[0020]
作为优选,在所述步骤(1)中,二碳酸二叔丁酯通过滴加的方式加入到精氨酸甲酯二盐酸盐与碱a以及反应溶剂a所形成的混合溶液中,体系温度控制在10℃以下,滴毕,体系温度升至40-60℃。其中,在该步骤中,本发明通过对反应温度的控制以及将二碳酸二叔丁酯通过滴加的方式加入至反应体系中,最大程度上避免了副反应的产生。
[0021]
作为优选,原料精氨酸甲酯二盐酸盐与二碳酸二叔丁酯的投料摩尔比为 1:3.3-1:10。在此范围内精氨酸甲酯二盐酸盐与二碳酸二叔丁酯反应最完全,收率最高。
[0022]
更进一步地,在所述步骤(2)中,溶剂b选自乙醚、乙二醇二甲醚、甲基叔丁基醚、四氢呋喃、甲基四氢呋喃或1,4-二氧六环。
[0023]
作为优选,在所述步骤(2)中,乙基溴化镁溶液通过滴加的方式加入到式 i所示化合物与反应溶剂b所形成的混合溶液中,体系温度控制在-20℃以下,滴毕,体系温度升至20-30℃。其中,在该步骤中,本发明通过对反应温度的控制以及将乙基溴化镁溶液通过滴加的方式加入至反应体系中,最大程度上避免了副反应的产生。
[0024]
作为优选,在所述步骤(2)中,式i所示化合物与乙基溴化镁溶液的投料摩尔比为1:5-1:20。
[0025]
更进一步地,在所述步骤(3)中,溶剂c选自甲醇、乙醇、异丙醇、正丁醇、1,4-二氧六环、乙酸乙酯或二氯甲烷。
[0026]
作为优选,在所述步骤(3)中,酸a为盐酸、硫酸、磷酸、三氟乙酸和三氯乙酸中的一种。
[0027]
更进一步地,所述步骤(4)具体为:将式iii所示化合物加入到有机溶剂d 中,加入碱b,加毕,体系温度控制在85-95℃;所述有机溶剂d选自甲醇、乙醇、异丙醇、正丁醇、1,4-二氧六环、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜和四氢呋喃中的一种。
[0028]
作为优选,在所述步骤(4)中,碱b选自氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸氢钠、三乙胺或n,n-二异丙基乙胺。
[0029]
作为优选,在所述步骤(4)中,式iii所示化合物与碱b的投料摩尔比为 1:1-1:3。
[0030]
作为优选,所述步骤(1)、(2)和步骤(4)中的保护气为氮气。
[0031]
本发明同现有技术相比具有以下优点及效果:在本发明以精氨酸甲酯二盐酸盐为
主要原料,避免了昂贵的原料鸟氨酸的使用,从原料上就引入胍基,最大程度上降低了生产所需的原料成本。本发明采用的氰胺关环方法,方法简单,反应活性更高,反应收率高。因此,本发明所述的制备方法新颖,原料廉价易得,反应步骤短,选择性好,总体制备成本低、收率高。
具体实施方式
[0032]
下面结合实施例对本发明做进一步的详细说明,以下实施例是对本发明的解释而本发明并不局限于以下实施例。
[0033]
实施例1:一种1-[3-(2-氨基-4-乙基-1h-咪唑-5-基)丙基]胍的制备方法,合成路线如下:
[0034][0035]
具体包括以下步骤:
[0036]
(1)式i所示化合物的制备:精氨酸甲酯二盐酸盐与二碳酸二叔丁酯的投料摩尔比为1:5,在氮气保护下,向2l四口瓶中,加入精氨酸甲酯二盐酸盐(100 g,1eq),无水乙醇(500ml),n,n-二异丙基乙胺(296.9g,6eq),控制温度不高于10度,滴加二碳酸二叔丁酯(417.9g,5eq),完毕后50度搅拌48h。 hplc检测反应完毕后,旋转蒸发仪浓缩至干,加入200ml水,乙酸乙酯萃取,水洗,干燥,减压浓缩至干,得到式i所示化合物三boc精氨酸甲酯168.4g,产率:90.0%。
[0037]
其氢核磁共振图谱如下:
[0038]1h nmr(400mhz,cdcl3,δppm):1.44(s,9h),1.50(s,9h),1.52(s, 9h),1.60-1.90(m,4h),3.74(s,3h),3.80-4.00(m,2h),4.25-4.40(m, 1h),5.41(d,1h),9.19(s,1h),9.36(s,1h)。
[0039]
其中,上述保护反应中,为了使底物精氨酸甲酯二盐酸盐与碱充分溶解参与反应,所用溶剂应选择质子性溶剂或者极性较大的非质子性溶剂,因此乙醇还可替换为甲醇、异丙醇、叔丁醇、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、四氢呋喃、1,4-二氧六
环或乙腈。
[0040]
所述碱n,n-二异丙基乙胺在反应初期起到中和底物精氨酸甲酯二盐酸盐中的盐酸的作用,而反应过程中起到缚酸剂作用,可选择常用的有机碱或无机碱,因此n,n-二异丙基乙胺还可替换为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸氢钠、磷酸钠、三乙胺、n-甲基吗啉、吡啶或哌啶。
[0041]
(2)式ii所示化合物的制备:式i所示化合物与乙基溴化镁溶液的投料摩尔比为1:8,在氮气保护下,向2l四口瓶中,加入式i所示化合物(100g,1eq),四氢呋喃(500ml),降温,控制-20℃以下滴加乙基溴化镁溶液(2m,819ml, 8eq),完毕后缓慢升温至20-30℃搅拌24h。hplc检测反应完毕后,降温至5~10 度,向反应体系中,加入0.1m盐酸(250ml),搅拌1h后,减压浓缩至剩余400ml 体系,乙酸乙酯萃取,水洗,5%碳酸氢钠水溶液洗,水洗,干燥,减压浓缩至干,得到式ii所示化合物三boc保护(s)-1-(4-氨基-5-氧代庚基)胍61.8g,产率:62.1%。
[0042]
其氢核磁共振图谱如下:
[0043]1h nmr(400mhz,cdcl3,δppm):1.07(t,3h),1.43(s,9h),1.48(s, 9h),1.49(s,9h),1.55-1.90(m,4h),2.53(m,2h),3.43(m,2h),4.34 (dd,1h),5.38(d,1h),8.35(s,1h),11.48(s,1h).
[0044]
其中,由于反应中用到格式试剂,因此反应溶剂应选择能够与该试剂形成稳定络合物的醚类溶剂,该反应所使用溶剂四氢呋喃还可替换为乙醚、乙二醇二甲醚、甲基叔丁基醚、甲基四氢呋喃或1,4-二氧六环。
[0045]
(3)式iii所示化合物的制备:
[0046]
向1l四口瓶中,加入式ii所示化合物(100g,1eq),甲醇(400ml),控温0℃以下滴加入盐酸(100ml),完毕后40度搅拌3h。hplc检测反应完毕后,减压浓缩至干,加入乙酸乙酯(200ml),搅拌1h,过滤,乙酸乙酯(100 ml)淋洗,所得滤饼在40度真空干燥,得到式iii所示化合物:(s)-1-(4-氨基-5-氧代庚基)胍三盐酸盐51.6g,产率:85.0%。
[0047]
其氢核磁共振图谱如下:
[0048]1h nmr(400mhz,cd3od,δppm):1.10(t,3h),1.59-2.04(m,4h), 2.59-2.70(m,2h),3.24(td,2h),4.19(dd,1h).
[0049]
其中,该反应为酸性条件下脱boc反应,反应溶剂应选择对底物ii溶解较好的质子性或非质子性溶剂,所以反应溶剂甲醇可替换为乙醇、异丙醇、正丁醇、1,4-二氧六环、乙酸乙酯或二氯甲烷。
[0050]
所述盐酸在反应中提供质子催化boc保护基分解,因此还可替换为硫酸、磷酸、三氟乙酸或三氯乙酸等能够提供质子并催化boc保护基分解的酸。
[0051]
(4)式iv所示化合物的制备:
[0052]
向1l四口瓶中,加入式iii所示化合物(100g,1eq),乙醇(400ml)he 氢氧化钠(33.8g,2.5eq),搅拌下加入氰胺(71.1,5eq),完毕后95度搅拌3h。 hplc检测反应完毕后,减压浓缩至干,加入乙酸乙酯(200ml)溶解,干燥,过滤,乙酸乙酯(100ml)淋洗,所得滤液减压浓缩干,乙醇重结晶,得到式 iv所示化合物:1-[3-(2-氨基-4-乙基-1h-咪唑-5-基)丙基]胍51.2g,产率: 71.9%。
[0053]
其氢核磁共振图谱如下:
[0054]1h nmr(400mhz,cd3od,δppm):1.17(t,3h),1.83(quin,2h),2.48 (q,2h),2.53(t,2h),3.18(t,2h).
[0055]
其中,反应溶剂应选择对底物和碱有较好溶解性的质子性溶剂或极性较大的非质子性溶剂,因此反应溶剂乙醇可替换为甲醇、异丙醇、正丁醇、1,4-二氧六环、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜或四氢呋喃。
[0056]
所述氢氧化钠起到中和底物iii中的盐酸,因此还可替换为氢氧化钾、碳酸钠、碳酸钾、碳酸氢钠、三乙胺或n,n-二异丙基乙胺。
[0057]
此外,需要说明的是,本说明书中所描述的具体实施例,其零、部件的形状、所取名称等可以不同。凡依本发明专利构思所述的构造、特征及原理所做的等效或简单变化,均包括于本发明专利的保护范围内。本发明所属技术领域的技术人员可以对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,只要不偏离本发明的结构或者超越本权利要求书所定义的范围,均应属于本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。