1.本发明涉及一种四取代联烯衍生物及其合成方法,属于化学有机合成技术领域。
背景技术:
2.取代的联烯是一种常见的重要合成砌块,广泛存在于天然产物、药物和分子材料中。其独特的正交累积π共轭体系赋予的活性也使它们在有机合成中具有高度的通用性和实用性,因此高效合成四取代联烯就显得至关重要。目前其合成策略包括通过氢化铝试剂合成(synthesis 2007,6,795)、烯炔重排反应(catal.sci.technol.2017,7,4570)、1,2-消除反应(chem.commun.2011,47,5384)、β-消除反应(nat.commun.2021,12,728)等。然而这些方法存在一定的缺点,反应条件苛刻,试剂非常活泼,合成步骤复杂,底物的适用范围也十分有限,因此发展四取代联烯一步催化的合成方法仍然非常必要。
技术实现要素:
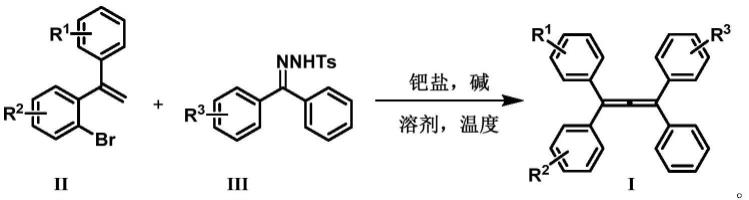
3.本发明的目的在于以易于制备、具有结构多样性和反应效率高的邻溴二苯乙烯ii和二苯酮对甲苯磺酰腙iii为原料,通过1,4-钯迁移/卡宾插入/β-氢消除,一步实现了联烯烯骨架的构建,加以调控邻溴二苯乙烯和二苯酮对甲苯磺酰腙中r1、r2、r3取代基,合成一系列不同结构的四取代联烯衍生物。
4.本发明提供一种四取代联烯衍生物,其分子结构式i如下:
[0005][0006]
r1,r2,r3分别独立选自氢、甲基、乙基、叔丁基、甲氧基、三氟甲基、氟、氯、三氟甲氧基、氰基,取代基的个数为1-5个。
[0007]
本发明提供一种四取代联烯衍生物i的合成方法,以邻溴二苯乙烯ii为起始原料,过渡金属钯盐为催化剂,在碱性条件下,在溶剂中与二苯酮对甲苯磺酰腙iii,通过1,4-钯迁移/卡宾插入/β-氢消除过程,一步生成四取代联烯衍生物i。
[0008]
邻溴二苯乙烯ii的分子结构式如下:
[0009][0010]
r1,r2独立选自氢、甲基、乙基、叔丁基、甲氧基、三氟甲基、氟、氯、三氟甲氧基、氰基,取代基的个数为1-5个;
[0011]
二苯酮对甲苯磺酰腙iii的分子结构式如下:
[0012][0013]
r3选自氢、甲基、乙基、叔丁基、甲氧基、三氟甲基、氟、氯、三氟甲氧基、氰基,取代基的个数为1-5个;
[0014]
合成路线如下述反应式所示:
[0015][0016]
其中:过渡金属钯盐选自氯化钯(pdcl2)、醋酸钯、四苯基膦钯(pd(pph3)4)、三氟乙酸钯、双三苯基膦二氯化钯的一种或两种以上,邻溴二苯乙烯ii与催化剂的摩尔比为1:0.01-1:0.5;
[0017]
邻溴二苯乙烯ii与二苯酮对甲苯磺酰腙iii的摩尔比为1:1-1:3;
[0018]
碱选自碳酸锂、碳酸钠、碳酸钾、碳酸铯、磷酸钾、醋酸钠、醋酸钾、醋酸铯、叔丁醇钾、叔丁醇锂中的一种或两种以上,邻溴二苯乙烯ii与碱的摩尔比为1:0.1-1:5;
[0019]
反应溶剂选自n,n-二甲基甲酰胺(dmf)、二甲基亚砜(dmso)、三氟甲苯、乙腈、甲苯(phme)、1,4-二氧六环、四氢呋喃(thf)中的一种或两种以上的混合物,邻溴二苯乙烯ii在反应溶剂中的摩尔浓度为0.05-1.0m;
[0020]
反应气氛为空气、氧气、氮气、氩气中的一种或两种以上;反应时间为0.1-48小时;反应温度为0-130℃。
[0021]
进一步地,在上述技术方案中,过渡金属钯盐最好是pdcl2。
[0022]
进一步地,在上述技术方案中,反应所用的碱最好是
t
buoli。
[0023]
进一步地,在上述技术方案中,反应最好在氮气气氛中进行。
[0024]
进一步地,在上述技术方案中,邻溴二苯乙烯ii与二苯酮对甲苯磺酰腙iii生成i的反应最佳反应时间为1-4小时。
[0025]
进一步地,在上述技术方案中,最佳反应温度是40-80℃。
[0026]
进一步地,在上述技术方案中,反应最好在非质子极性溶剂二甲基亚砜中进行。
[0027]
进一步地,在上述技术方案中,邻溴二苯乙烯ii与二苯酮对甲苯磺酰腙iii的优选摩尔比为1:2.0。
[0028]
进一步地,在上述技术方案中,邻溴二苯乙烯ii与催化剂(钯盐)的优选摩尔比为1:0.1。
[0029]
进一步地,在上述技术方案中,邻溴二苯乙烯ii与碱的优选摩尔比为1:1-1:4,更优选为1:2.0。
[0030]
本发明以邻溴二苯乙烯和二苯酮对甲苯磺酰腙为起始原料,过渡金属钯盐为催化剂,在碱性条件下,通过1,4-钯迁移及卡宾插入过程,一步生成四取代联烯衍生物。与已报
道的合成四取代联烯衍生物的方法相比较,本发明仅需一步反应、操作简便、条件温和、合成反应效率高,收率在25%-84,优选为41%-88%,且产物具有很好的区域选择性及官能团多样性。本发明合成的四取代联烯广泛存在于天然产物、药物和分子材料中。其独特的正交累积π共轭体系赋予的活性也使它们在有机合成中具有高度的通用性和实用性,同时骨架结构可以作为药物及多种化工用品结构的合成砌块。
[0031]
本发明具有以下优点:
[0032]
1)合成子邻溴二苯乙烯ii和二苯酮对甲苯磺酰腙iii具有结构多样性,可以用来合成不同类型和结构的四取代联烯衍生物i。
[0033]
2)合成子iii易于制备,成本低廉,易于工业化生产。
[0034]
3)四取代联烯衍生物i的合成反应使用价格较低的pdcl2作为催化剂。
[0035]
4)四取代联烯衍生物i合成反应仅需一步构建联烯骨架,产物收率高,最高可达到88%。
[0036]
5)四取代联烯衍生物i合成反应条件较温和,温度范围为40-80℃。
[0037]
6)四取代联烯衍生物i产物有好的立体选择性,及官能团多样性,具有广泛的应用性。
[0038]
总之,本发明利用邻溴二苯乙烯ii和二苯酮对甲苯磺酰腙iii的结构多样性与多反应中心来高效合成不同类型和结构的四取代联烯衍生物i,原料便宜易得,仅需一步反应,得到一系列四取代联烯衍生物结构,操作简便,条件温和,目标产物收率高。
具体实施方式
[0039]
在氮气下,在四氢呋喃溶剂中,邻溴二苯甲酮a与甲基三苯基溴化磷b反应生成邻溴二苯乙烯ii。式a中r1、r2中定义同式ii。
[0040][0041]
具体过程为:将甲基三苯基溴化磷b(10.0mmol)和叔丁醇钾(16.5mmol)加入反应瓶中,氮气下加入10ml四氢呋喃,室温下缓慢滴加邻溴二苯甲酮a(2.0mmol)的四氢呋喃溶液(1m),室温搅拌反应过夜。饱和氯化铵溶液淬灭,乙酸乙酯萃取,饱和食盐水洗涤有机相,无水硫酸钠干燥,减压下除去挥发组份,然后用硅胶柱层析分离(洗脱液为石油醚(60-90℃)/乙酸乙酯,v/v=50:1),得到目标产物ii。目标产物通过核磁共振谱和高分辨质谱测定得到确认。
[0042]
下述实施例的原料2a采用如下文献方法制备得到:
[0043]
q.n.wang,r.j.chen,j.lou,d.h.zhang,y.g.zhou,and z.k.yu,acs catal.2019,9,11669-11675.
[0044]
在乙醇溶液中,二苯乙酮c与对甲苯磺酰肼d反应生成二苯乙酮对甲苯磺酰腙iii。式c中r3中定义同式iii。
[0045][0046]
具体过程为:向100ml单口瓶中加入二苯乙酮(10mmol),tsnhnh2(10mmol,1.8623g),乙醇50ml,65℃下回流2h。停止反应后取出磁子,减压浓缩至20ml左右,放入冰箱降温结晶。重结晶后,抽滤,pe洗涤,干燥得目标产物白色固体iii。目标产物通过核磁共振谱得到确认。
[0047]
下述实施例的原料3a、3b用如下文献方法制备得到:
[0048]
f.huang,z.q.liu,q.n.wang,j.lou,z.k.yu,org.lett.2017,19,3660-3663.
[0049]
通过下述实施例有助于进一步理解本发明,但本发明的内容并不仅限于此。
[0050]
实施例1
[0051][0052]
在手套箱中,依次称取pdcl2(0.03mmol),
t
buli(0.6mmol),二苯酮对甲苯磺酰腙3a(0.6mmol),n2下加入邻溴二苯甲酮2a(0.3mmol),二甲基亚砜3ml,放入60℃的油浴中反应2小时。反应结束后,将混合物冷却至室温,然后用硅胶柱层析分离(洗脱液为石油醚(60-90℃)/乙酸乙酯,v/v=50:1),得到白色固体目标产物1a(86mg,收率84%)。目标产物通过核磁共振谱测定得到确认。
[0053]
化合物表征数据
[0054]
1,1,3,3-四苯基-1,2-丙二烯衍生物(1a),无色液体.1h nmr(400mhz,cdcl3)δ7.54
–
7.46(m,8h),7.44
–
7.32(m,12h).
13
c{1h}nmr(100mhz,cdcl3)δ208.6,136.4,128.6,128.5,127.6,112.7.
[0055]
实施例2
[0056][0057]
在手套箱中,依次称取pdcl2(0.03mmol),
t
buli(0.6mmol),3-甲基二苯酮对甲苯磺酰腙3b(0.6mmol),n2下加入邻溴二苯甲酮2a(0.3mmol),二甲基亚砜3ml,放入60℃的油浴中反应2小时。反应结束后,将混合物冷却至室温,然后用硅胶柱层析分离(洗脱液为石油醚(60-90℃)/乙酸乙酯,v/v=50:1),得到白色固体目标产物1b(77mg,收率72%)。目标产物通过核磁共振谱测定得到确认。
[0058]
化合物表征数据
[0059]
1,1,3-三苯基-3-间甲苯基-1,2-丙二烯衍生物(1b),白色固体.1h nmr(400mhz,cdcl3)δ7.50
–
7.43(m,6h),7.41
–
7.30(m,11h),7.20(d,j=8.0hz,2h),2.40(s,3h).
13
c{1h}nmr(100mhz,cdcl3)δ208.5,137.4,136.6,136.5,133.4,129.3,128.6,128.5,128.4,
127.5,112.6,112.6,21.3.
[0060]
实施例3
[0061][0062]
在手套箱中,依次称取pdcl2(0.03mmol),
t
buli(0.6mmol),二苯酮对甲苯磺酰腙3c(0.6mmol),n2下加入邻溴二苯甲酮2a(0.3mmol),二甲基亚砜3ml,放入60℃的油浴中反应2小时。反应结束后,将混合物冷却至室温,然后用硅胶柱层析分离(洗脱液为石油醚(60-90℃)/乙酸乙酯,v/v=50:1),得到白色固体目标产物1c(74mg,收率66%)。目标产物通过核磁共振谱测定得到确认。
[0063]
化合物表征数据
[0064]
1,1,3-三苯基-3-对甲氧苯基-1,2-丙二烯衍生物(1c),无色液体.1h nmr(400mhz,cdcl3)δ7.49
–
7.44(m,6h),7.42
–
7.35(m,8h),7.32
–
7.30(m,3h),6.95
–
6.91(m,2h),3.85(s,3h).
13
c{1h}nmr(100mhz,cdcl3)δ208.4,159.2,136.7,136.6,129.7,128.6,128.6,128.5,127.5,127.5,114.1,112.5,112.3,77.4,77.1,76.8,55.4.
[0065]
实施例4
[0066][0067]
在手套箱中,依次称取pdcl2(0.03mmol),
t
buli(0.6mmol),二苯酮对甲苯磺酰腙3d(0.6mmol),n2下加入邻溴二苯甲酮2a(0.3mmol),二甲基亚砜3ml,放入60℃的油浴中反应2小时。反应结束后,将混合物冷却至室温,然后用硅胶柱层析分离(洗脱液为石油醚(60-90℃)/乙酸乙酯,v/v=50:1),得到白色固体目标产物1d(87mg,收率88%)。目标产物通过核磁共振谱测定得到确认。
[0068]
化合物表征数据
[0069]
1,1,3-三苯基-3-对氟苯基-1,2-丙二烯衍生物(1d),无色液体.1h nmr(400mhz,cdcl3)δ7.53
–
7.29(m,17h),7.13
–
7.05(m,2h).
13
c{1h}nmr(100mhz,cdcl3)δ208.4,208.4,163.6,161.2,136.3,132.4,132.4,130.2,130.1,128.7,128.7,128.5,128.4,127.7,127.7,115.7,115.5,112.9,111.9.
[0070]
实施例5
[0071]
反应步骤与操作同实施例1,与实施例1不同之处在于,2a与pdcl2的摩尔比为1:0.05。停止反应,经后处理得到目标产物1a(44mg,收率43%)。
[0072]
实施例6
[0073]
反应步骤与操作同实施例1,与实施例1不同之处在于,dmso改为thf。停止反应,经后处理得到目标产物1a(41mg,收率40%)。
[0074]
实施例7
[0075]
反应步骤与操作同实施例1,与实施例1不同之处在于,dmso改为phme。停止反应,经后处理得到目标产物1a(31mg,收率30%)。
[0076]
实施例8
[0077]
反应步骤与操作同实施例1,与实施例1不同之处在于,pdcl2改为pd(pph3)4。停止反应,经后处理得到目标产物1a(26mg,收率25%)。
[0078]
实施例9
[0079]
反应步骤与操作同实施例1,与实施例1不同之处在于,pdcl2改为pd(oac)2。停止反应,经后处理得到目标产物1a(46mg,收率45%)。
[0080]
实施例10
[0081]
反应步骤与操作同实施例1,与实施例1不同之处在于,n2改为o2。停止反应,经后处理得到目标产物1a(36mg,收率35%)。
[0082]
实施例11
[0083]
反应步骤与操作同实施例1,与实施例1不同之处在于,n2改为空气。停止反应,经后处理得到目标产物1a(41mg,收率40%)。
[0084]
实施例12
[0085]
反应步骤与操作同实施例1,与实施例1不同之处在于,60℃改为40℃。停止反应,经后处理得到目标产物1a(62mg,收率60%)。
[0086]
本发明方法原料易得,操作简便,合成反应条件温和、反应效率高,其官能团具有多样性。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。