1.本发明属于电池材料技术领域,具体涉及链状卤代亚磷酸酯及其制备方法,电解液添加剂、电解液和二次电池。
背景技术:
2.锂离子电池是90年代开始发展起来的一种新型高能二次电池,锂离子电池与其他电池相比,具有能量密度高、体积小质量轻、放电速率快、自放电率低、循环寿命长、无记忆效应优点,在数码产品、动力、以及储能领域得到广泛的应用。
3.由于诸多领域对电池的能量密度要求越来越高,快速充电越发成为趋势,电池材料必然朝着更高镍、更高电压、更大倍率方向发展。目前行业的难点主要集中在以下几个方面:
4.1.体相结构控制:在电压较高情况下,正极由于过度脱锂,层状结构发生剧烈变化,同时伴随着相变及应力的产生,而过度的应力会使材料颗粒开裂粉化或破碎,破坏了材料的体相结构,劣化了循环性能。这一问题一定程度上可以通过元素共掺杂来抵消一定的应力,来达到抑制材料相变的目的;
5.2.界面结构的控制:主要通过引入新的界面包覆来优化界面结构,抑制过渡金属溶出,抑制界面结构变化,从而提高循环寿命;
6.3.抑制界面氧的活性:氧析出往往伴随着产气和过渡金属溶出。通过界面处理和电解液的组合使用,可以降低材料界面产气,从而达到提高材料高温状态下的稳定性,提高循环和存储性能。
7.随着社会需求的不断发展,锂离子电池的使用寿命、高低温性能、安全性能、倍率性能等已不能满足动力电池发展的要求。提升动力电池性能有多种途径,其中添加剂对于电池的电化学性能起着至关重要的作用。到目前为止,人们已开发出数目众多的新型添加剂来改善电池性能。添加剂可以在电极材料表面形成固态电解质界面膜(sei膜和cei膜),锂二次电池的性能很大程度上取决于sei膜,负极界面形成的sei膜抑制了电解液在负极界面的副反应,同时还防止了电解液溶剂共嵌入到负极材料中,减轻了负极材料结构坍塌,同时还可以发挥锂离子通道的作用。
8.在锂二次电池的充放电过程中,正极活性材料在结构上坍塌或发生相变,同时金属离子从正极溶出并在负极被还原,使电池性能劣化。高温会加剧电池性能的劣化。现有研究表明电池热失控的起点来自电解质界面膜(sei膜)的分解,随后电解质不断在正负极材料界面分解放出大量的热,导致安全问题的产生,诸多热失控因素都与电解液直接相关,即电解液是保证二次电池安全性的重要环节。
9.二次电池发展向高能量密度方向发展,正极高镍化是发展趋势,但是在研究和实际应用中发现,高能量密度二次电池在循环过程中易出现高镍材料劣化严重,产气严重以及化学稳定性低的问题。虽然目前公开报道了电解液添加剂,如包括改善电解液稳定性和抑制二次电池在循环过程中产气作用的添加剂,但是在研究和实际应用中发现,目前公开
的添加剂本身的热稳定性和对电解液热稳定的改善作用不理想,导致其提高电解液稳定和抑制产气作用的相关添加剂的作用依然不理想,特别是对于高能量密度二次电池循环过程中易出现严重的产气以及化学稳定性降低等问题的改善效果不理想。从而导致高镍正极在高温储存或循环期间大量产气,导致电池鼓胀同时高温性能变差。
10.鉴于以上情况,需要开发新型的添加剂,以稳定正负极材料结构并降低电池阻抗,同时保证锂二次电池在高温循环和高温存储过程中的稳定性。
技术实现要素:
11.本发明的目的在于克服现有技术的上述不足,提供一种链状卤代亚磷酸酯及其制备方法,以解决现有添加剂热稳定性和其提高电解液高温稳定性和抑制产气作用的效果不理想的技术问题。
12.本发明的另一目的在于提供一种添加剂、含有该添加剂的电解液和含有电解液的二次电池,以解决现有电解液由于热稳定性不理想而导致电池如高温循环性和储存性能不理想的技术问题。
13.为了实现上述发明目的,本发明的一方面,提供了一种链状卤代亚磷酸酯。本发明链状卤代亚磷酸酯的分子结构式如下述通式ⅰ所示:
[0014][0015]
其中,通式ⅰ中的r1选自碳原子数为1~10的烷基、碳原子数为1~10的烯基、碳原子数为1~10的炔基、碳原子数为1~10的链状烷氧基、碳原子数为2~10的链状烯氧基、碳原子数为2~10的链状炔氧基、碳原子数为3~10的环状烷氧基、碳原子数为3~10的环状烯氧基、三甲基硅基、三甲基硅氧基、含卤素的烷基、苯基、联苯基、萘基、吡啶基、噻吩基、卤代苯基、卤代联苯、苯酚基、含烷基的苯酚基、含烯基的苯酚基、含炔基的苯酚基、含腈基的苯酚基、卤代苯酚基、卤代萘酚基中的任一种;
[0016]
r2、r3、r4、r5独立的选自氢原子、卤素原子、碳原子数为6~10的芳香基、碳原子数为6~10的卤代芳香基、碳原子数为1~10的烷基、碳原子数为1~10的卤代烷基、碳原子数为1~10的烯基、碳原子数为1~10的卤代烯基、碳原子数为1~10的炔基、碳原子数为1~10的卤代炔基、碳原子数为2~10的链状烷氧基、碳原子数为2~10的链状烯氧基、碳原子数为2~10的链状炔氧基、碳原子数为3~10的环状烷氧基、碳原子数为3~10的环状烯氧基、碳原子数为3~20的三烷基硅基、碳原子数为3~20的三烷基硅氧基、含芳基硅基、含芳基硅氧基、苯基、联苯基、萘基、吡啶基、噻吩基、苯酚基、含烷基的苯酚基、含烯基的苯酚基、含炔基的苯酚基、含腈基的苯酚基、卤代苯酚基、卤代萘酚基中的任一种;
[0017]
r6、r7独立的选自卤素原子;
[0018]
n为0~10的整数。
[0019]
本发明链状卤代亚磷酸酯所含的亚磷酸基团具有正极成膜功能,可以显著改善正极材料的循环稳定性。亚磷酸酯基团还可以与正极材料析出的单线态氧反应,减少电解质在正极界面的氧化分解。亚磷酸酯基团氧化生成的磷酸酯也能够起到成膜添加剂作用。链状卤代亚磷酸酯所含的其他基团能够起到对亚磷酸基团的成膜增效作用,提高链状卤代亚
磷酸酯正负极界面的稳定性。所含的卤代基团和/或卤素原子赋予链状卤代亚磷酸酯良好浸润性。因此,本发明链状卤代亚磷酸酯能够显著改善电解液的热稳定性,提高其成膜性能和浸润性能。
[0020]
进一步地,r1至r5中的至少一基团为链状基团,链状基团包括直链基团或支链基团。该些链状基团均能够调节电解质界面的膜组分和成膜能力,同时能够提高其与亚磷酸酯基团之间的成膜增效作用,以提升链状卤代亚磷酸酯的热稳定作用。
[0021]
进一步地,r1至r5中的至少一基团为链状基团,且链状基团包含卤素原子、氧原子或不饱和键官能团中的至少一种。
[0022]
具体地,链状基团包含不饱和键官能团时,不饱和键官能团包括碳碳双键、碳碳三键、碳氮双键、碳氮三键、碳氧双键、硫氧双键、磷氧双键、酰胺、酰亚胺、磺酰胺、磺酰亚胺、磷酰胺、磷酰亚胺、羧酸酯、磺酸酯和磷酸酯中的至少一种。
[0023]
具体地,链状基团包含不饱和键官能团时,不饱和键官能团的位置在端基或/和者内侧。
[0024]
该些链状基团能够进一步提高对亚磷酸基团的成膜增效作用,提高链状卤代亚磷酸酯正负极界面的稳定性,提高链状卤代亚磷酸酯正负极界面的稳定性。
[0025]
进一步地,r1至r5中的至少一基团为卤代基团,卤代基团为部分取代或全取代。
[0026]
进一步地,r6至r7所示的卤素原子为氟、氯、溴、碘原子中的至少一种。
[0027]
该些卤代基团和r6至r7所代表卤素原子的提高了链状卤代亚磷酸酯的浸润性。
[0028]
进一步地,链状卤代亚磷酸酯包括如下分子结构式ⅰ1
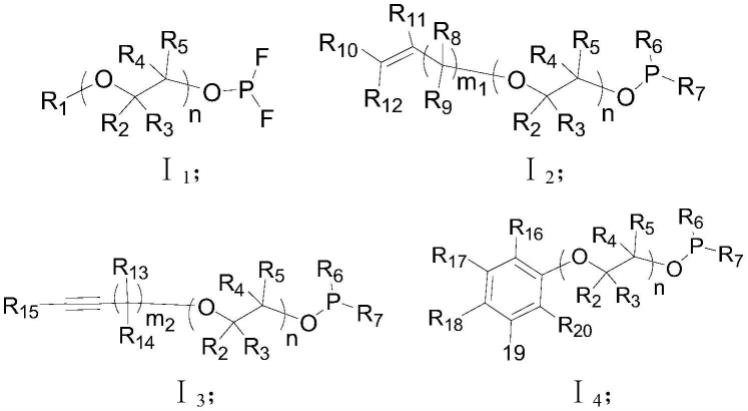
至结构式ⅰ4
中至少一种:
[0029][0030]
其中,通式ⅰ2
至ⅰ4
中的r8、r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
、r
16
、r
17
、r
18
、r
19
、r
20
独立的选自氢原子、卤素原子、碳原子数为1~10的烷基、碳原子数为1~10的烯基、碳原子数为1~10的炔基、碳原子数为1~10的链状烷氧基、碳原子数为2~10的链状烯氧基、碳原子数为2~10的链状炔氧基、碳原子数为3~10的环状烷氧基、碳原子数为3~10的环状烯氧基、三甲基硅基、三甲基硅氧基、含卤素的烷基、苯基、联苯基、萘基、吡啶基、噻吩基、卤代苯基、卤代联苯、苯酚基、含烷基的苯酚基、含烯基的苯酚基、含炔基的苯酚基、含腈基的苯酚基、卤代苯酚基、卤代萘酚基中的一种;m1、m2独立的为0~10的整数。
[0031]
具体实施例中,链状卤代亚磷酸酯包含如下式1至式24所示化合物中的至少一种:
[0032][0033]
结构式ⅰ1
至结构式ⅰ4
所示具体如式1至式24所示的链状卤代亚磷酸酯具有更优异的正极成膜功能和浸润性,热稳定性更优异。
[0034]
本发明的另一方面,提供了本发明链状卤代亚磷酸酯的制备方法。本发明链状卤代亚磷酸酯的制备方法包括如下步骤:
[0035]
将如下结构式ⅰa
所示的反应物a与如下结构式ⅰb
所示的反应物b于第一非水溶液中进行第一取代反应,生成如下结构式ⅰ所示的链状卤代亚磷酸酯产物;
[0036][0037]
其中,式ⅰa
和ⅰ中的r1选自碳原子数为1~10的烷基、碳原子数为1~10的烯基、碳原子数为1~10的炔基、碳原子数为1~10的链状烷氧基、碳原子数为2~10的链状烯氧基、碳原子数为2~10的链状炔氧基、碳原子数为3~10的环状烷氧基、碳原子数为3~10的环状烯氧基、三甲基硅基、三甲基硅氧基、含卤素的烷基、苯基、联苯基、萘基、吡啶基、噻吩基、卤代苯基、卤代联苯、苯酚基、含烷基的苯酚基、含烯基的苯酚基、含炔基的苯酚基、含腈基的苯酚基、卤代苯酚基、卤代萘酚基中的任一种;r2、r3、r4、r5分别独立的选自氢原子、卤素原子、碳原子数为6~10的芳香基、碳原子数为6~10的卤代芳香基、碳原子数为1~10的烷基、碳原子数为1~10的卤代烷基、碳原子数为1~10的烯基、碳原子数为1~10的卤代烯基、碳原
子数为1~10的炔基、碳原子数为1~10的卤代炔基、碳原子数为2~10的链状烷氧基、碳原子数为2~10的链状烯氧基、碳原子数为2~10的链状炔氧基、碳原子数为3~10的环状烷氧基、碳原子数为3~10的环状烯氧基、碳原子数为3~20的三烷基硅基、碳原子数为3~20的三烷基硅氧基、含芳基硅基、含芳基硅氧基、苯基、联苯基、萘基、吡啶基、噻吩基、苯酚基、含烷基的苯酚基、含烯基的苯酚基、含炔基的苯酚基、含腈基的苯酚基、卤代苯酚基、卤代萘酚基中的任一种;r6、r7、rb独立的选自卤素原子;n为0~10的整数。
[0038]
本发明链状卤代亚磷酸酯制备方法制备的目标产物链状卤代亚磷酸酯含有亚磷酸酯基团等功能基团,赋予制备的链状卤代亚磷酸酯具有优异成膜性能和浸润性能,其热稳定性高。另外,链状卤代亚磷酸酯制备方法通过一步反应生成目标产物,目标产物得率高,副反应少,工艺条件易控,其制备获得的链状卤代亚磷酸酯的得率和性能稳定。同时,采用非水溶液作为反应溶剂,还能够降低对目标产物的纯化难度。
[0039]
进一步地,反应物a与反应物b是按照摩尔比为1:(1~6)的比例混合于第一非水溶液中并进行第一取代反应。
[0040]
进一步地,反应物a与第一非水溶液的质量比为1:(1~6)。
[0041]
通过反应物a与反应物b的比例和浓度的调整,提高第一取代反应正向反应速率,提高第一取代反应的效率,并节约反应物用量降低合成成本。
[0042]
进一步地,第一取代反应的温度为-20~40℃。通过对取代反应的温度控制和优化,提高取代反应的效率。
[0043]
具体地,第一取代反应包括先进行前段取代反应再进行后段取代反应的步骤;其中,前段取代反应是将反应物a逐渐加入至含有反应物b的第一非水溶液中直至添加完毕后继续反应1-2小时的取代反应阶段,后段取代反应为反应物a添加完毕并继续反应1-2小时后直至取代反应结束的阶段;且前段取代反应的温度为-20~0℃;后段取代反应的温度为0~40℃。通过将取代反应设置为两阶段,提高第一取代反应的效率,降低副产物的生成,提高目标产物的得率。
[0044]
进一步地,反应物a包括如下结构式a1至a3中的至少一种:
[0045][0046]
其中,通式a1至a3中的r8、r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
、r
16
、r
17
、r
18
、r
19
、r
20
独立的选自氢原子、卤素原子、碳原子数为1~10的烷基、碳原子数为1~10的烯基、碳原子数为1~10的炔基、碳原子数为1~10的链状烷氧基、碳原子数为2~10的链状烯氧基、碳原子数为2~10的链状炔氧基、碳原子数为3~10的环状烷氧基、碳原子数为3~10的环状烯氧基、三甲基硅基、三甲基硅氧基、含卤素的烷基、苯基、联苯基、萘基、吡啶基、噻吩基、卤代苯基、卤代联苯、苯酚基、含烷基的苯酚基、含烯基的苯酚基、含炔基的苯酚基、含腈基的苯酚基、卤代苯酚基、卤代萘酚基中的一种;m1、m2独立的为0~10的整数。该些反应物a能够与反应物b生成上文结构式ⅰ2
至结构式ⅰ4
所示的链状卤代亚磷酸酯,而且能够提高第二取代反应的效率和目标产物的得率。
[0047]
进一步地,第一非水溶液选自乙腈、丙腈、1,3-二氧戊环、四氢呋喃、2-甲基四氢呋喃、2,5-二甲基四氢呋喃、1,4-二氧六环、乙二醇二甲醚、乙二醇二乙醚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、甲酰胺、六甲基磷酰三胺、六甲基亚磷酰三胺、六乙基磷酰三胺、六乙基亚磷酰三胺、二甲基亚砜、二乙基亚砜、二氯甲烷、三氯甲烷、乙醚、丙醚、甲基叔丁基醚、乙基叔丁基醚、乙酸甲酯、乙酸乙酯、丙酸乙酯、乙酸丙酯、碳酸二甲酯、碳酸二乙酯、碳酸甲乙酯、正己烷、正庚烷、环己烷、苯、甲苯、二甲苯中的至少一种。该些非水溶液能够有效溶解两反应物,而且能够有效减少副产物的生成,提高目标产物的得率。
[0048]
进一步地,r6、r7独立的选自氯、溴、碘原子中的任一种时,还包括如下步骤:
[0049]
将经第一取代反应生成的结构式ⅰ所示的链状卤代亚磷酸酯产物与氟化物于第二非水溶液中进行第二取代反应,生成如下述通式ⅰ1
所示的链状二氟亚磷酸酯产物;
[0050][0051]
通过对第一取代反应生成产物进行氟取代反应,使得生成的链状二氟亚磷酸酯产物具有更加优异的浸润性,而且添加电解液中后不附带杂质元素或杂质元素少,从而提高相应电解质或电解液纯度和相应的化学性能。
[0052]
更进一步地,结构式ⅰ所示的链状卤代亚磷酸酯产物与氟化物是按照摩尔比为1:(1~6)的比例混合于第二非水溶液中并进行第二取代反应。
[0053]
更进一步地,结构式ⅰ所示的链状卤代亚磷酸酯产物与第二非水溶液的质量比为1:(1~10)。
[0054]
通过结构式ⅰ所示的链状卤代亚磷酸酯产物与氟化物的比例和浓度的调整,提高第二取代反应的效率,并节约反应物用量降低合成成本。
[0055]
更进一步地,第二取代反应的温度为-20~80℃。通过对取代反应的温度控制和优化,提高取代反应的效率。
[0056]
更进一步地,氟化物选自氟化氢、三乙胺氟化氢、吡啶氟化氢、氟化钾、氟化钠、氟化镁、氟化锌、氟化铝、三氟化锑、五氟化锑、四氟化硫、六氟化硫中的至少一种。该些氟化物能够有效与结构式ⅰ所示的链状卤代亚磷酸酯发生取代反应,提高第二取代反应的效率和提高链状二氟亚磷酸酯产物的得率。
[0057]
更进一步地,第二非水溶液选自乙腈、丙腈、1,3-二氧戊环、四氢呋喃、2-甲基四氢呋喃、2,5-二甲基四氢呋喃、1,4-二氧六环、乙二醇二甲醚、乙二醇二乙醚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、甲酰胺、六甲基磷酰三胺、六甲基亚磷酰三胺、六乙基磷酰三胺、六乙基亚磷酰三胺、二甲基亚砜、二乙基亚砜、二氯甲烷、三氯甲烷、乙醚、丙醚、甲基叔丁基醚、乙基叔丁基醚、乙酸甲酯、乙酸乙酯、丙酸乙酯、乙酸丙酯、碳酸二甲酯、碳酸二乙酯、碳酸甲乙酯、正己烷、正庚烷、环己烷、苯、甲苯、二甲苯中的至少一种。该些非水溶液能够有效溶解两反应物,而且能够有效减少副产物的生成,提高目标产物的得率。
[0058]
本发明的再一方面,提供了电解液添加剂。本发明电解液添加剂含有本发明链状卤代亚磷酸酯或由链状卤代亚磷酸酯制备方法制备的链状卤代亚磷酸酯。由于电解液添加剂含有本发明链状卤代亚磷酸酯,因此,电解液添加剂能够显著改善电解液的热稳定性,提高其成膜性能和浸润性能。
[0059]
本发明的又一方面,提供了一种电解液。本发明电解液包括添加剂,且添加剂为本发明电解液添加剂。电解液由于含有本发明电解液添加剂,也即是含有本发明链状卤代亚磷酸酯,因此,本发明电解液的成膜性能和浸润性能以及热稳定性好,可以显著提升二次电池高低温、循环和存储等性能,提高了二次电池安全性能和综合性能。
[0060]
本发明的还一方面,提供了一种二次电池。本发明二次电池包括电解液,电解液包括添加剂,添加剂为本发明电解液添加剂,链状卤代亚磷酸酯在电解液中的质量浓度为1%-15%。由于本发明二次电池包括本发明电解液。因此,二次电池在高低温下均具有良好的循环性能和存储性能,安全性高,综合化学性能好,使用寿命更长。
具体实施方式
[0061]
为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0062]
本技术中,术语“和/或”,描述关联对象的关联关系,表示可以存在三种关系,例如,a和/或b,可以表示:单独存在a,同时存在a和b,单独存在b的情况。其中a,b可以是单数或者复数。字符“/”一般表示前后关联对象是一种“或”的关系。
[0063]
本技术中,“至少一个”是指一个或者多个,“多个”是指两个或两个以上。“以下至少一项(个)”或其类似表达,是指的这些项中的任意组合,包括单项(个)或复数项(个)的任意组合。例如,“a,b,或c中的至少一项(个)”,或,“a,b,和c中的至少一项(个)”,均可以表示:a,b,c,a-b(即a和b),a-c,b-c,或a-b-c,其中a,b,c分别可以是单个,也可以是多个。
[0064]
应理解,在本技术的各种实施例中,上述各过程的序号的大小并不意味着执行顺序的先后,部分或全部步骤可以并行执行或先后执行,各过程的执行顺序应以其功能和内在逻辑确定,而不应对本技术实施例的实施过程构成任何限定。
[0065]
在本技术实施例中使用的术语是仅仅出于描述特定实施例的目的,而非旨在限制本技术。在本技术实施例和所附权利要求书中所使用的单数形式的“一种”和“该”也旨在包括多数形式,除非上下文清楚地表示其他含义。
[0066]
本技术实施例说明书中所提到的相关成分的重量不仅仅可以指代各组分的具体含量,也可以表示各组分间重量的比例关系,因此,只要是按照本技术实施例说明书相关组分的含量按比例放大或缩小均在本技术实施例说明书公开的范围之内。具体地,本技术实施例说明书中的质量可以是μg、mg、g、kg等化工领域公知的质量单位。
[0067]
一方面,本发明实施例提供了一种链状卤代亚磷酸酯。本发明实施例链状卤代亚磷酸酯的分子结构式如下述通式ⅰ所示:
[0068][0069]
其中,通式ⅰ中的r1选自碳原子数为1~10的烷基、碳原子数为1~10的烯基、碳原子数为1~10的炔基、碳原子数为1~10的链状烷氧基、碳原子数为2~10的链状烯氧基、碳原子数为2~10的链状炔氧基、碳原子数为3~10的环状烷氧基、碳原子数为3~10的环状烯氧基、三甲基硅基、三甲基硅氧基、含卤素的烷基、苯基、联苯基、萘基、吡啶基、噻吩基、卤代
苯基、卤代联苯、苯酚基、含烷基的苯酚基、含烯基的苯酚基、含炔基的苯酚基、含腈基的苯酚基、卤代苯酚基、卤代萘酚基中的任一种;
[0070]
通式ⅰ中的r2、r3、r4、r5独立的选自氢原子、卤素原子、碳原子数为6~10的芳香基、碳原子数为6~10的卤代芳香基、碳原子数为1~10的烷基、碳原子数为1~10的卤代烷基、碳原子数为1~10的烯基、碳原子数为1~10的卤代烯基、碳原子数为1~10的炔基、碳原子数为1~10的卤代炔基、碳原子数为2~10的链状烷氧基、碳原子数为2~10的链状烯氧基、碳原子数为2~10的链状炔氧基、碳原子数为3~10的环状烷氧基、碳原子数为3~10的环状烯氧基、碳原子数为3~20的三烷基硅基、碳原子数为3~20的三烷基硅氧基、含芳基硅基、含芳基硅氧基、苯基、联苯基、萘基、吡啶基、噻吩基、苯酚基、含烷基的苯酚基、含烯基的苯酚基、含炔基的苯酚基、含腈基的苯酚基、卤代苯酚基、卤代萘酚基中的任一种;
[0071]
通式ⅰ中的r6、r7独立的选自卤素原子;
[0072]
通式ⅰ中的n为0~10的整数。
[0073]
本发明实施例链状卤代亚磷酸酯所含的亚磷酸基团具有正极成膜功能,可以显著改善正极材料的循环稳定性。亚磷酸酯基团还可以与正极材料析出的单线态氧反应,减少电解质在正极界面的氧化分解。亚磷酸酯基团氧化生成的磷酸酯也能够起到成膜添加剂作用。链状卤代亚磷酸酯所含的其他基团能够起到对亚磷酸基团的成膜增效作用,提高链状卤代亚磷酸酯正负极界面的稳定性。所含的卤代基团赋予链状卤代亚磷酸酯良好浸润性。因此,本发明实施例链状卤代亚磷酸酯用于电解液中后,能够显著改善电解液的热稳定性,提高电解液成膜性能和浸润性能。
[0074]
实施例中,通式ⅰ中的r1至r5中的至少一基团为链状基团时,该链状基团包括直链基团或/和支链基团。该些含有或为直链基团或/和支链基团的链状基团能够提高其与链状卤代亚磷酸酯所含的亚磷酸酯基团之间的成膜增效作用,以提升链状卤代亚磷酸酯的热稳定作用。
[0075]
在上述r1至r5中的至少一基团为链状基团时,实施例中,该链状基团包含卤素原子、氧原子或不饱和键官能团中的至少一种。具体实施例中,链状基团包含不饱和键官能团时,不饱和键官能团包括碳碳双键、碳碳三键、碳氮双键、碳氮三键、碳氧双键、硫氧双键、磷氧双键、酰胺、酰亚胺、磺酰胺、磺酰亚胺、磷酰胺、磷酰亚胺、羧酸酯、磺酸酯和磷酸酯中的至少一种。另些具体实施例中,该些不饱和键官能团的位置在端基或/和者内侧。该些链状基团能够进一步提高对亚磷酸基团的成膜增效作用,提高链状卤代亚磷酸酯正负极界面的稳定性,提高链状卤代亚磷酸酯正负极界面的稳定性。
[0076]
实施例中,当上述r1至r5中的至少一基团为卤代基团时,卤代基团为部分取代或全取代。具体实施例中,该卤代基团中的卤原子可以是氟、氯、溴或碘中的至少一种。该卤代基团能够进一步提高链状卤代亚磷酸酯产物的浸润性。
[0077]
通式ⅰ中的r6至r7为卤素原子,以改善链状卤代亚磷酸酯的浸润性,实施例中,r6至r7所示的卤素原子为氟、氯、溴、碘原子中的至少一种,进一步实施例中,r6至r7所示的卤素原子为氟,此时,链状卤代亚磷酸酯如下述结构式ⅰ1
所示的链状二氟亚磷酸酯,以进一步提高了链状卤代亚磷酸酯的浸润性,而且添加电解液中后不附带杂质元素或杂质元素少,从而提高相应电解质或电解液纯度和相应的化学性能。
[0078]
实施例中,链状卤代亚磷酸酯至少包括如下分子结构式ⅰ1
至结构式ⅰ4
中至少一种:
[0079][0080][0081]
其中,通式ⅰ2
至ⅰ4
中的r8、r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
、r
16
、r
17
、r
18
、r
19
、r
20
独立的选自氢原子、卤素原子、碳原子数为1~10的烷基、碳原子数为1~10的烯基、碳原子数为1~10的炔基、碳原子数为1~10的链状烷氧基、碳原子数为2~10的链状烯氧基、碳原子数为2~10的链状炔氧基、碳原子数为3~10的环状烷氧基、碳原子数为3~10的环状烯氧基、三甲基硅基、三甲基硅氧基、含卤素的烷基、苯基、联苯基、萘基、吡啶基、噻吩基、卤代苯基、卤代联苯、苯酚基、含烷基的苯酚基、含烯基的苯酚基、含炔基的苯酚基、含腈基的苯酚基、卤代苯酚基、卤代萘酚基中的一种;m1、m2独立的为0~10的整数。该些类型的链状卤代亚磷酸酯具有更优异的正极成膜功能和浸润性,热稳定性更优异。
[0082]
其中,上述通式ⅰ2
至ⅰ4
中的r8、r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
、r
16
、r
17
、r
18
、r
19
、r
20
中的至少一基团为链状基团时,该链状基团包括直链基团或/和支链基团。该些含有或为直链基团或/和支链基团的链状基团能够提高其与链状卤代亚磷酸酯所含的亚磷酸酯基团之间的成膜增效作用,以提升链状卤代亚磷酸酯的热稳定作用。
[0083]
在上述通式ⅰ2
至ⅰ4
中的r8、r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
、r
16
、r
17
、r
18
、r
19
、r
20
中的至少一基团为链状基团时,实施例中,该链状基团包含卤素原子、氧原子或不饱和键官能团中的至少一种。具体实施例中,当该链状基团包含不饱和键官能团时,该不饱和键官能团包括碳碳双键、碳碳三键、碳氮双键、碳氮三键、碳氧双键、硫氧双键、磷氧双键、酰胺、酰亚胺、磺酰胺、磺酰亚胺、磷酰胺、磷酰亚胺、羧酸酯、磺酸酯和磷酸酯中的至少一种。另些具体实施例中,该些不饱和键官能团的位置在端基或/和者内侧。该些链状基团能够进一步提高对亚磷酸基团的成膜增效作用,提高链状卤代亚磷酸酯正负极界面的稳定性,提高链状卤代亚磷酸酯正负极界面的稳定性。
[0084]
实施例中,当上述通式ⅰ2
至ⅰ4
中的r8、r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
、r
16
、r
17
、r
18
、r
19
、r
20
中的至少一基团为卤代基团时,卤代基团为部分取代或全取代。具体实施例中,该卤代基团中的卤原子可以是氟、氯、溴或碘中的至少一种。该卤代基团能够进一步提高链状卤代亚磷酸酯产物的浸润性。
[0085]
基于上述链状卤代亚磷酸酯所含的各基团,具体实施例中,上文链状卤代亚磷酸酯具体至少可以是包含如下式1至式24所示化合物中的至少一种:
[0086][0087][0088]
式1至式24所示的链状卤代亚磷酸酯具有更优异的正极成膜功能和浸润性,热稳定性更优异。当然,式1至式24仅仅是上文结构式ⅰ所示链状卤代亚磷酸酯的部分举例,上文结构式ⅰ所示链状卤代亚磷酸酯还可以是基于上述r1至r
20
所示基团范围内的其他化合物,而且其他化合物也具有优异的正极成膜功能和浸润性以及热稳定性。
[0089]
另一方面,本发明实施例提供了上文本发明实施例链状卤代亚磷酸酯的制备方法。本发明实施例链状卤代亚磷酸酯的制备方法包括如下步骤:
[0090]
s01:将如下结构式ⅰa
所示的反应物a与如下结构式ⅰb
所示的反应物b于第一非水溶液中进行第一取代反应,生成如下结构式ⅰ所示的链状卤代亚磷酸酯产物;根据反应物a、反应物b,该第一取代反应的化学反应式(1)所示:
[0091][0092]
经第一取代反应生成的结构式ⅰ所示的链状卤代亚磷酸酯产物为上文本发明实施例结构式ⅰ所示的链状卤代亚磷酸酯。因此,步骤s01中的式ⅰa
和ⅰ中的r1至r5如上文结构式ⅰ所示的本发明实施例链状卤代亚磷酸酯所含的r1至r5。具体的如r1选自碳原子数为1~10的
烷基、碳原子数为1~10的烯基、碳原子数为1~10的炔基、碳原子数为1~10的链状烷氧基、碳原子数为2~10的链状烯氧基、碳原子数为2~10的链状炔氧基、碳原子数为3~10的环状烷氧基、碳原子数为3~10的环状烯氧基、三甲基硅基、三甲基硅氧基、含卤素的烷基、苯基、联苯基、萘基、吡啶基、噻吩基、卤代苯基、卤代联苯、苯酚基、含烷基的苯酚基、含烯基的苯酚基、含炔基的苯酚基、含腈基的苯酚基、卤代苯酚基、卤代萘酚基中的任一种。r2、r3、r4、r5分别独立的选自氢原子、卤素原子、碳原子数为6~10的芳香基、碳原子数为6~10的卤代芳香基、碳原子数为1~10的烷基、碳原子数为1~10的卤代烷基、碳原子数为1~10的烯基、碳原子数为1~10的卤代烯基、碳原子数为1~10的炔基、碳原子数为1~10的卤代炔基、碳原子数为2~10的链状烷氧基、碳原子数为2~10的链状烯氧基、碳原子数为2~10的链状炔氧基、碳原子数为3~10的环状烷氧基、碳原子数为3~10的环状烯氧基、碳原子数为3~20的三烷基硅基、碳原子数为3~20的三烷基硅氧基、含芳基硅基、含芳基硅氧基、苯基、联苯基、萘基、吡啶基、噻吩基、苯酚基、含烷基的苯酚基、含烯基的苯酚基、含炔基的苯酚基、含腈基的苯酚基、卤代苯酚基、卤代萘酚基中的任一种。n为0~10的整数。
[0093]
基于上述结构式ⅰa
所示的反应物a以及其所含的基团r1所示的基团种类,实施例中,反应物a至少包括如下结构式a1至a3中的至少一种:
[0094][0095]
其中,通式a1至a3中的r8、r9、r
10
、r
11
、r
12
、r
13
、r
14
、r
15
、r
16
、r
17
、r
18
、r
19
、r
20
独立的选自氢原子、卤素原子、碳原子数为1~10的烷基、碳原子数为1~10的烯基、碳原子数为1~10的炔基、碳原子数为1~10的链状烷氧基、碳原子数为2~10的链状烯氧基、碳原子数为2~10的链状炔氧基、碳原子数为3~10的环状烷氧基、碳原子数为3~10的环状烯氧基、三甲基硅基、三甲基硅氧基、含卤素的烷基、苯基、联苯基、萘基、吡啶基、噻吩基、卤代苯基、卤代联苯、苯酚基、含烷基的苯酚基、含烯基的苯酚基、含炔基的苯酚基、含腈基的苯酚基、卤代苯酚基、卤代萘酚基中的一种;m1、m2独立的为0~10的整数。该些反应物a能够与反应物b生成上文结构式ⅰ2
至结构式ⅰ4
所示的链状卤代亚磷酸酯,而且能够提高第二取代反应的效率和目标产物的得率。
[0096]
进一步实施例中,如上文本发明实施例结构式ⅰ所示的链状卤代亚磷酸酯,当上述r1至r5、r8至r
20
中的至少一基团为链状基团时,该链状基团包括直链基团或支链基团。该些链状基团能够提高其与亚磷酸酯基团之间的成膜增效作用,以提升链状卤代亚磷酸酯的热稳定作用。
[0097]
进一步地,当上述r1至r5、r8至r
20
中的至少一基团为链状基团时,该链状基团包含卤素原子、氧原子或不饱和键官能团中的至少一种。具体实施例中,当该链状基团包含不饱和键官能团时,不饱和键官能团包括碳碳双键、碳碳三键、碳氮双键、碳氮三键、碳氧双键、硫氧双键、磷氧双键、酰胺、酰亚胺、磺酰胺、磺酰亚胺、磷酰胺、磷酰亚胺、羧酸酯、磺酸酯和磷酸酯中的至少一种。另些具体实施例中,该链状基团包含不饱和键官能团时,不饱和键官能团的位置在端基或/和者内侧。该些链状基团特别是含该些含不饱和基团时,其能够进一
步提高对亚磷酸基团的成膜增效作用,提高链状卤代亚磷酸酯正负极界面的稳定性,提高链状卤代亚磷酸酯正负极界面的稳定性。
[0098]
实施例中,当上述r1至r5、r8至r
20
中的至少一基团为卤代基团时,卤代基团为部分取代或全取代。具体实施例中,该卤代基团中的卤原子可以是氟、氯、溴或碘中的至少一种。该卤代基团能够进一步提高链状卤代亚磷酸酯产物的浸润性。
[0099]
实施例中,步骤s01中的化合物a可以按照如下方法制备获得:
[0100]
结构ia所示化合物a的制备方法反应式如下:
[0101][0102]
当然,结构ia所示化合物a还可以通过其他合成路线进行合成获得或者市购。当合成化合物a时,具体实施例中,以炔丙基二乙二醇为例说明化合物a的合成,其合成路线的反应式如下所示:
[0103][0104]
按照上述炔丙基二乙二醇的合成反应式,其具体的制备方法如下:
[0105]
sa1:室温条件下向反应釜中加入thf 50ml,然后加入炔丙醇钠7.8g(0.1mol),0℃条件下搅拌0.5h;在第二反应釜中将环氧丙烷的thf溶液(环氧丙烷有效含量为4.5g),在-20℃条件下冷却搅拌0.5h;将炔丙醇钠的thf溶液加入到环氧丙烷的thf溶液中,加入过程全程搅拌并保持温度在-20℃,控制滴加速度保持温度平稳,烯丙醇钠的thf溶液溶液加入完毕后保持温度不变继续反应3h;
[0106]
sa2:向第三个反应釜中加入环氧丙烷的thf溶液(环氧丙烷有效含量为4.5g),在-20℃条件下冷却搅拌0.5h;,然后加入上述第二个反应釜的溶液,加入过程全程搅拌并保持温度在-20℃,控制滴加速度保持温度平稳,加入完毕后保持温度不变继续反应3-6h;反应完成后向反应釜中加入水2ml,缓慢升至室温,常压蒸馏除去溶剂thf,然后加入50ml dmc萃取,过滤后将dmc溶液使用无水硫酸镁干燥12h后常压蒸馏除去溶剂dmc,然后减压蒸馏得到粘稠状无色液体13.3g,产率为92%。即为炔丙基二乙二醇。
[0107]
sa3:在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,分析结果显示gc-ms(esi)calcd for c7h
12
o3[m]
为144.25。1h nmr(400mhz,cdcl3)δ:5.4(br,1h),4.15(s,2h),3.70(t,2h),5.54(t,2h),3.52(m,4h),3.32(s,1h);
13
c nmr(100mhz,cdcl3)δ:78.7,76.4,70.3,69.5,69.2,61.3,60.3。证明所得无色液体为炔丙基二乙二醇。通过卡氏水分测定仪和电位滴定仪测定水分为21ppm,酸度为12ppm,氯离子浓度为2ppm。
[0108]
步骤s01中的式ⅰb
和ⅰ中的r6、r7如上文结构式ⅰ所示的本发明实施例链状卤代亚磷酸酯所含的r6至r7,式ⅰb
中的rb为卤素原子,因此,r6、r7、rb独立的为卤素原子。实施例中,该卤代基团中的卤原子可以是氟、氯、溴或碘中的至少一种。因此,具体实施例中,式ⅰb
所示的
化合物b可以是三卤化磷如pf3、pcl3、pbr3、pi3中的至少一种。
[0109]
实施例中,在化学反应式(1)所示的第一取代反应体系中,控制反应物a与反应物b是按照摩尔比为1:(1~6)进一步为1:(1~2)的比例混合于第一非水溶液中。具体实施例中,反应物a与反应物b的摩尔比为1:1、1:1.5、1:2、1:2.5、1:3、1:3.5、1:4、1:4.5、1:5、1:5.5、1:6等典型而非限制性的比例。
[0110]
另些实施例中,在化学反应式(1)所示的第一取代反应体系中,控制反应物a与第一非水溶液的质量比为1:(1~6)。具体实施例中,反应物a与第一非水溶液的质量比为1:1、1:1.5、1:2、1:2.5、1:3、1:3.5、1:4、1:4.5、1:5、1:5.5、1:6等典型而非限制性的比例。
[0111]
通过反应物a与反应物b的比例和浓度的调整,提高第一取代反应正向反应速率,提高第一取代反应的效率。同时使反应物完全反应、减少杂质的产生,并节约反应物用量降低合成成本。
[0112]
实施例中,第一取代反应的温度为-20~40℃。具体可以是-20℃、-15℃、-10℃、-5℃、0℃、5℃、10℃、15℃、20℃、25℃、30℃、35℃、40℃等典型而非限制性的温度。.通过对取代反应的温度控制和优化,提高取代反应的效率。
[0113]
为了提高第一取代反应的效率,实施例中,在将反应物a与反应物b混合之前,理想是先将反应物a溶解于第一非水溶液中,配制反应物a的第一非水溶液;将反应物b溶解于第一非水溶液中,配制反应物b的第一非水溶液;然后将反应物a的第一非水溶液加入至反应物b的第一非水溶液中进行第一取代反应。
[0114]
进一步实施例中,步骤s01中的第一取代反应包括先进行前段取代反应再进行后段取代反应的步骤;其中,前段取代反应是将反应物a逐渐加入至含有反应物b的第一非水溶液中直至添加完毕后继续反应1-2小时的取代反应阶段,后段取代反应为反应物a添加完毕并继续反应1-2小时后直至取代反应结束的阶段。其中,前段取代反应的温度为-20~0℃,具体可以是-20℃、-15℃、-10℃、-5℃、0℃等典型而非限制性的温度。后段取代反应的温度为0~40℃,具体可以是0℃、5℃、10℃、15℃、20℃、25℃、30℃、35℃、40℃等典型而非限制性的温度。后段取代反应应该是充分的,如直至第一取代反应无任何气体产生为止。通过将取代反应设置为两阶段,提高第一取代反应的效率,降低副产物的生成,提高目标产物的得率。同时有利于避免反应过于剧烈,并吸收反应释放的热量。
[0115]
实施例中,第一非水溶液选自乙腈、丙腈、1,3-二氧戊环、四氢呋喃、2-甲基四氢呋喃、2,5-二甲基四氢呋喃、1,4-二氧六环、乙二醇二甲醚、乙二醇二乙醚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、甲酰胺、六甲基磷酰三胺、六甲基亚磷酰三胺、六乙基磷酰三胺、六乙基亚磷酰三胺、二甲基亚砜、二乙基亚砜、二氯甲烷、三氯甲烷、乙醚、丙醚、甲基叔丁基醚、乙基叔丁基醚、乙酸甲酯、乙酸乙酯、丙酸乙酯、乙酸丙酯、碳酸二甲酯、碳酸二乙酯、碳酸甲乙酯、正己烷、正庚烷、环己烷、苯、甲苯、二甲苯中的至少一种。该些非水溶液能够有效溶解两反应物,而且能够有效减少副产物的生成,提高目标产物的得率。而且便于对目标产物的纯化,如采用浓缩纯化得到纯度高的链状卤代亚磷酸酯。
[0116]
另外,步骤s01的在进行第一取代反应过程中所生成的副产物气体可以用水吸收,以提高环保性或副产物的再利用。
[0117]
经步骤s01中的第一取代反应结束后,还包括对结构式ⅰ所示的链状卤代亚磷酸酯产物进行分离纯化处理的步骤,该纯化处理包括将经第一取代反应后的混合物除去第一非
水溶液后,对粗品进行蒸馏处理或者采用非水溶液进行重结晶并干燥处理。具体实施例中,当粗品流动性差如粘滞挂壁,将粗品溶解于第三非水溶液中进行重结晶处理、固液分离处理并进行干燥处理,获得纯的链状卤代亚磷酸酯产物。其中,干燥为真空干燥,如真空干燥温度为0~80℃,干燥时长为2~6h。当粗品流动性好如不出现粘滞挂壁现象,将粗品直接进行减压蒸馏处理得到获得纯的链状卤代亚磷酸酯产物。如减压蒸馏温度为20~300℃。
[0118]
具体实施例中,用于重结晶的第三非水溶液为乙腈、丙腈、1,3-二氧戊环、四氢呋喃、2-甲基四氢呋喃、2,5-二甲基四氢呋喃、1,4-二氧六环、乙二醇二甲醚、乙二醇二乙醚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、甲酰胺、六甲基磷酰三胺、六甲基亚磷酰三胺、六乙基磷酰三胺、六乙基亚磷酰三胺、二甲基亚砜、二乙基亚砜、二氯甲烷、三氯甲烷、乙醚、丙醚、甲基叔丁基醚、乙基叔丁基醚、乙酸甲酯、乙酸乙酯、丙酸乙酯、乙酸丙酯、碳酸二甲酯、碳酸二乙酯、碳酸甲乙酯、正己烷、正庚烷、环己烷、苯、甲苯、二甲苯中的至少一种。
[0119]
当步骤s01中的结构式ⅰb
中的r6、r7独立的选自氯、溴、碘原子中的任一种也即是反应物b为氯、溴、碘中的至少一种的三卤化磷具体如pcl3、pbr3、pi3时,上述链状卤代亚磷酸酯的制备方法还包括如下步骤s02:
[0120]
步骤s02:将经第一取代反应生成的结构式ⅰ所示的链状卤代亚磷酸酯产物与氟化物于第二非水溶液中进行第二取代反应,生成如下述通式ⅰ1
所示的链状二氟亚磷酸酯产物。
[0121]
具体地,根据结构式ⅰ所示的链状卤代亚磷酸酯产物,该第二取代反应的化学反应式(2)所示,需要说明的是,化学反应式(2)所示结构式ⅰ中的r6、r7均非氟原子:
[0122][0123]
通过对第一取代反应生成产物进行氟取代反应,使得生成的链状二氟亚磷酸酯产物具有更加优异的浸润性,而且添加电解液中后不附带杂质元素或杂质元素少,从而提高相应电解质或电解液纯度和相应的化学性能。为提高链状卤代亚磷酸酯制备方法的效率,在化学反应式(2)所示的第二取代反应中,结构式ⅰ所示的链状卤代亚磷酸酯产物也可不经纯化直接用的粗品或直接向经第一取代反应后的混合物溶液中添加。
[0124]
实施例中,结构式ⅰ所示的链状卤代亚磷酸酯产物与氟化物是按照摩尔比为1:(1~6)的比例混合于第二非水溶液中并进行第二取代反应。具体实施例中,结构式ⅰ所示的链状卤代亚磷酸酯产物与氟化物的摩尔比为1:1、1:1.5、1:2、1:2.5、1:3、1:3.5、1:4、1:4.5、1:5、1:5.5、1:6等典型而非限制性的比例。
[0125]
另些实施例中,结构式ⅰ所示的链状卤代亚磷酸酯产物与第二非水溶液的质量比为1:(1~10)。具体实施例中,链状卤代亚磷酸酯产物与第二非水溶液的质量比为1:1、1:2、1:3、1:4、1:5、1:6、1:7、1:8、1:9、1:10等典型而非限制性的比例。
[0126]
通过上述化学反应式(2)中结构式ⅰ所示的链状卤代亚磷酸酯产物与氟化物的比例和浓度的调整,提高第二取代反应的效率。同时使反应物完全反应、减少杂质的产生,并节约反应物用量降低合成成本。
[0127]
实施例中,第二取代反应的温度为-20~80℃。具体可以是-20℃、-15℃、-10℃、-5
℃、0℃、5℃、10℃、15℃、20℃、25℃、30℃、35℃、40℃、45℃、50℃、55℃、60℃、65℃、70℃、75℃、80℃等典型而非限制性的温度。.通过对取代反应的温度控制和优化,提高取代反应的效率。通过对取代反应的温度控制和优化,提高取代反应的效率。基于该取代反应的温度,第二取代反应应该是充分的,如反应时长为3~12h,具体可以是3h、4h、5h、6h、7h、8h、9h、10h、11h、12h等典型而非限制性的反应时间。
[0128]
实施例中,氟化物选氟化氢(hf)、三乙胺氟化氢(et3n
·
3hf)、吡啶氟化氢(py
·
hf)、氟化钾(kf)、氟化钠(naf)、氟化镁(mgf2)、氟化锌(znf2)、氟化铝(alf3)、三氟化锑(sbf3)、五氟化锑(sbf5)、四氟化硫(sf4)、六氟化硫(sf6)中的至少一种。该些氟化物能够有效与结构式ⅰ所示的链状卤代亚磷酸酯发生取代反应,提高第二取代反应的效率和提高链状二氟亚磷酸酯产物的得率。
[0129]
实施例中,第二非水溶液选自乙腈、丙腈、1,3-二氧戊环、四氢呋喃、2-甲基四氢呋喃、2,5-二甲基四氢呋喃、1,4-二氧六环、乙二醇二甲醚、乙二醇二乙醚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、甲酰胺、六甲基磷酰三胺、六甲基亚磷酰三胺、六乙基磷酰三胺、六乙基亚磷酰三胺、二甲基亚砜、二乙基亚砜、二氯甲烷、三氯甲烷、乙醚、丙醚、甲基叔丁基醚、乙基叔丁基醚、乙酸甲酯、乙酸乙酯、丙酸乙酯、乙酸丙酯、碳酸二甲酯、碳酸二乙酯、碳酸甲乙酯、正己烷、正庚烷、环己烷、苯、甲苯、二甲苯中的至少一种。该些非水溶液能够有效溶解两反应物,而且能够有效减少副产物的生成,提高目标产物的得率。
[0130]
另外,待步骤s02中的第二取代反应结束后,也还包括对结构式ⅰ1
所示的链状二氟亚磷酸酯产物进行分离纯化处理的步骤,该纯化处理包括将经第二取代反应后的混合物除去第二非水溶液后,对粗品进行蒸馏处理或者采用非水溶液进行重结晶并干燥处理。具体实施例中,当粗品流动性差如粘滞挂壁,将粗品溶解于第四非水溶液中进行重结晶处理、固液分离处理并进行干燥处理,获得纯的链状卤代亚磷酸酯产物。其中,干燥为真空干燥,如真空干燥温度为0~80℃,干燥时长为2~6h。当粗品流动性好如不出现粘滞挂壁现象,将粗品直接进行减压蒸馏处理得到获得纯的链状卤代亚磷酸酯产物。如减压蒸馏温度为20~300℃。
[0131]
具体实施例中,用于重结晶的第四非水溶液为乙腈、丙腈、1,3-二氧戊环、四氢呋喃、2-甲基四氢呋喃、2,5-二甲基四氢呋喃、1,4-二氧六环、乙二醇二甲醚、乙二醇二乙醚、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、甲酰胺、六甲基磷酰三胺、六甲基亚磷酰三胺、六乙基磷酰三胺、六乙基亚磷酰三胺、二甲基亚砜、二乙基亚砜、二氯甲烷、三氯甲烷、乙醚、丙醚、甲基叔丁基醚、乙基叔丁基醚、乙酸甲酯、乙酸乙酯、丙酸乙酯、乙酸丙酯、碳酸二甲酯、碳酸二乙酯、碳酸甲乙酯、正己烷、正庚烷、环己烷、苯、甲苯、二甲苯中的至少一种。
[0132]
由上述本发明实施例链状卤代亚磷酸酯制备方法制备的目标产物链状卤代亚磷酸酯含有亚磷酸酯基团等功能基团,赋予制备的链状卤代亚磷酸酯具有优异成膜性能和浸润性能,其热稳定性高。另外,链状卤代亚磷酸酯制备方法通过一步反应生成目标产物,目标产物得率高,副反应少,工艺条件易控,其制备获得的链状卤代亚磷酸酯的得率和性能稳定。同时,采用非水溶液作为反应溶剂,还能够降低对目标产物的纯化难度。另外,还能够有效通过调节反应条件提高反应效率和提高目标产物的得率。
[0133]
再一方面,基于上文本发明实施例链状卤代亚磷酸酯及其制备方法,本发明实施例还提供一种电解液添加剂。本发明实施例电解液添加剂含有上文本发明实施例链状卤代
亚磷酸酯。当然,该电解质盐还可以进一步含有其添加剂,也即是该电解液添加剂可以单独含有上文链状卤代亚磷酸酯,也可以与其添加剂进行复配形成混合添加剂。其他添加剂可以是电解液领域的添加剂,并可以根据需要进行选择其他添加剂的种类和与上文链状卤代亚磷酸酯的配比量。由于电解液添加剂含有上文链状卤代亚磷酸酯,因此,本发明实施例电解液添加剂能够显著改善电解液的热稳定性,提高其成膜性能和浸润性能。
[0134]
基于上文链状卤代亚磷酸酯及其制备方法和电解液添加剂,本发明实施例还提供一种电解液。该电解液含有上文电解液添加剂,也即是含有上文链状卤代亚磷酸酯。因此,本发明实施例电解液的成膜性能和浸润性能以及热稳定性好,可以显著提升二次电池高低温、循环和存储等性能,提高了二次电池安全性能和综合性能。
[0135]
实施例中,以该电解液的总质量为100%计,上文链状卤代亚磷酸酯在电解液中的质量浓度为1%-15%,具体可以是1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%等典型而非限制性的含量。通过控制和调节上文链状卤代亚磷酸酯在电解液中的浓度,一方面充分发挥上文链状卤代亚磷酸酯对电解液热稳定性改善作用,提高其成膜性能和浸润性能;另一方面,可以与电解液中的其它成分起到功能互补的作用,进一步提高电解液的稳定性和安全性以及电化学性能。
[0136]
另外,电解液除了含有上文链状卤代亚磷酸酯之外,还包括电解液必含的组分如溶剂、电解质锂盐或进一步还可以含有应用于电解液中的其他组分,其他组分可以单独发挥作用,也可以与上文链状卤代亚磷酸酯起到增效协同作用。
[0137]
其中,电解液所含的溶剂为非水溶液,定义为第五非水溶液,在一些实施例中,该第五非水溶液可以但不仅仅为碳酸酯类溶剂,其中碳酸酯为链状或环状的碳酸酯。在一些具体实施例中,碳酸酯为链状或环状的碳酸酯。在一些具体实施例中,环状碳酸酯选自碳酸乙烯酯(ec)、碳酸亚乙烯酯(vc)、乙烯基碳酸乙烯酯(vec)、碳酸丙烯酯(pc)、γ-丁内酯中的至少一种;链状碳酸酯选自碳酸二甲酯(dmc)、碳酸二乙酯(dec)、碳酸二丙酯、碳酸甲乙酯(emc)、碳酸甲丙酯、碳酸乙丙酯、甲酸甲酯、甲酸乙酯、甲酸丙酯、乙酸甲酯、乙酸乙酯、乙酸丙酯、丙酸甲酯、丙酸乙酯、丙酸丙酯中的至少一种。
[0138]
具体实施例中,电解质锂盐包括lipf6、libf4、liclo4、lisbf6、liasf6、litdi、lin(so2c2f5)2、lin(so2cf3)2、lin(so3c2f5)2、lin(so2f)2、lin(so2c6f5)2、lin(so3c6f5)2、liso3cf3、liso3c2f5、liso3c4f9、liso3c6h5、liso3c6f5中的一种或多种。
[0139]
具体实施例中,其他添加剂包括但不仅限于1,3-丙磺酸内酯(ps)、1,3-丙烯磺酸内酯(pes)、亚硫酸乙烯酯(es)、硫酸乙烯酯(dtd)、二氟草酸硼酸锂(liodfb)、双草酸硼酸锂(libob)中的一种或多种。
[0140]
另外,上述电解液可以是锂离子电池电解液或锂金属电池电解液。
[0141]
基于上文电解液,本发明实施例还提供了一种二次电池。本发明实施例二次电池包括正极、负极等必要的部件,还包电解液,各部件和电解液按照锂离子电池组装要求进行组装。
[0142]
其中,二次电池所含的电解液为上文本发明实施例电解液。因此,本发明实施例二次电池的在高低温下均具有良好的循环性能和存储性能,安全性高,综合化学性能好,使用寿命更长。
[0143]
二次电池所含的正极可以是常规正极结构,如包括正极集流体和结合在正极集流
体表面的正极活性层,其中,正极活性层包括正极活性材料、粘结剂、导电剂以及增稠剂(如需要)等组分。实施例中,正极活性物质可选自liacoo2(0.5《a《1.3)、lianio2(0.5《a《1.3)、liamno2(0.5《a《1.3)、liamn2o4(0.5《a《1.3)、lia(ni
x
coymnz)o2(0.5《a《1.3,0《x《1,0《y《1,0《z《1,x y z=1)、liani
1-x
co
x
o2(0.5《a《1.3,0《x《1)、liaco
1-x
mn
x
o2(0.5《a《1.3,0≤x《1)、liani
1-x
mn
x
o2(0.5《a《1.3,0≤x《1)、lia(ni
x
coymnz)o4(0.5《a《1.3,0《x《2,0《y《2,0《z《2,x y z=2)、liamn
2-xnx
o4(0.5《a《1.3,0《x《2)、liamn
2-xnx
o4(0.5《a《1.3,0《y《2)、和lianpo4(0.5《a《1.3)中的任何一种或两种以上的混合物;n选自fe、ni、co、mn、zn、al、cr、mg、zr、mo、w、v、ti、b、f和y中的一种或多种。正极活性物质还可以是li-ni/co/mn氧化物;更进一步为lia(ni
x
coymnz)o2,其中0.90≤a≤1.10,0.3≤x≤0.9,0.05≤y《0.5,0.05≤z《0.5,且x y z=1;更进一步为li(ni
x
coymnz)o2,其中0.3≤x≤0.9,0.05≤y《0.5,0.05≤z《0.5,并且x y z=1。具体实施例中,正极活性物质可以为lini
0.6
co
0.2
mn
0.2
o2、lini
0.5
co
0.2
mn
0.3
o2、lini
0.8
co
0.1
mn
0.1
o2、lini
1/3
co
1/3
mn
1/3
o2中的一种或多种的混合。且正极活性物质的质量占正极活性浆料的质量的88%-98%。
[0144]
二次电池所含的负极可以是常规负极结构,如包括负极集流体和结合在负极集流体表面的负极活性层,其中,负极活性层包括能够使锂离子嵌入和脱出的负极活性物质、导电剂、粘结剂、增稠剂(如需要)等组分。实施例中,负极活性物质可选自碳材料(诸如结晶碳、无定形碳、碳复合物和碳纤维)、锂金属、锂与其他元素的合金等能够发生锂离子嵌入和脱出的至少一种。结晶碳的非限制实例包括石墨基材料,如人造石墨、天然石墨、石墨化焦炭、石墨化mcmb、石墨化mpcf等。无定形碳的非限制实例可包括软碳(低温焙烧碳)、硬碳、焦炭、中间相碳微球(mcmb)、中间相沥青基碳纤维(mpcf)等。与锂金属形成合金的其他元素,包括铝、锌、铋、镉、锑、硅、铅、锡、镓或铟等元素中的一种或多种。且负极活性物质的质量占负极活性浆料的质量的90%-96%。
[0145]
需要说明的是,上述正极集流体(或负极集流体)和正极活性材料层(或负极活性材料层)仅提供了一种常用的位置关系,即将正极活性浆料(或负极活性浆料)涂覆于正极集流体(或负极集流体)表面形成正极活性材料层(或负极活性材料层),不应理解为其是对本发明实施例所提供的二次电池的限制。根据实际情况,结合对二次电池性能的要求可对集流体和活性材料进行改变,如将正极活性物质(或负极活性物质)及助剂的混合粉料填充在空心正极集流体(或空心负极集流体)的内部等各种方式。
[0146]
实施例中,形成正极活性成的正极活性浆料和形成负极活性成的负极活性浆料时还需要加入溶剂,该溶剂作用为分散电极活性材料、粘合剂、导电材料等,可以为非水溶液或含水溶剂。为高纯去离子水时,高纯度去离子水的电导率≤3us/cm,非水溶液可包括n-甲基-2-吡咯烷酮(nmp)、二甲基甲酰胺、二甲基乙酰胺、n,n-二甲基氨基丙胺、环氧乙烷、四氢呋喃等,其中的水分含量≤100ppm。
[0147]
实施例中,正极、负极所用导电剂可选自石墨基导电剂、炭黑基导电剂、金属基或金属化合物基导电剂。石墨基导电剂的应用实例包括人造石墨、天然石墨等。炭黑基导电剂的应用实例包括乙炔黑、科琴黑(ketjen black)、超导乙炔炭黑(denka black)、热裂炭黑(thermal black)、槽法炭黑(channel black)等。金属基或金属化合物基的导电剂的应用实例包括锡、氧化锡、磷酸锡、氧化钛、钛酸钾、钙钛矿材料例如lasrcoo3或lasrmno3等中的至少一种,且正、负极导电剂的质量分别占正、负极活性浆料的质量的0.1%-6%。当导电剂
的质量比小于0.1%时,会导致电化学性能劣化,当含量大于6%时,会减少正负极活性材料的含量,导致电池的能量密度变低。需要说明的是导电剂可以提高材料导电性,任何在电池体系中不会发生化学反应且是电子导体的材料皆可作为导电剂使用。
[0148]
实施例中,正、负极所用粘结剂可以选自聚偏二氟乙烯(pvdf)、聚六氟丙烯-聚偏二氟乙烯(hfp/pvdf)的共聚物、聚醋酸乙烯酯、聚乙烯醇、聚环氧乙烷、聚乙烯吡咯烷酮、烷基化聚环氧乙烷、聚乙烯基醚、聚甲基丙烯酸甲酯、聚丙烯酸乙酯、聚四氟乙烯、聚氯乙烯、聚丙烯腈、聚乙烯基吡啶、丁苯橡胶、丙烯腈-丁二烯橡胶等中的至少一种,且正、负极粘结剂的质量分别占正、负极活性浆料质量的1%-6%。粘结剂含量过低,正负极活性材料与集流体之间的粘接强度不足,粘结剂含量太高,粘接强度会增强,但是会减少正负极活性材料的含量,不利于提高电池的能量密度。
[0149]
实施例中,负极增稠剂可以使用羟甲基纤维素、羟乙基纤维素、羟丙基纤维素等,其质量占负极活性浆料的质量的1%-4%。增稠剂没有特别限制,只要可用于调节负极活性材料浆料的粘度即可。
[0150]
另外,正极所含的正极集流体和负极所含的负极集流体可以但不仅仅为铝、铝合金等,形貌可以但不限于箔(foil)或网(mesh)等。
[0151]
二次电池所含的隔膜主要为离子提供迁移所需的离子通道,同时将正极、负极隔离开防止正极和负极发生短路,一般使用烯烃聚合物膜(例如聚丙烯、聚乙烯、聚乙烯/聚丙烯、聚乙烯/聚丙烯/聚乙烯和聚丙烯/聚乙烯/聚丙烯)或者多层膜(multiple-film)、微孔膜以及它们的织造和非织造物。具体可采用三层复合隔膜,其厚度为12μm-36μm,孔隙率为30%-70%。为提升隔膜热稳定性,可以在隔膜表面涂覆一层或多层结构稳定的树脂或陶瓷材料。
[0152]
实施例中,二次电池可以为方形,圆柱或软包等形式。另外,二次电池可以锂离子电池,也可以是锂金属电池。
[0153]
为使本发明上述实施细节和操作能清楚地被本领域技术人员理解,以及本发明实施例链状卤代亚磷酸酯及其制备方法、电解液和二次电池的进步性能显著的体现,以下通过多个实施例来举例说明上述技术方案。
[0154]
a.链状卤代亚磷酸酯及其制备方法实施例
[0155]
实施例a1
[0156]
本实施例提供一种乙基乙二醇二氯亚磷酸酯、乙基乙二醇二氟亚磷酸酯(上文式1)及其制备方法。
[0157]
本实施例乙基乙二醇二氯亚磷酸酯制备方法具体如下:
[0158]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入乙基乙二醇90g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0159]
s2:将乙基乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,乙基乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到乙基乙二醇二氯亚磷酸酯。
[0160]
本实施例乙基乙二醇二氟亚磷酸酯制备方法具体如下:
[0161]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,60℃减压蒸馏得到无色液体130g,产率为85%。
[0162]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,分析结果显示gc-ms(esi)calcd for c4h9o2pf2[m]
为158.22。1h nmr(400mhz,cdcl3)δ:3.70(t,2h),3.54(t,2h),3.46(q,2h),1.05(t,3h);
13
c nmr(100mhz,cdcl3)δ:70.9,66.6,61.0,15.2。证明所得无色液体为乙基乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为72ppm,酸度为65ppm,氯离子浓度为15ppm。
[0163]
实施例a2
[0164]
本实施例提供一种三氟乙基乙二醇二氯亚磷酸酯、三氟乙基乙二醇二氟亚磷酸酯(上文式2)及其制备方法。
[0165]
本实施例三氟乙基乙二醇二氯亚磷酸酯制备方法具体如下:
[0166]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入三氟乙基乙二醇144g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0167]
s2:将三氟乙基乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,三氟乙基乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到三氟乙基乙二醇二氯亚磷酸酯。
[0168]
本实施例三氟乙基乙二醇二氟亚磷酸酯制备方法具体如下:
[0169]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,60℃减压蒸馏得到无色液体182g,产率为86%。
[0170]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,分析结果显示gc-ms(esi)calcd for c4h6o2pf5[m]
为212.24。1h nmr(400mhz,cdcl3)δ:3.89(q,2h),3.70(t,2h),3.54(q,2h);
13
c nmr(100mhz,cdcl3)δ:122.8,70.9,70.1,61.0。证明所得无色液体为三氟乙基乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为62ppm,酸度为67ppm,氯离子浓度为21ppm。
[0171]
实施例a3
[0172]
本实施例提供一种丙基乙二醇二氯亚磷酸酯、丙基乙二醇二氟亚磷酸酯(上文式3)及其制备方法。
[0173]
本实施例丙基乙二醇二氯亚磷酸酯制备方法具体如下:
[0174]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入丙基乙二醇104g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0175]
s2:将丙基乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,丙基乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到丙基乙二醇二氯亚磷酸酯。
[0176]
本实施例丙基乙二醇二氟亚磷酸酯制备方法具体如下:
[0177]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,70℃减压蒸馏得到无色液体151.4g,产率为88%。
[0178]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,分析结果显示gc-ms(esi)calcd for c5h
11
o2pf2[m]
为172.16。1h nmr(400mhz,cdcl3)δ:3.70(m,2h),3.54(t,2h),3.35(q,2h),1.49(m,2h),0.99(t,3h);
13
c nmr(100mhz,cdcl3)δ:71.2,71.0,61.0,27.3,10.4。证明所得无色液体为丙基乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为55ppm,酸度为71ppm,氯离子浓度为22ppm。
[0179]
实施例a4
[0180]
本实施例提供一种五氟丙基乙二醇二氯亚磷酸酯、五氟丙基乙二醇二氟亚磷酸酯(上文式4)及其制备方法。
[0181]
本实施例五氟丙基乙二醇二氯亚磷酸酯制备方法具体如下:
[0182]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入五氟丙基乙二醇104g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0183]
s2:将五氟丙基乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,五氟丙基乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到五氟丙基乙二醇二氯亚磷酸酯。
[0184]
本实施例五氟丙基乙二醇二氟亚磷酸酯制备方法具体如下:
[0185]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,70℃减压蒸馏得到无色液体228g,产率为87%。
[0186]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,分析结果显示gc-ms(esi)calcd for c5h6o2pf7[m]
为262.14。1h nmr(400mhz,cdcl3)δ:3.70(m,2h),3.67(m,2h),3.54(t,2h);
13
c nmr(100mhz,cdcl3)δ:128.4,123.9,71.2,65.8,61.0。证明所得无色液体为五氟丙基乙二醇二氟亚磷酸酯。通过
卡氏水分测定仪和电位滴定仪测定水分为51ppm,酸度为55ppm,氯离子浓度为19ppm。
[0187]
实施例a5
[0188]
本实施例提供一种烯丙酸单乙二醇酯二氯亚磷酸酯、烯丙酸单乙二醇酯二氟亚磷酸酯(上文式5)及其制备方法。
[0189]
本实施例烯丙酸单乙二醇酯二氯亚磷酸酯制备方法具体如下:
[0190]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入烯丙酸单乙二醇116.1g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0191]
s2:将烯丙酸单乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,烯丙酸单乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到烯丙酸单乙二醇酯二氯亚磷酸酯。
[0192]
本实施例烯丙酸单乙二醇酯二氟亚磷酸酯制备方法具体如下:
[0193]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,85℃减压蒸馏得到无色液体158.3g,产率为86%。
[0194]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,分析结果显示gc-ms(esi)calcd for c5h7o3pf2[m]
为184.14。1h nmr(400mhz,cdcl3)δ:6.41(m,1h),6.12(m,1h),5.83(m,1h),4.29(t,2h),3.81(m,2h);
13
c nmr(101mhz,cdcl3)δ:166.5,131.3,128.2,65.6,60.1。证明所得无色液体为烯丙基乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为45ppm,酸度为41ppm,氯离子浓度为17ppm。
[0195]
实施例a6
[0196]
本实施例提供一种二乙二醇单烯丙酸酯二氯亚磷酸酯、二乙二醇单烯丙酸酯二氟亚磷酸酯(上文式6)及其制备方法。
[0197]
本实施例二乙二醇单烯丙酸酯二氯亚磷酸酯制备方法具体如下:
[0198]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入二乙二醇单烯丙酸酯160g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0199]
s2:将二乙二醇单烯丙酸酯的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,二乙二醇单烯丙酸酯的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到二乙二醇单烯丙酸酯二氯亚磷酸酯。
[0200]
本实施例二乙二醇单烯丙酸酯二氟亚磷酸酯制备方法具体如下:
[0201]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,100℃减压蒸馏得到无色液体201g,产率为88%。
[0202]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,分析结果显示gc-ms(esi)calcd for c7h
11
o4pf2[m]
为228.13。1h nmr(400mhz,cdcl3)δ:6.41(m,1h),6.12(m,1h),5.83(m,1h),4.27(t,2h),3.70(m,2h),3.63(t,2h),3.54(m,4h);
13
c nmr(101mhz,cdcl3)δ:166.5,131.3,128.2,70.9,69.4,64.8,61.0。证明所得无色液体为二乙二醇单烯丙酸酯二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为45ppm,酸度为51ppm,氯离子浓度为16ppm。
[0203]
实施例a7
[0204]
本实施例提供一种三乙二醇单烯丙酸酯二氯亚磷酸酯、三乙二醇单烯丙酸酯二氟亚磷酸酯(上文式7)及其制备方法。
[0205]
本实施例三乙二醇单烯丙酸酯二氯亚磷酸酯制备方法具体如下:
[0206]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入三乙二醇单烯丙酸酯204.2g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0207]
s2:将三乙二醇单烯丙酸酯的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,三乙二醇单烯丙酸酯的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到三乙二醇单烯丙酸酯二氯亚磷酸酯。
[0208]
本实施例三乙二醇单烯丙酸酯二氟亚磷酸酯制备方法具体如下:
[0209]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,120℃减压蒸馏得到无色液体231.4g,产率为85%。
[0210]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,分析结果显示gc-ms(esi)calcd for c9h
15
o5pf2[m]
为272.22。1h nmr(400mhz,cdcl3)δ:6.41(m,1h),6.12(m,1h),5.83(m,1h),4.27(t,2h),3.70(m,2h),3.63(t,2h),3.54(t,2h),3.52(m,4h);
13
c nmr(101mhz,cdcl3)δ:166.5,131.3,128.2,71.2,70.4,70.1,69.4,64.8 61.1。证明所得无色液体为三乙二醇单烯丙酸酯二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为52ppm,酸度为47ppm,氯离子浓度为21ppm。
[0211]
实施例a8
[0212]
本实施例提供一种炔丙基乙二醇二氯亚磷酸酯、炔丙基乙二醇二氟亚磷酸酯(上
文式8)及其制备方法。
[0213]
本实施例炔丙基乙二醇二氯亚磷酸酯制备方法具体如下:
[0214]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入炔丙基乙二醇100.1g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0215]
s2:将炔丙基乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,炔丙基乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到三乙二醇单烯丙酸酯二氯亚磷酸酯。
[0216]
本实施例炔丙基乙二醇二氟亚磷酸酯制备方法具体如下:
[0217]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,70℃减压蒸馏得到无色液体141.2g,产率为84%。
[0218]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c5h7o2pf2[m]
为168.14。1h nmr(400mhz,cdcl3)δ:4.15(s,2h),,3.70(m,2h),3.54(t,2h),3.32(s,1h);
13
c nmr(101mhz,cdcl3)δ:78.7,76.4,70.1,60.3,60.1。证明所得无色液体为炔丙基乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为35ppm,酸度为41ppm,氯离子浓度为18ppm。
[0219]
实施例a9
[0220]
本实施例提供一种(2-甲基炔丙基)乙二醇二氯亚磷酸酯、(2-甲基炔丙基)乙二醇二氟亚磷酸酯(上文式9)及其制备方法。
[0221]
本实施例(2-甲基炔丙基)乙二醇二氯亚磷酸酯制备方法具体如下:
[0222]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入2-甲基炔丙基乙二醇114.1g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0223]
s2:将2-甲基炔丙基乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,2-甲基炔丙基乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到(2-甲基炔丙基)乙二醇二氯亚磷酸酯。
[0224]
本实施例(2-甲基炔丙基)乙二醇二氟亚磷酸酯制备方法具体如下:
[0225]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,70℃减压蒸馏得到无色液体154.8g,产率为85%。
[0226]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c6h9o2pf2[m]
为182.16。1h nmr(400mhz,cdcl3)δ:4.44(q,1h),3.70(m,2h),3.56(m,3h),1.43(d,3h);
13
c nmr(101mhz,cdcl3)δ:84.0,72.2,71.0,67.5,60.4,22.2。证明所得无色液体为2-甲基炔丙基乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为35ppm,酸度为37ppm,氯离子浓度为18ppm。
[0227]
实施例a10
[0228]
本实施例提供一种(2,2-二甲基炔丙基)乙二醇二氯亚磷酸酯、(2,2-二甲基炔丙基)乙二醇二氟亚磷酸酯(上文式10)及其制备方法。
[0229]
本实施例(2,2-二甲基炔丙基)乙二醇二氯亚磷酸酯制备方法具体如下:
[0230]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入2,2-二甲基炔丙基乙二醇128.1g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0231]
s2:将2,2-二甲基炔丙基乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,2,2-二甲基炔丙基乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到(2,2-二甲基炔丙基)乙二醇二氯亚磷酸酯。
[0232]
本实施例(2,2-二甲基炔丙基)乙二醇二氟亚磷酸酯制备方法具体如下:
[0233]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,75℃减压蒸馏得到无色液体168.6g,产率为86%。
[0234]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c7h
11
o2pf2[m]
为196.12。1h nmr(400mhz,cdcl3)δ:4.15(d,2h),3.70(m,2h),3.54(m,3h),3.52(m,4h);
13
c nmr(101mhz,cdcl3)δ:78.7,76.4,71.2,69.5,69.2,61.0,60.3。证明所得无色液体为2,2-二甲基炔丙基乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为37ppm,酸度为35ppm,氯离子浓度为16ppm。
[0235]
实施例a11
[0236]
本实施例提供一种炔丙基二乙二醇二氯亚磷酸酯、炔丙基二乙二醇二氟亚磷酸酯(上文式11)及其制备方法。
[0237]
本实施例炔丙基二乙二醇二氯亚磷酸酯制备方法具体如下:
[0238]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入炔丙基二乙二醇144.2g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0239]
s2:将炔丙基二乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程
全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,炔丙基二乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到炔丙基二乙二醇二氯亚磷酸酯。
[0240]
本实施例炔丙基二乙二醇二氟亚磷酸酯制备方法具体如下:
[0241]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,100℃减压蒸馏得到无色液体168.6g,产率为86%。
[0242]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c7h
11
o3pf2[m]
为212.16。1h nmr(400mhz,cdcl3)δ:3.70(m,2h),3.56(s,1h),3.54(m,2h),1.46(s,6h);
13
c nmr(101mhz,cdcl3)δ:88.3,84.5,72.1,65.0,60.7,29.6。分析结果显示gc-ms(esi)[]-,证明所得无色液体为炔丙基二乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为31ppm,酸度为42ppm,氯离子浓度为21ppm。
[0243]
实施例a12
[0244]
本实施例提供一种(2-甲基炔丙基)二乙二醇二氯亚磷酸酯、(2-甲基炔丙基)二乙二醇二氟亚磷酸酯(上文式12)及其制备方法。
[0245]
本实施例(2-甲基炔丙基)二乙二醇二氯亚磷酸酯制备方法具体如下:
[0246]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入2-甲基炔丙基二乙二醇158.2g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0247]
s2:将2-甲基炔丙基二乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,2-甲基炔丙基二乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到(2-甲基炔丙基)二乙二醇二氯亚磷酸酯。
[0248]
本实施例(2-甲基炔丙基)二乙二醇二氟亚磷酸酯制备方法具体如下:
[0249]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,100℃减压蒸馏得到无色液体189.9g,产率为84%。
[0250]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c8h
13
o3pf2[m]
为226.12。1h nmr(400mhz,cdcl3)δ:4.44(q,1h),3.70(m,2h),3.56(m,1h),3.54(m,6h),1.43(d,3h);
13
c nmr(101mhz,
cdcl3)δ:84.0,72.2,71.2,71.0,69.8,66.7,61.0,22.2。证明所得无色液体为2-甲基炔丙基二乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为38ppm,酸度为44ppm,氯离子浓度为22ppm。
[0251]
实施例a13
[0252]
本实施例提供一种(2,2-二甲基炔丙基)二乙二醇二氯亚磷酸酯、(2,2-二甲基炔丙基)二乙二醇二氟亚磷酸酯(上文式13)及其制备方法。
[0253]
本实施例(2,2-二甲基炔丙基)二乙二醇二氯亚磷酸酯制备方法具体如下:
[0254]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入2,2-二甲基炔丙基二乙二醇172.1g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0255]
s2:将2,2-二甲基炔丙基二乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,2,2-二甲基炔丙基二乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到(2,2-二甲基炔丙基)二乙二醇二氯亚磷酸酯。
[0256]
本实施例(2,2-二甲基炔丙基)二乙二醇二氟亚磷酸酯制备方法具体如下:
[0257]
s3:向上述步骤s2中向上述粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,105℃减压蒸馏得到无色液体204.2g,产率为85%。
[0258]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c9h
15
o3pf2[m]
为240.23。1h nmr(400mhz,cdcl3)δ:3.70(m,2h),3.56(m,1h),3.54(t,2h),3.52(m,4h),1.46(s,6h);
13
c nmr(101mhz,cdcl3)δ:88.3,84.5,72.1,71.2,70.1,64.2,61.0,29.5。证明所得无色液体为2,2-二甲基炔丙基二乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为37ppm,酸度为46ppm,氯离子浓度为24ppm。
[0259]
实施例a14
[0260]
本实施例提供一种炔丙基三乙二醇二氯亚磷酸酯、炔丙基三乙二醇二氟亚磷酸酯(上文式14)及其制备方法。
[0261]
本实施例炔丙基三乙二醇二氯亚磷酸酯制备方法具体如下:
[0262]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入炔丙基三乙二醇188.2g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0263]
s2:将炔丙基三乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,炔丙基三乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到炔丙基三乙二醇
二氯亚磷酸酯。
[0264]
本实施例炔丙基三乙二醇二氟亚磷酸酯制备方法具体如下:
[0265]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,120℃减压蒸馏得到无色液体220.3g,产率为86%。
[0266]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c9h
15
o4pf2[m]
为256.24。1h nmr(400mhz,cdcl3)δ:4.15(s,2h),3.70(m,2h),3.54(t,2h),3.52(m,8h),3.32(m,1h);
13
c nmr(101mhz,cdcl3)δ:78.7,76.4,71.2,70.4,69.5,69.2,61.0,60.3。证明所得无色液体为炔丙基三乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为35ppm,酸度为47ppm,氯离子浓度为23ppm。
[0267]
实施例a15
[0268]
本实施例提供一种(2-甲基炔丙基)三乙二醇二氯亚磷酸酯、(2-甲基炔丙基)三乙二醇二氟亚磷酸酯(上文式15)及其制备方法。
[0269]
本实施例(2-甲基炔丙基)三乙二醇二氯亚磷酸酯制备方法具体如下:
[0270]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入2-甲基炔丙基三乙二醇202.3g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0271]
s2:将2-甲基炔丙基三乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,2-甲基炔丙基三乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到(2-甲基炔丙基)三乙二醇二氯亚磷酸酯。
[0272]
本实施例(2-甲基炔丙基)三乙二醇二氟亚磷酸酯制备方法具体如下:
[0273]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,120℃减压蒸馏得到无色液体224.3g,产率为83%。
[0274]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c
10h17
o4pf2[m]
为270.25。1h nmr(400mhz,cdcl3)δ:4.44(q,1h),3.70(m,2h),3.56(m,1h),3.54(m,6h),3.52(m,4h),1.43(d,3h);
13
c nmr(101mhz,cdcl3)δ:84.0,72.2,71.2,71.0,70.4,69.8,66.7,61.0,22.2。证明所得无色液体为2-甲基炔丙基三乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为38ppm,酸度为45ppm,氯离子浓度为22ppm。
[0275]
实施例a16
[0276]
本实施例提供一种(2,2-二甲基炔丙基)三乙二醇二氯亚磷酸酯、(2,2-二甲基炔丙基)三乙二醇二氟亚磷酸酯(上文式16)及其制备方法。
[0277]
本实施例(2,2-二甲基炔丙基)三乙二醇二氯亚磷酸酯制备方法具体如下:
[0278]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入2,2-二甲基炔丙基三乙二醇216.3g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0279]
s2:将2,2-二甲基炔丙基三乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,2,2-二甲基炔丙基三乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到(2,2-二甲基炔丙基)三乙二醇二氯亚磷酸酯。
[0280]
本实施例(2,2-二甲基炔丙基)三乙二醇二氟亚磷酸酯制备方法具体如下:
[0281]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,120℃减压蒸馏得到无色液体250.1g,产率为88%。
[0282]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c
11h19
o4pf2[m]
为284.21。1h nmr(400mhz,cdcl3)δ:3.70(m,2h),3.56(m,1h),3.54(m,2h),3.52(m,8h),1.46(d,6h);
13
c nmr(101mhz,cdcl3)δ:88.3,84.5,72.1,71.2,70.4,70.1,64.2,61.0,29.6。证明所得无色液体为2,2-二甲基炔丙基三乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为48ppm,酸度为52ppm,氯离子浓度为17ppm。
[0283]
实施例a17
[0284]
本实施例提供一种(4-氟苯基)乙二醇二氯亚磷酸酯、(4-氟苯基)乙二醇二氟亚磷酸酯(上文式17)及其制备方法。
[0285]
本实施例(4-氟苯基)乙二醇二氯亚磷酸酯制备方法具体如下:
[0286]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入(4-氟苯基)乙二醇156.2g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0287]
s2:将(4-氟苯基)乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,(4-氟苯基)乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到(4-氟苯基)乙二醇二氯亚磷酸酯。
[0288]
本实施例(4-氟苯基)乙二醇二氟亚磷酸酯制备方法具体如下:
[0289]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶
液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,150℃减压蒸馏得到无色液体188.2g,产率为84%。
[0290]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c8h8o2pf3[m]
为224.12。1h nmr(400mhz,cdcl3)δ:7.19(m,2h),7.09(m,2h),4.33(t,2h),3.95(t,2h);
13
c nmr(101mhz,cdcl3)δ:155.0,154.5,116.1,116.0,70.1,60.6。证明所得无色液体为(4-氟苯基)乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为45ppm,酸度为50ppm,氯离子浓度为20ppm。
[0291]
实施例a18
[0292]
本实施例提供一种(3,5-二氟苯基)乙二醇二氯亚磷酸酯、(3,5-二氟苯基)乙二醇二氟亚磷酸酯(上文式18)及其制备方法。
[0293]
本实施例(3,5-二氟苯基)乙二醇二氯亚磷酸酯制备方法具体如下:
[0294]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入(3,5-二氟苯基)乙二醇174.1g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0295]
s2:将(3,5-二氟苯基)乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,(3,5-二氟苯基)乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到(3,5-二氟苯基)乙二醇二氯亚磷酸酯。
[0296]
本实施例(3,5-二氟苯基)乙二醇二氟亚磷酸酯制备方法具体如下:
[0297]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,150℃减压蒸馏得到无色液体205.8g,产率为85%。
[0298]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c8h7o2pf4[m]
为242.16。1h nmr(400mhz,cdcl3)δ:6.90(m,2h),6.36(m,1h),4.33(t,2h),3.95(t,2h);
13
c nmr(101mhz,cdcl3)δ:165.1,160.6,98.1,96.3,70.1,60.6。证明所得无色液体为(3,5-二氟苯基)乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为37ppm,酸度为22ppm,氯离子浓度为14ppm。
[0299]
实施例a19
[0300]
本实施例提供一种(五氟苯基)乙二醇二氯亚磷酸酯、(五氟苯基)乙二醇二氟亚磷酸酯(上文式19)及其制备方法。
[0301]
本实施例(五氟苯基)乙二醇二氯亚磷酸酯制备方法具体如下:
[0302]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入(五氟苯基)乙二醇228.1g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0303]
s2:将(五氟苯基)乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,(五氟苯基)乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到(五氟苯基)乙二醇二氯亚磷酸酯。
[0304]
本实施例(五氟苯基)乙二醇二氟亚磷酸酯制备方法具体如下:
[0305]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,150℃减压蒸馏得到无色液体260.6g,产率为88%。
[0306]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c8h4o2pf7[m]
为296.14。1h nmr(400mhz,cdcl3)δ:4.33(m,2h),3.95(m,2h);
13
c nmr(101mhz,cdcl3)δ:144.2,142.6,134.9,131.4,70.1,60.6。证明所得无色液体为(五氟苯基)乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为35ppm,酸度为32ppm,氯离子浓度为18ppm。
[0307]
实施例a20
[0308]
本实施例提供一种(4-氟苯基)二乙二醇二氯亚磷酸酯、(4-氟苯基)二乙二醇二氟亚磷酸酯(上文式20)及其制备方法。
[0309]
本实施例(4-氟苯基)二乙二醇二氯亚磷酸酯制备方法具体如下:
[0310]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入(4-氟苯基)二乙二醇200.2g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0311]
s2:将(4-氟苯基)二乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,(4-氟苯基)二乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到(4-氟苯基)二乙二醇二氯亚磷酸酯。
[0312]
本实施例(4-氟苯基)二乙二醇二氟亚磷酸酯制备方法具体如下:
[0313]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,180℃减压蒸馏得到无色液体230.7g,产率为86%。
[0314]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机
滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c
10h12
o3pf3[m]
为268.22。1h nmr(400mhz,cdcl3)δ:7.19(m,2h),7.09(m,2h),4.31(t,2h),3.77(t,2h),3.70(t,2h),3.54(t,2h);
13
c nmr(101mhz,cdcl3)δ:155.0,154.5,116.1,116.0,71.2,70.0,69.3,61.0。证明所得无色液体为(4-氟苯基)二乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为32ppm,酸度为34ppm,氯离子浓度为20ppm。
[0315]
实施例a21
[0316]
本实施例提供一种(3,5-二氟苯基)二乙二醇二氯亚磷酸酯、(3,5-二氟苯基)二乙二醇二氟亚磷酸酯(上文式21)及其制备方法。
[0317]
本实施例(4-氟苯基)二乙二醇二氯亚磷酸酯制备方法具体如下:
[0318]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入(3,5-二氟苯基)二乙二醇218.2g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0319]
s2:将(3,5-二氟苯基)二乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,(3,5-二氟苯基)二乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到(3,5-二氟苯基)二乙二醇二氯亚磷酸酯。
[0320]
本实施例(3,5-二氟苯基)二乙二醇二氟亚磷酸酯制备方法具体如下:
[0321]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,180℃减压蒸馏得到无色液体243.3g,产率为85%。
[0322]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c
10h11
o3pf4[m]
为286.23。1h nmr(400mhz,cdcl3)δ:6.90(m,2h),6.36(m,1h),4.31(t,2h),3.77(t,2h),3.70(m,2h),3.54(t,2h);
13
c nmr(101mhz,cdcl3)δ:165.1,160.6,98.1,96.3,71.2,70.0,69.3,61.0。证明所得无色液体为(3,5-二氟苯基)二乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为35ppm,酸度为32ppm,氯离子浓度为16ppm。
[0323]
实施例a22
[0324]
本实施例提供一种(五氟苯基)二乙二醇二氯亚磷酸酯、(五氟苯基)二乙二醇二氟亚磷酸酯(上文式22)及其制备方法。
[0325]
本实施例(五氟苯基)二乙二醇二氯亚磷酸酯制备方法具体如下:
[0326]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入(五氟苯基)二乙二醇272.2g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0327]
s2:将(五氟苯基)二乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入
过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,(五氟苯基)二乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到(五氟苯基)二乙二醇二氯亚磷酸酯。
[0328]
本实施例(五氟苯基)二乙二醇二氟亚磷酸酯制备方法具体如下:
[0329]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,180℃减压蒸馏得到无色液体295.9g,产率为87%。
[0330]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c
10
h8o3pf7[m]
为340.14。1h nmr(400mhz,cdcl3)δ:4.31(t,2h),3.77(m,2h),3.70(m,2h),3.54(m,2h);
13
c nmr(101mhz,cdcl3)δ:144.2,142.6,134.9,131.4,71.2,70.0,69.3,61.0。证明所得无色液体为(五氟苯基)二乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为41ppm,酸度为35ppm,氯离子浓度为18ppm。
[0331]
实施例a23
[0332]
本实施例提供一种甲基丙烯酸乙二醇二氯亚磷酸酯、甲基丙烯酸乙二醇二氟亚磷酸酯(上文式23)及其制备方法。
[0333]
本实施例甲基丙烯酸乙二醇二氯亚磷酸酯制备方法具体如下:
[0334]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入甲基丙烯酸乙二醇130.1g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0335]
s2:将甲基丙烯酸乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,甲基丙烯酸乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到甲基丙烯酸乙二醇二氯亚磷酸酯。
[0336]
本实施例甲基丙烯酸乙二醇二氟亚磷酸酯制备方法具体如下:
[0337]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,85℃减压蒸馏得到无色液体166.4g,产率为84%。
[0338]
在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c6h9o3pf2[m]
为198.11。1h nmr(400mhz,cdcl3)δ:6.48(s,1h),6.40(s,1h),4.29(t,2h),3.81(m,2h),2.01(s,3h);
13
c nmr(101mhz,
cdcl3)δ:167.2,136.0,125.2,65.8,60.0,17.9。证明所得无色液体为甲基丙烯酸乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为32ppm,酸度为36ppm,氯离子浓度为15ppm。
[0339]
实施例a24
[0340]
本实施例提供一种(1-丁炔基)乙二醇二氯亚磷酸酯、(1-丁炔基)乙二醇二氟亚磷酸酯(上文式24)及其制备方法。
[0341]
本实施例(1-丁炔基)乙二醇二氯亚磷酸酯制备方法具体如下:
[0342]
s1:室温条件下向反应釜中加入二氯甲烷200ml,然后加入1-丁炔基乙二醇116.1g(1mol),室温搅拌0.5h;在另一反应釜中将二氯甲烷200ml与150g pcl3在0℃条件下混合;
[0343]
s2:将1-丁炔基乙二醇的二氯甲烷溶液加入到pcl3的二氯甲烷溶液中,加入过程全程搅拌并保持温度在0℃,控制滴加速度保持产气速度平稳,1-丁炔基乙二醇的二氯甲烷溶液加入完毕后保持温度不变继续反应至体系无气体产生为反应终点,反应产生的hcl气体使用纯净水吸收得到盐酸,盐酸可以作为工业原料出售;反应完成后室温常压蒸馏除去二氯甲烷,然后室温减压蒸馏除去过量的pcl3得到粘稠状黄色液体,得到(1-丁炔基)乙二醇二氯亚磷酸酯。
[0344]
本实施例(1-丁炔基)乙二醇二氟亚磷酸酯制备方法具体如下:
[0345]
s3:向上述步骤s2中粘稠状黄色液体中加入200ml乙腈后室温搅拌得到淡黄色溶液,向溶液中分批次加入kf150g然后升温至60℃反应3h;反应过程中无气体放出,然后静置沉降冷却至室温后常压过滤得到淡黄色溶液,常温减压蒸馏除去乙腈得到黄色液体,80℃减压蒸馏得到无色液体156.6g,产率为86%。在手套箱中,取所得无色液体0.1ml,加入2ml无水乙腈中使其完全溶解,使用有机滤膜过滤除去悬浮物,取少量滤液使用注射器进样,通过气质联用(thermo fisher scientific)进行分析,gc-ms(esi)calcd for c6h9o2pf2[m]
为182.13。1h nmr(400mhz,cdcl3)δ:3.70(m,2h),3.54(m,4h),3.06(m,1h),2.20(t,2h);
13
c nmr(101mhz,cdcl3)δ:81.4,70.3,69.7,68.4,61.0,19.6。证明所得无色液体为1-丁炔基乙二醇二氟亚磷酸酯。通过卡氏水分测定仪和电位滴定仪测定水分为46ppm,酸度为42ppm,氯离子浓度为17ppm。
[0346]
b.非水电解液实施例
[0347]
本实施例提供非水电解液电解液。
[0348]
非水电解液电解液配制:
[0349]
在氩气氛围的手套箱中(手套箱中h2o、o2含量低于0.1ppm)配置实施例非水电解液,将碳酸乙烯酯(ec)、碳酸甲乙酯(emc)和碳酸二乙酯(dec)以质量比为30:50:20的比例进行混合得到电解液溶液,然后向电解液溶液中加入六氟磷酸锂(lipf6),使六氟磷酸锂(lipf6)浓度为1.0mol/l。以非水电解液的总重量为100%计,加入二氟磷酸锂(lio2pf2)、1,3-丙磺酸内酯(ps)和硫酸乙烯酯(dtd),使其质量分数都为1%,同时加入碳酸亚乙烯酯(vc)、丙烯基-1,3-磺酸内酯(pes),使其质量分数都为0.5%,得到对照电解液样品,编号记为(1);
[0350]
在氩气氛围的手套箱中(手套箱中h2o、o2含量低于0.1ppm)配置实施例非水电解液,将碳酸乙烯酯(ec)、碳酸甲乙酯(emc)和碳酸二乙酯(dec)以质量比为30:50:20的比例进行混合得到电解液溶液,然后向电解液溶液中加入六氟磷酸锂(lipf6),使六氟磷酸锂
(lipf6)浓度为1.0mol/l,以非水电解液的总重量为100%计,加入二氟磷酸锂(lio2pf2)、1,3-丙磺酸内酯(ps)和硫酸乙烯酯(dtd),使其质量分数都为1%,同时加入碳酸亚乙烯酯(vc)、丙烯基-1,3-磺酸内酯(pes),使其质量分数都为0.5%,然后分别加入式1~式24所示添加剂,使其质量分数都为1%。分别得到编号为(2)~(25)的电解液。
[0351]
上述编号(1)-(25)非水电解液所含的组分如下表1中所示:
[0352]
表1非水电解液主要成分
[0353]
[0354][0355]
c.二次电池实施例
[0356]
实施例c1(对比例c1)
[0357]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0358]
正极极片制备:
[0359]
以镍钴锰酸锂(lini
0.8
co
0.1
mn
0.1
o2,简称ncm811)三元材料作为正极活性物质,正极活性浆料按质量比97%正极活性物质 1.8%pvdf粘合剂 1.2%super p导电炭黑溶于溶剂n-甲基吡咯烷酮中混合得到。然后将正极活性浆料均匀涂布在集流体铝箔上,涂布量为305g/m2,随后在80℃下烘干后进行冷压、切边、裁片、分条后,在80℃真空条件下干燥4h,焊接极耳,得到正极片。
[0360]
负极极片制备:
[0361]
以人造石墨作为负极活性物质,负极活性浆料按质量比96%负极活性物质 2%cmc/sbr粘合剂 2%super p导电炭黑混合后加入去离子水中搅拌均匀得到,然后将负极活性浆料均匀涂布在集流体铜箔上,涂布量为215g/m2,随后在85℃下烘干后进行冷压、切边、裁片、分条后,在110℃真空条件下干燥4h,焊接极耳,得到负极片。
[0362]
软包锂离子电池的制备:
[0363]
将正极片、负极片以及陶瓷涂覆的pe隔膜经过叠片工艺制作成软包电芯,并在75℃下真空烘烤10h,使用上述编号为(1)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a1)。
[0364]
实施例c2
[0365]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0366]
除了在软包锂离子电池的制备过程中使用上述编号为(2)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a2)。其它都与实施例c1相同。
[0367]
实施例c3
[0368]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0369]
除了在软包锂离子电池的制备过程中使用上述编号为(3)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a3)。其它都与实施例c1相同。
[0370]
实施例c4
[0371]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0372]
除了在软包锂离子电池的制备过程中使用上述编号为(4)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a4)。其它都与实施例c1相同。
[0373]
实施例c5
[0374]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0375]
除了在软包锂离子电池的制备过程中使用上述编号为(5)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a5)。其它都与实施例c1相同。
[0376]
实施例c6
[0377]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0378]
除了在软包锂离子电池的制备过程中使用上述编号为(6)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a6)。其它都与实施例c1相同。
[0379]
实施例c7
[0380]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0381]
除了在软包锂离子电池的制备过程中使用上述编号为(7)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a7)。其它都与实施例c1相同。
[0382]
实施例c8
[0383]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0384]
除了在软包锂离子电池的制备过程中使用上述编号为(8)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a8)。其它都与实施例c1相同。
[0385]
实施例c9
[0386]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0387]
除了在软包锂离子电池的制备过程中使用上述编号为(9)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a9)。其它都与实施例c1相同。
[0388]
实施例c10
[0389]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0390]
除了在软包锂离子电池的制备过程中使用上述编号为(10)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a10)。
其它都与实施例c1相同。
[0391]
实施例c11
[0392]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0393]
除了在软包锂离子电池的制备过程中使用上述编号为(11)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a11)。其它都与实施例c1相同。
[0394]
实施例c12
[0395]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0396]
除了在软包锂离子电池的制备过程中使用上述编号为(12)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a12)。其它都与实施例c1相同。
[0397]
实施例c13
[0398]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0399]
除了在软包锂离子电池的制备过程中使用上述编号为(13)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a13)。其它都与实施例c1相同。
[0400]
实施例c14
[0401]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0402]
除了在软包锂离子电池的制备过程中使用上述编号为(14)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a14)。其它都与实施例c1相同。
[0403]
实施例c15
[0404]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0405]
除了在软包锂离子电池的制备过程中使用上述编号为(15)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a15)。其它都与实施例c1相同。
[0406]
实施例c16
[0407]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0408]
除了在软包锂离子电池的制备过程中使用上述编号为(16)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a16)。其它都与实施例c1相同。
[0409]
实施例c17
[0410]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0411]
除了在软包锂离子电池的制备过程中使用上述编号为(17)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a17)。其它都与实施例c1相同。
[0412]
实施例c18
[0413]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0414]
除了在软包锂离子电池的制备过程中使用上述编号为(18)的电解液对软包电芯
进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a18)。其它都与实施例c1相同。
[0415]
实施例c19
[0416]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0417]
除了在软包锂离子电池的制备过程中使用上述编号为(19)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a19)。其它都与实施例c1相同。
[0418]
实施例c20
[0419]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0420]
除了在软包锂离子电池的制备过程中使用上述编号为(20)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a20)。其它都与实施例c1相同。
[0421]
实施例c21
[0422]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0423]
除了在软包锂离子电池的制备过程中使用上述编号为(21)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a21)。其它都与实施例c1相同。
[0424]
实施例c22
[0425]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0426]
除了在软包锂离子电池的制备过程中使用上述编号为(22)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a22)。其它都与实施例c1相同。
[0427]
实施例c23
[0428]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0429]
除了在软包锂离子电池的制备过程中使用上述编号为(23)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a23)。其它都与实施例c1相同。
[0430]
实施例c24
[0431]
本实施例提供一种锂二次电池。该锂二次电池按照如下方法组装:
[0432]
除了在软包锂离子电池的制备过程中使用上述编号为(24)的电解液对软包电芯进行注液,注液后静置24h,经过化成老化、夹具、分容等工序,得到软包电池编号为(a24)。其它都与实施例c1相同。
[0433]
锂离子电池性能测试
[0434]
为了保持实验的一致性,所有小软包电芯都使用相同体积的电解液。然后对制备好的小软包电芯进行充放电测试,使用land充放电测试系统对组装好的电芯进行如下性能测试。
[0435]
在正极极片制备过程中以镍钴锰酸锂(lini
0.8
co
0.1
mn
0.1
o2,简称ncm811)三元材料作为正极活性物质制作的软包电池测试如下:
[0436]
1常温循环性能测试
[0437]
将化成后的电池置于恒温25℃的烘箱中,使用1c恒流恒压(cc cv)充电至电压为4.2v,截至电流为0.01c,然后用1c恒流(cc)放电至电压为3.0v。如此充/放电n次循环后,记录第一次和第n次循环后容量的保持率,以评估其常温循环性能。
[0438]
25℃1c循环n次容量保持率计算公式如下:
[0439]
第n次循环容量保持率(%)=(第n次循环放电容量/第一次循环放电量)
×
100%。
[0440]
2.高温循环性能测试
[0441]
将化成后的电池置于恒温45℃的烘箱中,使用1c恒流恒压(cc cv)充电至电压为4.2v,截至电流为0.01c,然后用1c恒流(cc)放电至电压为3.0v。如此充/放电n次循环后,记录第一次和第n次循环后容量的保持率,以评估其高温循环性能。
[0442]
45℃1c循环n次容量保持率计算公式如下:
[0443]
第n次循环容量保持率(%)=(第n次循环放电容量/第一次循环放电量)
×
100%。
[0444]
3.室温储存性能测试
[0445]
将化成后的电芯在常温下用1c恒流恒压(cc cv)充电至电压为4.2v,截至电流为0.01c,再用1c恒流(cc)放电至电压为3.0v,测量电池初始放电容量,再用1c恒流恒压(cc cv)充电至电压为4.2v,截至电流为0.01c,测量电池的初始厚度,然后将电池在室温条件下储存n天后,测量电池的厚度,再以1c恒流(cc)放电至电压为3.0v,测量电池的保持容量,再用1c恒流恒压(cc cv)充电至电压为4.2v,截至电流为0.01c,然后用1c恒流(cc)放电至电压为3.0v,测量恢复容量。
[0446]
容量保持率、容量恢复率的计算公式如下:
[0447]
电池容量保持率(%)=保持容量/初始容量
×
100%;
[0448]
电池容量恢复率(%)=恢复容量/初始容量
×
100%;
[0449]
4.60℃高温储存性能测试
[0450]
将化成后的电芯在常温下用1c恒流恒压(cc cv)充电至电压为4.2v,截至电流为0.01c,再用1c恒流(cc)放电至电压为3.0v,测量电池初始放电容量,再用1c恒流恒压(cc cv)充电至电压为4.2v,截至电流为0.01c,测量电池的初始厚度,然后将电池在60℃条件下储存n天后,测量电池的厚度,再以1c恒流(cc)放电至电压为3.0v,测量电池的保持容量,再用1c恒流恒压(cc cv)充电至电压为4.2v,截至电流为0.01c,然后用1c恒流(cc)放电至电压为3.0v,测量恢复容量。
[0451]
容量保持率、容量恢复率的计算公式如下:
[0452]
电池容量保持率(%)=保持容量/初始容量
×
100%;
[0453]
电池容量恢复率(%)=恢复容量/初始容量
×
100%;
[0454]
5.60℃高温储存电池厚度膨胀率
[0455]
将化成后的电芯在常温下以1c恒流(cc)充电至电压为4.2v,然后以4.2v恒压(cv)充电至电流为0.05c,此时测试锂离子电池的厚度并记为h0;之后将锂离子电池放入60℃的恒温箱,存储n天后取出,测试此时锂离子电池的厚度并记为h1。
[0456]
电池厚度膨胀率(%)=(n天后的厚度h
1-初始厚度h0)/初始厚度
×
100%。
[0457]
电芯测试电压为3.0~4.2v,分别在室温以1c恒流(cc)充放电循环500周后测试电池的容量保持率;45℃条件下以1c恒流(cc)充放电循环300周后测试电池的容量保持率;分别在室温下储存100天和在60℃下存储21天后的容量保持率和恢复率;以及在60℃下存储
30天后的电芯体积膨胀率。
[0458]
具体数据如表2所示。
[0459]
表2电池性能测试结果
[0460][0461][0462]
通过表2的测试结果可知,使用本发明实施例链状卤代亚磷酸酯作为添加剂使用有利于提升电池的常温循环性能、高温循环性能、常温存储性能和高温存储性能,同时可以抑制电池产气,减小电池的体积膨胀,提升电池安全性和使用寿命。
[0463]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。