1.本发明涉及一种以光学各向异性薄膜、可见光反射薄膜、隔热薄膜等各种光学部件的材料为首的,在各种用途中有用的化合物(尤其液晶取向促进剂)和包含该化合物的液晶组合物及固化物、以及利用这些的薄膜。
背景技术:
2.若液晶涂布在实施了取向处理的膜(取向膜)上,则会有规则地取向。并且,通过将液晶夹入二片取向膜之间,能够控制液晶的取向状态。因此,在包括由棒状液晶性分子和用于封入棒状液晶性分子的二片基板构成的液晶单元及用于对棒状液晶性分子施加电压的电极层的液晶显示装置中,成为棒状液晶性分子注入在形成于二片基板上的取向膜的间隙的状态,因此能够比较容易地控制棒状液晶性分子的取向状态。
3.另一方面,以扩大液晶显示装置的视场角或消除着色为目的,在液晶单元与偏振片之间,配置光学补偿片(相位差板)。在这种情况下,将在透明支撑体上具有由液晶性分子形成的光学各向异性层的光学各向异性元件用作光学补偿片。光学各向异性层通过使液晶性分子取向并固定该取向状态而形成。此时,液晶性分子通过设置于透明支撑体与光学各向异性层之间的一片取向膜而取向。但是,用一片取向膜难以使液晶性分子从取向膜界面均匀地取向(单畴取向)到空气界面。这是因为,在未实施取向处理的界面(空气界面)侧没有取向限制力,因此导致液晶的取向紊乱。若液晶性分子未均匀地取向,则会发生因向错引起的光散射,形成不透明的膜。从提高液晶显示装置的视觉辨认度的观点而言,不优选这种膜。
4.鉴于这种必要性,开发如下技术:在未实施取向处理的界面(空气界面)侧,即使不具有取向膜也对液晶赋予取向限制力,从而使其均匀地取向(例如,参考专利文献1及专利文献2。)。这里,通过添加液晶取向促进剂来控制液晶性分子的取向。然后,通过使用液晶取向促进剂,提供液晶性分子容易均匀地取向的液晶组合物。
5.以往技术文献
6.专利文献
7.专利文献1:日本特开2002-129162号公报
8.专利文献2:日本特开2013-47204号公报
技术实现要素:
9.发明要解决的技术课题
10.本发明人等明确了专利文献1中所记载的液晶取向促进剂的可使用浓度范围或溶解性并不充分,扔存在改善的余地。
11.并且,本发明人等还明确了,关于专利文献2中所记载的液晶取向促进剂,可以包括被指出对环境有害的pfoa(全氟辛酸)及其类似物,因此存在改善的余地。
12.因此,本发明的课题在于解决现有技术中的上述課題,并提供显示充分的溶解性、
可使用的浓度范围广、并具有环境适应性且显示优异的液晶取向促进性的化合物。
13.并且,本发明的课题在于提供一种能够通过上述化合物而将液晶性分子容易均匀地取向的液晶组合物及其固化物。
14.并且,本发明的课题在于提供一种利用上述液晶组合物或上述固化物的薄膜(例如,光学各向异性薄膜、可见光反射薄膜、隔热薄膜等)。
15.用于解决技术课题的手段
16.本发明人等为了达成上述课题而进行了深入研究,结果发现在分子内具有规定的环结构及规定的含氟末端结构,且在分子内具有2个以上规定的含氟末端结构的化合物显示充分的溶解性、可使用的浓度范围广,并且具有环境适应性且显示优异的液晶取向促进性,从而完成了本发明。
17.即,本发明人等发现了通过以下结构能够解决上述课题。
18.[1]一种化合物,其在分子内具有下述a所表示的结构a及下述b所表示的结构b,
[0019]
在分子内具有2个以上上述结构b。
[0020]
a:具有至少1个以上的芳香族性的环结构,且分子内的芳香族性的环结构的π电子数的合计为8以上的结构,但是,后述式(i)~(iv)所表示的芳基胺结构除外。
[0021]
b:后述式(b-1)~(b-5)中的任一个所表示的含氟末端结构。
[0022]
[2]根据[1]所述的化合物,其中,
[0023]
上述结构a为后述式(1)或式(2)所表示的结构。
[0024]
[3]根据[1]或[2]所述的化合物,其中,
[0025]
上述结构a为后述式(1-1)~(1-10)中的任一个所表示的结构。
[0026]
[4]根据[3]所述的化合物,其中,
[0027]
后述式(1-1)~(1-10)中,l1、l2、l3及l4分别独立地为单键、或-c(=o)o-、-oc(=o)-、-o-、-nh-c(=o)-及-nh-中的任一种2价的连结基团。
[0028]
[5]根据[1]或[2]所述的化合物,其中,
[0029]
上述结构a为后述式(2-1)或式(2-2)所表示的结构。
[0030]
[6]根据[1]至[5]中任一项所述的化合物,其用于液晶取向促进剂。
[0031]
[7]一种液晶组合物,其含有[1]至[6]中任一项所述的化合物及聚合性液晶化合物。
[0032]
[8]根据[7]所述的液晶组合物,其中,
[0033]
上述聚合性液晶化合物为棒状液晶化合物或圆盘状液晶化合物。
[0034]
[9]根据[7]或[8]所述的液晶组合物,其含有至少1种手性化合物。
[0035]
[10]一种固化物,其使[7]至[9]中任一项所述的液晶组合物聚合而成。
[0036]
[11]一种薄膜,其含有[10]所述的固化物中的至少1种。
[0037]
[12]一种薄膜,其将[7]至[9]中任一项所述的液晶组合物的胆甾醇型液晶相固定而成。
[0038]
[13]根据[11]或[12]所述的薄膜,其显示光学各向异性。
[0039]
[14]根据[11]至[13]中任一项所述的薄膜,其显示选择反射特性。
[0040]
[15]根据[14]所述的薄膜,其在红外线波长区域显示选择反射特性。
[0041]
[16]根据[14]所述的薄膜,其在可见光波长区域显示选择反射特性。
[0042]
发明效果
[0043]
根据本发明,能够提供一种显示充分的溶解性、可使用的浓度范围广、并且具有环境适应性且显示优异的液晶取向促进性的化合物。
[0044]
并且,根据本发明,能够提供一种能够通过上述化合物而将液晶性分子容易均匀地取向的液晶组合物及其固化物。
[0045]
并且,根据本发明,能够提供一种利用上述液晶组合物或上述固化物的薄膜(例如,光学各向异性薄膜、可见光反射薄膜、隔热薄膜等)。
具体实施方式
[0046]
以下,对本发明进行详细说明。
[0047]
以下所记载的构成要件的说明有时根据本发明的代表性实施方式或具体例进行,但是本发明并不限定于这种实施方式或具体例。
[0048]
另外,在本说明书中,使用“~”表示的数值范围是指将记载于“~”的前后的数值作为下限值及上限值而包含的范围。
[0049]
并且,在本说明书中,各成分可以单独使用1种对应于各成分的物质,也可以并用2种以上。其中,关于各成分,在并用2种以上的物质的情况下,只要没有特别指定,则关于该成分的含量是指所并用的物质的合计含量。
[0050]
并且,在本说明书中,“(甲基)丙烯酸酯”为表示“丙烯酸酯”或“甲基丙烯酸酯”的标记,“(甲基)丙烯酸”为表示“丙烯酸”或“甲基丙烯酸”的标记,“(甲基)丙烯酰基”为表示“丙烯酰基”或“甲基丙烯酰基”的标记。
[0051]
[化合物]
[0052]
本发明的化合物是在分子内具有下述a所表示的结构a及下述b所表示的结构b,并且在分子内具有2个以上结构b的化合物。
[0053]
a:具有至少1个以上的芳香族性的环结构,且分子内的芳香族性的环结构的π电子数的合计为8以上的结构,但是,下述式(i)~(iv)所表示的芳基胺结构除外。
[0054]
b:下述式(b-1)~(b-5)中的任一个所表示的含氟末端结构。
[0055]
[化学式1]
[0056][0057]
[化学式2]
[0058][0059]
上述式(i)~(iv)中,ar1、ar2、ar3、ar4、ar5、ar6及ar7分别独立地表示经取代或未经取代的碳原子数6~20的芳基。
[0060]
并且,上述式(b-1)~(b-5)中,*表示键合位置。
[0061]
由于本发明的化合物在分子内具有2个以上上述结构b(含氟末端结构),因此具有作为液晶取向促进剂的功能。
[0062]
其中,在末端具有氟化烷基的化合物作为取向促进剂是有效的,但是现有已知的取向促进剂由于使用浓度范围窄、溶解性低及使用有可能对环境有害的结构,因此用途受到限制。
[0063]
相对于此,本发明的化合物以更广的浓度范围且良好的溶解性显示同等以上的取向性能,并且不具有有可能对环境有害的长链的全氟烷基结构,因此包含这些的组合物具有在制造中容易使用的优点。
[0064]
并且,由于在可固化的聚合性液晶组合物中具有取向性能,因此在光学部件等各
种用途中有用。
[0065]
〔结构a〕
[0066]
如上所述,本发明的化合物具有下述a所表示的结构a。
[0067]
a:具有至少1个以上的芳香族性的环结构,且分子内的芳香族性的环结构的π电子数的合计为8以上的结构,但是,上述式(i)~(iv)所表示的芳基胺结构除外。
[0068]
作为上述结构a,从显示更优异的液晶取向促进性的理由出发,优选下述式(1)或式(2)所表示的结构。
[0069]
[化学式3]
[0070][0071]
上述式(1)中,m表示0或1,n表示1或2,p表示0~3的整数。在p为2或3的情况下,多个la、cy2、m、lb、cy3及n分别可以相同也可以不同。在n为2的情况下,多个lb及cy3分别可以相同也可以不同。
[0072]
并且,上述式(1)中,la及lb分别独立地表示单键或2价的连结基团。
[0073]
并且,上述式(1)中,cy1、cy2及cy3分别独立地表示下述式(cy-1)~(cy-23)中的任一个所表示的环结构。但是,cy1、cy2及cy3中,至少1个为具有芳香族性的环结构,cy1、cy2及cy3中,表示芳香族性的环结构的π电子数的合计为8以上。
[0074]
上述式(2)中,q表示3或4,多个cy4分别可以相同也可以不同。
[0075]
并且,上述式(2)中,lc表示q价的连结基团。
[0076]
上述式(2)中,cy4表示下述式(cy-1)~(cy-23)中的任一个所表示的环结构。但是,多个cy4中,至少1个为具有芳香族性的环结构,多个cy4中,表示芳香族性的环结构的π电子数的合计为8以上。
[0077]
[化学式4]
中,表示芳香族性的环结构的π电子数的合计为8以上。
[0085]
并且,上述式(2)中,多个cy4中,至少1个为具有芳香族性的环结构,多个cy4中,表示芳香族性的环结构的π电子数的合计为8以上。
[0086]
其中,具有芳香族性的环结构是指π电子数为4n 2(n为0以上的整数)的环结构,可以优选举出苯基、萘基、三亚苯基及其稠环结构。
[0087]
另外,上述式(cy-1)~(cy-23)中,作为具有芳香族性的环结构的上述式(cy-1)~(cy-5)及上述式(cy-8)~(cy-23)的π电子数如下所述。
[0088]
π电子数为6的环:式(cy-1)~(cy-5)、(cy-11)~(cy-16)、(cy-19)及(cy-20)
[0089]
π电子数为10的环:式(cy-8)~(cy-10)、(cy-17)及(cy-18)
[0090]
π电子数为14的环:式(cy-21)及(cy-22)
[0091]
π电子数为18的环:式(cy-23)
[0092]
关于上述式(1)及(2)所表示的结构中所含的芳香族性的环结构,从提高液晶取向性的目的出发,优选具有2个以上苯环(即,上述式(cy-1)),更优选具有3个以上。
[0093]
上述式(1)中,la及lb分别独立地表示单键或2价的连结基团。
[0094]
其中,作为2价的连结基团,例如,可以举出由碳原子、氧原子、氮原子、硫原子、c=c、c=o、c=n、n=n、c≡c的组合构成的2价的基团,碳原子及氮原子可以任意地被氢原子取代。
[0095]
并且,作为2价的连结基团,例如,优选为组合,选自包括碳原子数1~18的直链状的亚烷基、碳原子数3~18的支链状或环状的亚烷基、可以具有取代基的碳原子数6~12的亚芳基、醚基(-o-)、羰基(-c(=o)-)及可以具有取代基的亚氨基(-nh-)的组中的至少2个以上的基团的2价的连结基团。
[0096]
本发明中,作为la及lb,优选为包含单键及-o-的2价的连结基团。
[0097]
上述式(2)中,lc表示q价(即,3价或4价)的连结基团。
[0098]
其中,作为3价或4价的连结基团,例如,可以举出由碳原子、氧原子、氮原子、硫原子、c=c、c=o、c=n、n=n、c≡c的组合构成的3~4价的基团,碳原子及氮原子可以任意地被氢原子取代。
[0099]
并且,作为3价的连结基团,可以举出由脂肪族烃基、芳香族烃基、杂环基及这些组合构成的基团、以及将选自脂肪族烃基、芳香族烃基及杂环基中的至少1种与选自-o-、-co-、-coo-、-oco-及-nh-中的至少1种组合而成的基团,优选为脂肪族烃基。脂肪族烃基的碳原子数优选为1~30,更优选为1~20,进一步优选为1~15。
[0100]
并且,作为4价的连结基团,可以举出4价的亚芳基、下述式所表示的基团等。另外,下述式中,*表示键合位置。
[0101]
[化学式5]
[0102][0103]
上述式(1)中,m表示0或1,n表示1或2,p表示0~3的整数。p优选为1~3的整数,更优选为2或3。
[0104]
上述式(2)中,q表示3或4。
[0105]
在本发明中,从显示更优异的液晶取向促进性的理由出发,优选上述结构a尤其上述式(1)所表示的结构为下述式(1-1)~(1-10)中的任一个所表示的结构。
[0106]
[化学式6]
[0107][0108]
[0109]
上述式(1-1)~(1~10)中,构成环结构的碳原子所具有的氢原子可以被取代基或包含上述结构b的基团取代。另外,作为取代基,可以举出与可以取代上述式(cy-1)~(cy-23)中的氢原子的取代基相同的取代基,优选范围也相同。
[0110]
并且,上述式(1-1)~(1~10)中,l1、l2、l3及l4分别独立地表示单键或2价的连结基团。
[0111]
作为l1、l2、l3及l4的一方式所表示的2价的连结基团,例如,可以举出由碳原子、氧原子、氮原子、硫原子、c=c、c=o、c=n、n=n、c≡c的组合构成的2价的基团,碳原子及氮原子可以任意地被氢原子取代。
[0112]
并且,作为2价的连结基团,例如,优选为组合,选自包括碳原子数1~18的直链状的亚烷基、碳原子数3~18的支链状或环状的亚烷基、可以具有取代基的碳原子数6~12的亚芳基、醚基(-o-)、羰基(-c(=o)-)及可以具有取代基的亚氨基(-nh-)的组中的至少2个以上的基团的2价的连结基团。
[0113]
在本发明中,作为l1、l2、l3及l4,分别独立地优选为单键、或者包含氧原子或氮原子的2价的连结基团,更优选为单键、或者-c(=o)o-、-oc(=o)-、-o-、-nh-c(=o)-及-nh-中的任一种2价的连结基团。
[0114]
在本发明中,从显示更优异的液晶取向促进性的理由出发,优选上述结构a尤其上述式(2)所表示的结构为下述式(2-1)或式(2-2)所表示的结构。
[0115]
[化学式7]
[0116][0117]
上述式(2-1)~(2~2)中,构成环结构的碳原子所具有的氢原子可以被取代基或包含上述结构b的基团取代。另外,作为取代基,可以举出与可以取代上述式(cy-1)~(cy-23)中的氢原子的取代基相同的取代基,优选范围也相同。
[0118]
并且,上述式(2-1)中,l5表示3价的连结基团,上述式(2-2)中,l6表示4价的连结基团。
[0119]
其中,作为l5所表示的3价的连结基团,例如,可以举出具有3个原子以上亚甲基碳的直链或支链状的3价的结构,任意的亚甲基碳可以被氧原子、氮原子、硫原子、c=c、c=o取代,碳原子及氮原子可以任意地被氢原子取代。并且,作为亚甲基碳原子数,更优选为5以上,进一步优选为7以上。
[0120]
其中,作为l6所表示的4价的连结基团,例如,可以举出具有3个原子以上亚甲基碳的直链或支链状的4价的结构,任意的亚甲基碳可以被氧原子、氮原子、硫原子、c=c、c=o取代,碳原子及氮原子可以任意地被氢原子取代。并且,作为亚甲基碳原子数,更优选为5以上,进一步优选为7以上。
[0121]
〔结构b〕
[0122]
如上所述,本发明的化合物具有2个以上下述b所表示的结构b。
[0123]
b:下述式(b-1)~(b-5)中的任一个所表示的含氟末端结构。
[0124]
另外,下述式(b-1)~(b-5)中,*表示键合位置。
[0125]
[化学式8]
[0126][0127]
本发明的化合物在分子内具有2个以上上述结构b,优选具有4个以上。
[0128]
并且,本发明的化合物优选在分子内具有3个以上且10个以下的上述结构b,更优选具有4个以上且6个以下。
[0129]
并且,为了进一步提高提高液晶取向性的效果,至少2个以上的上述结构b优选取代为具有立体对称性。
[0130]
本发明的化合物中,从可使用的浓度范围变得更广的理由出发,作为上述结构b优选具有2个以上上述式(b-5)所表示的含氟末端结构,更优选具有4~6个上述式(b-5)所表示的含氟末端结构。
[0131]
并且,本发明的化合物中,从可使用的浓度范围进一步变得广的理由出发,优选作为上述结构b,具有4~6个上述式(b-5)所表示的含氟末端结构,且上述结构a中,作为位于分子的中心的环结构,具有上述式(cy-1)或(cy-8)所表示的环结构。
[0132]
对于连结上述结构a与上述结构b的连结基团(以下,省略为“连结基团x”。)并没有特别限制,但是可以举出单键、或者碳原子数1~30的直链或支链的亚烷基。
[0133]
其中,上述亚烷基中的任意的亚甲基可以被-o-、-s-、-nr-、-c=c-、-c=n-、-c≡c-或-c(=o)-取代。但是,r表示碳原子数1~3的烷基。
[0134]
这些之中,作为连结基团x,优选为碳原子数1~10的直链的亚烷基。更优选为碳原子数2~7的亚烷基。尤其进一步优选为上述亚烷基中的任意的亚甲基被-c(=o)-或-o-取代的结构。
[0135]
以下,示出本发明中优选的化合物的具体例。但是,本发明中能够采用的化合物不应被下述具体例限定性解释。
[0136]
[化学式9]
[0137][0138]
[化学式10]
[0139][0140]
[化学式11]
[0141][0142]
[化学式12]
[0143]
[0144][0145]
[化学式13]
[0146][0147]
[化学式14]
[0148][0149]
[化学式15]
[0150][0151]
[化学式16]
[0152]
[0153][0154]
[化学式17]
[0155][0156]
[化学式18]
[0157]
[0158][0159]
[化学式19]
[0160]
[0161][0162]
[化学式20]
[0163][0164]
[化学式21]
[0165][0166]
[化学式22]
[0167][0168]
[化学式23]
[0169][0170]
[化学式24]
[0171]
[0172][0173]
[化学式25]
[0174][0175]
[化学式26]
[0176][0177]
[化学式27]
[0178][0179]
[化学式28]
[0180][0181]
[化学式29]
[0182][0183]
[化学式30]
[0184][0185]
[化学式31]
[0186][0187]
[化学式32]
[0188][0189]
[化学式33]
[0190][0191]
[化学式34]
[0192][0193]
[化学式35]
[0194][0195]
[化学式36]
[0196][0197]
[化学式37]
[0198][0199]
[化学式38]
[0200][0201]
[化学式39]
[0202][0203]
[化学式40]
[0204]
[0205][0206]
[化学式41]
[0207][0208]
[化学式42]
[0209]
[0210][0211]
[化学式43]
[0212][0213]
[化学式44]
[0214][0215]
[化学式45]
[0216][0217]
[化学式46]
[0218][0219]
[化学式47]
[0220][0221]
[化学式48]
[0222][0223]
[化学式49]
[0224][0225]
[化学式50]
[0226]
[0227][0228]
[化学式51]
[0229]
[0230][0231]
[化学式52]
[0232][0233]
[化学式53]
[0234][0235]
[化学式54]
[0236][0237]
[化学式55]
[0238][0239]
[化学式56]
[0240]
[0241][0242]
[化学式57]
[0243][0244]
[化学式58]
[0245][0246]
[化学式59]
[0247][0248]
[化学式60]
[0249][0250]
关于本发明的化合物,能够通过适当选择日本特开2002-129162号公报及日本特开2013-47204号公报、以及在这些公报中所引用的文献中所记载的合成法并进行组合来合成。并且,其他公知的合成法也能够根据需要通过组合来合成。
[0251]
[液晶组合物]
[0252]
本发明的液晶组合物是含有上述的本发明的化合物及聚合性液晶化合物的液晶组合物。
[0253]
在本发明的液晶组合物中,上述的本发明的化合物能够用作液晶取向促进剂。
[0254]
并且,本发明的液晶组合物还可以并用1种以上的非聚合性的液晶化合物。
[0255]
并且,本发明的液晶组合物可以使用两种以上上述的本发明的化合物(液晶取向促进剂),也可以并用不属于本发明的化合物的液晶取向促进剂。
[0256]
其中,本发明的液晶组合物中所含的本发明的化合物(液晶取向促进剂)的含量相对于后述的聚合性液晶化合物100质量份,优选为0.01~20质量份,更优选为0.03~2质量份。
[0257]
〔聚合性液晶化合物〕
[0258]
聚合性液晶化合物是指具有聚合性基团的液晶化合物。
[0259]
关于聚合性液晶化合物的种类,并没有特别限制。
[0260]
通常,液晶化合物能够根据其形状而分类为棒状类型(棒状液晶化合物)和圆盘状类型(盘状液晶化合物)。此外,液晶化合物能够分类为低分子类型与高分子类型。高分子通常是指聚合度为100以上的化合物(高分子物理
·
相变动力学,土井正男著,2页,岩波书店,1992)。
[0261]
在本发明中,能够使用任一的液晶化合物,优选使用至少1种以上的聚合性棒状液晶化合物或聚合性圆盘状液晶化合物,更优选使用聚合性棒状液晶化合物。也可以使用2种以上的聚合性棒状液晶化合物、2种以上的聚合性圆盘状液晶化合物或聚合性棒状液晶化合物和聚合性圆盘状液晶化合物的混合物。
[0262]
作为聚合性圆盘状液晶化合物,例如,能够优选使用日本特开2002-129162号公报的0161~0171段、日本特开2007-108732号公报的0020~0067段、日本特开2010-244038号公报的0013~0108段等中所记载的化合物。
[0263]
作为聚合性棒状液晶化合物,优选使用甲亚胺类、氧化偶氮类、氰基联苯类、氰基苯酯类、苯甲酸酯类、环己烷羧酸苯酯类、氰基苯基环己烷类、氰基取代苯基嘧啶类、烷氧基取代苯基嘧啶类、苯基二噁烷类、二苯乙炔类及链烯基环己基苄腈类。
[0264]
作为聚合性棒状液晶化合物的具体例,例如,可以举出makromol.chem.,190卷、2255页(1989年)、advanced materials 5卷、107页(1993年)、美国专利4683327号、美国专利5622648号、美国专利5770107号、wo95/22586号、wo95/24455号、wo97/00600号、wo98/23580号、wo98/52905号、日本特开平1-272551号、日本特开平6-16616号、日本特开平7-110469号、日本特开平11-80081号、日本特表平11-513019号、日本特开2001-328973号、日本特开2005-289980号、日本特开2014-198815号及日本特开2014-198814号公报等中所记载的化合物。可以并用2种以上的聚合性液晶化合物。若并用2种以上的聚合性液晶化合物,则能够降低取向温度。
[0265]
关于本发明的液晶组合物中的聚合性液晶化合物的含量,并没有特别限制,相对于液晶组合物中的总固体成分的总质量,优选为50质量%以上,更优选为70质量%以上。关于上限,并没有特别限制,但95质量%以下的情况较多。
[0266]
另外,固体成分是指,去除溶剂后能够形成光学各向异性层的成分,其性状即使为液体状也设为固体成分。
[0267]
本发明的液晶组合物除了聚合性液晶化合物及液晶取向促进剂之外,还能够根据需要包含溶剂、后述的手性试剂及聚合性引发剂、或者其他的添加剂(例如,纤维素酯)。
[0268]
〔溶剂〕
[0269]
本发明的液晶组合物可以包含溶剂。
[0270]
作为溶剂,优选使用有机溶剂。
[0271]
作为有机溶剂,可以举出,酰胺(例如,n,n-二甲基甲酰胺等)、亚砜(例如,二甲基亚砜等)、烃(例如,甲苯、己烷等)、卤代烷(例如,氯仿、二氯甲烷等)、酯(例如,乙酸甲酯、乙酸丁酯、丙酸乙酯等)、酮(例如,丙酮、甲基乙基酮、环己酮、甲基异丁基酮、环茂酮等)、醚(例如,四氢呋喃、1,2-二甲氧基乙烷等)。
[0272]
这些有机溶剂中,优选酯及酮。
[0273]
并且,作为溶剂,可以并用二种以上的有机溶剂。
[0274]
〔手性试剂〕
[0275]
本发明的液晶组合物可以包含手性试剂。
[0276]
手性试剂具有诱发胆甾醇型液晶相的螺旋结构的功能。
[0277]
手性试剂根据其化合物结构而诱发的螺旋的扭曲方向或螺旋间距不同,因此根据目的选择即可。
[0278]
作为手性试剂,并没有特别限制,能够使用公知的化合物(例如记载于液晶装置手册、第3章4-3项、tn、stn用手性试剂、199页、日本学术振兴会第142委员会编、1989)、异山梨醇衍生物、异甘露糖醇衍生物、联萘酚衍生物等。
[0279]
手性试剂通常包含不对称碳原子,但不包含不对称碳原子的轴向不对称化合物或表面不对称化合物也能够用作手性试剂。
[0280]
轴向不对称化合物或表面不对称化合物的例中包括联萘、螺烯、对环芳烷及它们的衍生物。
[0281]
手性试剂可以具有聚合性基团。在手性试剂和液晶化合物均具有聚合性基团的情况下,可以通过聚合性手性试剂和聚合性液晶化合物的聚合反应来形成具有由聚合性液晶化合物衍生的重复单元和由手性试剂衍生的重复单元的聚合物。在该方式中,聚合性手性试剂所具有的聚合性基团优选为与聚合性液晶化合物所具有的聚合性基团相同种类的基团。因此,手性试剂的聚合性基团也优选为不饱和聚合性基团、环氧基或氮丙啶基,进一步优选为不饱和聚合性基团,尤其优选为烯属不饱和聚合性基团。
[0282]
并且,手性试剂可以为液晶化合物。
[0283]
在手性试剂具有光异构化基团的情况下,在涂布、取向之后,能够通过活化光线等光掩模照射来形成与发光波长相对应的所期望的反射波长的图案,因此优选。作为光异构化基团,优选为显示光致变色性的化合物的异构化部位、偶氮苯部位、肉桂酰基部位、α-氰基肉桂酰基部位、芪部位、查耳酮部位。作为具体的化合物,能够使用日本特开2002-80478号公报、日本特开2002-80851号公报、日本特开2002-179668号公报、日本特开2002-179669号公报、日本特开2002-179670号公报、日本特开2002-179681号公报、日本特开2002-179682号公报、日本特开2002-338575号公报、日本特开2002-338668号公报、日本特开2003-313189号公报、日本特开2003-313292号公报中所记载的化合物。
[0284]
作为手性试剂的具体例,可以举出以下的式(12)所表示的化合物。另外,式(12)中,x为2~5(整数)。
[0285]
[化学式61]
[0286][0287]
本发明的液晶组合物含有手性试剂时的手性试剂的含量,优选为聚合性液晶性化合物量的0.01摩尔%~200摩尔%,更优选为1摩尔%~30摩尔%。
[0288]
〔聚合引发剂〕
[0289]
本发明的液晶组合物可以包含聚合引发剂。
[0290]
本发明中优选的聚合反应是使用热聚合引发剂的热聚合反应和使用光聚合引发剂的光聚合反应,更优选光聚合反应。
[0291]
作为光聚合引发剂的例,包括α-羰基化合物(记载于美国专利2367661号、美国专利2367670号的各说明书)、偶姻醚(记载于美国专利2448828号说明书)、α-烃取代的芳香族偶姻化合物(记载于美国专利2722512号说明书)、多核醌化合物(记载于美国专利3046127号、美国专利2951758号的各说明书)、三芳基咪唑二聚物与对氨基苯基酮的组合(记载于美国专利3549367号说明书)、吖啶及吩嗪化合物(记载于日本特开昭60-105667号公报、美国专利4239850号说明书)、噁二唑化合物(记载于美国专利4212970号说明书)、酰基氧化膦化合物(记载于日本特公昭63-40799号公报、日本特公平5-29234号公报、日本特开平10-95788号公报、日本特开平10-29997号公报)、或肟酯化合物(例如omni公司制oxe-01、oxe-02或adeka corporation制nci-1919等)。
[0292]
本发明的液晶组合物含有聚合引发剂时的聚合引发剂的含量相对于液晶组合物的固体成分的总质量,优选为0.01~20质量%,进一步优选为0.4~8质量%。
[0293]
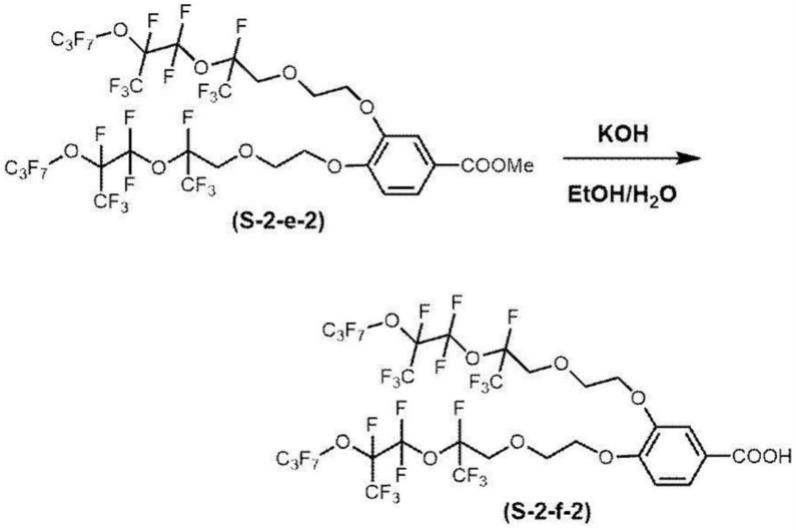
[固化物]
[0294]
本发明的固化物是使上述的本发明的液晶组合物聚合而成的固化物。
[0295]
本发明的固化物能够用作光学各向异性层。
[0296]
该光学各向异性层是使聚合性液晶化合物(聚合性棒状液晶化合物或聚合性圆盘状液晶化合物)取向之后,通过聚合固定而形成的层,形成为层之后不再需要显示液晶性。
[0297]
[薄膜]
[0298]
本发明的薄膜是含有上述本发明的固化物中的至少1种的薄膜。
[0299]
并且,本发明的薄膜是将上述本发明的液晶组合物的胆甾醇型液晶相固定而成的薄膜。
[0300]
本发明的薄膜能够通过利用涂布等方法将本发明的液晶组合物制膜来形成。能够通过将液晶组合物涂布于后述的取向膜上来形成液晶层而制作光学各向异性元件。
[0301]
液晶组合物的涂布能够通过公知的方法(例如,挤压涂布法、直接凹版涂布法、逆向凹版涂布法、模涂法、棒涂法等)来实施。
[0302]
液晶性分子优选维持取向状态而固定。
[0303]
并且,固定化优选通过导入到液晶性分子中的聚合性基团的聚合反应来实施。
[0304]
关于本发明的液晶组合物的聚合方法,优选通过对上述的光聚合引发剂进行光照射来进行光聚合,作为光源优选使用紫外线。照射能优选为5mj/cm2~100j/cm2,更优选为30~600mj/cm2,进一步优选为100~400mj/cm2。为了促进光聚合反应,可以在加热条件下实施
光照射。关于液晶层的厚度,优选为0.1~50μm,进一步优选为0.3~20μm,最优选为1~10μm。
[0305]
关于形成液晶层的液晶组合物(即,涂膜)中所含的本发明的化合物的涂布量,优选为0.005~0.5g/m2,更优选为0.01~0.45g/m2,进一步优选为0.02~0.4g/m2,最优选为0.03~0.35g/m2。
[0306]
〔取向膜〕
[0307]
取向膜能够利用,有机化合物(优选聚合物)的摩擦处理、无机化合物的倾斜蒸镀、具有微槽的层的形成或基于朗朗缪尔
·
布洛杰特法(lb膜)的有机化合物(例如,ω-二十三烷酸、双十八烷基甲基氯化铵、硬脂酸甲酯等)的累积等方法来设置。
[0308]
此外,还已知通过赋予电场、赋予磁场或光照射而产生取向功能的取向膜。其中,优选为通过光照射而产生取向功能的光取向膜。
[0309]
摩擦处理通过用纸或布沿规定方向擦拭数次聚合物层的表面来实施。
[0310]
取向膜中所使用的聚合物的种类根据液晶性分子的取向(尤其平均倾斜角)来确定。为了使液晶性分子水平(平均倾斜角:0~50
゜
)取向,使用不降低取向膜的表面能的聚合物(通常的取向膜用聚合物)。为了使液晶性分子垂直(平均倾斜角:50~90
゜
),使用降低取向膜的表面能的聚合物。为了降低取向膜的表面能,优选在聚合物的侧链导入碳原子数为10~100的烃基。
[0311]
对于具体的聚合物的种类,在关于使用与各种各样的显示模式相对应的液晶性分子的光学补偿片的文献中有所记载。
[0312]
取向膜的厚度优选为0.01~5μm,进一步优选为0.05~1μm。另外,可以使用取向膜而使光学各向异性层的液晶性分子取向之后,将液晶层转印在透明支撑体上。在取向状态下被固定的液晶性分子,即使没有取向膜也能够维持取向状态。
[0313]
并且,在平均倾斜角为小于5
゜
的取向的情况下,不需要进行摩擦处理,也不需要取向膜。但是,以改善液晶性分子与透明支撑体的密合性为目的,可以使用在界面与液晶性分子形成化学键的取向膜(日本特开平9-152509号公报中所记载)。在以改善密合性为目的使用取向膜的情况下,也可以不实施摩擦处理。在将两种液晶层设置于透明支撑体的同一侧的情况下,还能够使形成于透明支撑体上的液晶层作为设置于其上的液晶层的取向膜而发挥作用。
[0314]
〔透明支撑体〕
[0315]
本发明的薄膜或具有本发明的薄膜的光学各向异性元件可以具有透明支撑体。
[0316]
作为透明支撑体,可以使用玻璃板或聚合物薄膜,优选使用聚合物薄膜。支撑体为透明是指,透光率为80%以上。
[0317]
作为透明支撑体,通常使用光学各向同性的聚合物薄膜。具体而言,光学各向同性是指,面内延迟(re)优选小于10nm,进一步优选小于5nm。并且,在光学各向同性透明支撑体中,厚度方向的延迟(rth)也优选小于10nm,进一步优选小于5nm。透明支撑体的面内延迟(re)与厚度方向的延迟(rth),分别通过下述式来定义。
[0318]
re=(nx-ny)d
[0319]
rth=[{(nx ny)/2}-nz]d式中,nx及ny为透明支撑体的面内折射率,nz为透明支撑体的厚度方向的折射率,然后d为透明支撑体的厚度。
[0320]
有时作为透明支撑体而使用光学各向异性的聚合物薄膜。
[0321]
在这种情况下,透明支撑体优选具有光学单轴性或光学双轴性。在光学单轴性支撑体的情况下,在光学上可以为正(光轴方向的折射率比垂直于光轴的方向的折射率大),也可以为负(光轴方向的折射率比垂直于光轴的方向的折射率小)。在光学双轴性支撑体的情况下,上述式的折射率nx、ny及nz全部成为不同的值(nx≠ny≠nz)。
[0322]
光学各向异性透明支撑体的面内延迟(re)优选为10~1000nm,进一步优选为15~300nm,最优选为20~200nm。光学各向异性透明支撑体的厚度方向的延迟(rth)优选为10~1000nm,更优选为15~300nm,进一步优选为20~200nm。
[0323]
关于形成透明支撑体的材料,根据设为光学各向同性支撑体还是设为光学各向异性支撑体来确定。在光学各向同性支撑体的情况下,通常使用玻璃或纤维素酯。在光学各向异性支撑体的情况下,通常使用合成聚合物(例如,聚碳酸酯、聚砜、聚醚砜、聚丙烯酸酯、聚甲基丙烯酸酯、降冰片烯树脂)。但是,能够通过欧州专利0911656a2号说明书中所记载的(1)延迟上升剂的使用、(2)乙酸纤维素的乙酰化度的降低、或(3)基于冷却溶解法的薄膜的制造,来制造光学各向异性的(延迟高)纤维素酯薄膜。由聚合物薄膜构成的透明支撑体优选通过溶液流延法来形成。
[0324]
为了获得光学各向异性透明支撑体,优选对聚合物薄膜实施拉伸处理。在制造光学单轴性支撑体的情况下,实施通常的单轴拉伸处理或双轴拉伸处理即可。在制造光学双轴性支撑体的情况下,优选实施不平衡双轴拉伸处理。在不平衡双轴拉伸中,将聚合物薄膜沿某个方向拉伸一定倍率(例如3~100%,优选为5~30%),在与其垂直的方向上拉伸其以上的倍率(例如6~200%,优选为10~90%)。可以同时实施两个方向的拉伸处理。拉伸方向(不平衡双轴拉伸中为拉伸倍率高的方向)与拉伸后的薄膜的面内慢轴优选实质上成为相同方向。拉伸方向与慢轴的角度优选小于10
゜
,进一步优选小于5
゜
,最优选小于3
゜
。
[0325]
透明支撑体的厚度优选为10~500μm,进一步优选为50~200μm。为了改善透明支撑体与设置于其上的层(粘接层、取向膜或光学各向异性层)的粘接,可以对透明支撑体实施表面处理(例如,辉光放电处理、电晕放电处理、紫外线(uv)处理、火焰处理)。可以对透明支撑体添加紫外线吸收剂。可以在透明支撑体上设置粘接层(下涂层)。关于粘接层,在日本特开平7-333433号公报中有所记载。粘接层的厚度优选为0.1~2μm,进一步优选为0.2~1μm。透明支撑体可以在形成薄膜之后进行剥离。
[0326]
[用途]
[0327]
本发明的薄膜尤其将本发明的液晶组合物的胆甾醇型液晶相固定而成的薄膜,能够用作对规定的波长区域的光显示选择反射特性的层。
[0328]
固定胆甾醇型液晶相而成的层(胆甾醇型液晶层)在其选择反射波长区域中,作为选择性地反射右旋圆偏振光及左旋圆偏振光中的任一个,使另一个旋向的圆偏振光透过的圆偏振光选择反射层发挥作用。
[0329]
包含1层或2层以上胆甾醇型液晶层的薄膜能够用于各种用途中。在包含2层以上胆甾醇型液晶层的薄膜中,各胆甾醇型液晶层所反射的圆偏振光的旋向根据用途可以相同或相反。并且,各胆甾醇型液晶层的后述的选择反射的中心波长也可以根据用途相同或不同。
[0330]
另外,在本说明书中,当对于圆偏振光提及“旋向”时,是指是右旋圆偏振光或左旋
圆偏振光。关于圆偏振光的旋向,定义为如下:在以光朝向正前方行进的方式眺望的情况下,电场矢量的前端随着时间的增加而顺时针旋转的情况为右旋圆偏振光,逆时针旋转的情况为左旋圆偏振光。本说明书中,有时还会对胆甾醇型液晶的螺旋的扭曲方向使用术语“旋向”。关于基于胆甾醇型液晶的选择反射,在胆甾醇型液晶的螺旋的扭曲方向(旋向)为右的情况下,反射右旋圆偏振光,并透射左旋圆偏振光,在旋向为左的情况下,反射左旋圆偏振光,并透射右旋圆偏振光。
[0331]
例如,包含在可见光波长区域(波长400~750nm)显示选择反射特性的胆甾醇型液晶层的薄膜能够用作投影图像显示用的屏幕及半反射镜。
[0332]
并且,通过控制成在红外线波长区域中显示选择反射特性,能够用作隔热薄膜或用作传感器用的红外线截止滤波器。
[0333]
并且,上述反射层能够用于作为光学元件的构成要件的偏振元件、反射膜、防反射膜、视场角补偿膜、全息及取向膜等各种用途中。
[0334]
根据胆甾醇型液晶层的上述功能,能够在投射光中表现出选择反射的波长中,反射任意一个旋向的圆偏振光,从而形成投影图像。投影图像可以是显示在投影图像显示用部件表面上并被这样视觉辨认的投影图像,也可以是从观察者观察时浮现在投影图像显示用部件的前端上而看到的虚像。
[0335]
上述选择反射的中心波长λ依赖于胆甾醇型液晶相中的螺旋结构的间距p(=螺旋的周期),与胆甾醇型液晶层的平均折射率n遵从λ=n
×
p的关系。另外,其中,胆甾醇型液晶层所具有的选择反射的中心波长λ是指位于从胆甾醇型液晶层的法线方向测定的圆偏振光反射光谱的反射峰的重心位置上的波长。由上述式可知,通过调节螺旋结构的间距,能够调节选择反射的中心波长。即,调节n值与p值,例如,调节中心波长λ以相对于蓝色光选择性地反射右旋圆偏振光及左旋圆偏振光中的任一个,从而能够使表观上的选择反射的中心波长成为450~495nm的波长区域。另外,表观上的选择反射的中心波长是指,位于从实际使用时(作为投影图像显示用部件使用时)的观察方向测定的胆甾醇型液晶层的圆偏振光反射光谱的反射峰的重心位置上的波长。由于胆甾醇型液晶相的间距依赖于与液晶化合物一起使用的手性试剂的种类或其添加浓度,因此通过对它们进行调节能够获得所期望的间距。关于螺旋的旋向及间距的测定法,能够使用“液晶化学实验入门”日本液晶学会编西格玛(sigma)出版2007年出版、46页及“液晶便览”液晶便览编辑委员会丸善196页中所记载的方法。
[0336]
并且,分别制作在红色光波长区域、绿色光波长区域及蓝色光波长区域分别具有表观上的选择反射的中心波长的胆甾醇型液晶层,并将它们进行层叠,由此能够制作能够显示全彩的投影图像的投影图像显示用部件。
[0337]
通过根据投影中所使用的光源的发光波长区域及投影图像显示用部件的使用方式来调节各胆甾醇型液晶层的选择反射的中心波长,能够显示光利用效率良好且清晰的投影图像。特别是,通过分别根据投影中所使用的光源的发光波长区域等来分别调节胆甾醇型液晶层的选择反射的中心波长,能够显示光利用效率良好且清晰的彩色投影图像。
[0338]
并且,例如,通过将上述投影图像显示用部件设为相对于可见光区域的光具有透射性的结构,能够设为平视显示器的投影图像显示用的半反射镜。投影图像显示用半反射镜能够以可视觉辨认的方式显示从投影仪投影的图像,并且在从显示有图像的相同的面侧
观察投影图像显示用半反射镜时,能够同时观察位于相反的面侧上的信息或风景。
[0339]
实施例
[0340]
以下,举出实施例和比较例,对本发明的特征进行进一步具体的说明。以下的实施例中示出的材料、使用量、比例、处理内容、处理步骤等只要不脱离本发明的主旨则能够适当进行变更。因此,本发明的范围不应通过以下所示的具体例进行限定性解释。
[0341]
(化合物(a-101)的合成)
[0342]
通过下述途径合成了化合物(a-101)。
[0343]
[化学式62]
[0344][0345]
<二醇(101b)的合成>
[0346]
使由羟基苯甲酸及甲基氢醌组成的二苯酚(101a)(40mmol)和2-溴乙基苄基醚(85mmol),在碳酸钾(90mmol)的存在下,在二甲基乙酰胺(dmac)80ml中,在90℃下反应了3小时。加入乙酸乙酯和盐酸水进行分液处理,并过滤获取了添加甲醇所生成的固体。
[0347]
使用所有过滤获取到的固体,在乙酸乙酯200ml中,在钯催化剂(1.2g,5%钯/活性炭,degussa类型e 101o/w 5%pd,wako公司制)的存在下,与氢反应。
[0348]
反应结束后,通过硅藻土过滤去除了钯催化剂,过滤获取了加入甲醇而生成的固体。使用乙酸乙酯和甲醇进行重结晶,获得了二醇(101b)(两步工序产率82%)。
[0349]
<化合物(a-101)的合成>
[0350]
使二醇(101b)(10mmol)与三乙基胺(24mmol)溶解于dmac50ml,滴加氟化合物(101c)(26mmol),并在40℃下反应了4小时。
[0351]
之后,加入乙酸乙酯和盐酸水进行分液处理之后,用蒸发器浓缩并进行管柱纯化,从而得到了化合物(a-101)(产率65%)。
[0352]
以下示出所得到的化合物(a-101)的1h-nmr(nuclear magnetic resonance)数据。
[0353]1h-nmr(400mhz,cdcl3)δ(ppm):2.3(s,3h)、4.3-4.6(m,8h)、7.1-7.3(m,7h)、8.1(d、2h)
[0354]
(化合物(a-410)的合成)
[0355]
通过下述途径合成了化合物(a-410)。
[0356]
[化学式63]
[0357][0358]
<酯(410b)的合成>
[0359]
向二氯甲烷100ml中加入醇(410a)(100mmol),并向其中加入了三乙基胺(105mmol)。将该溶液浸渍在冰水中,以内温成为20℃以下的方式滴加三氟甲磺酸酐(105mmol),并在冰冷下反应了1小时。对反应液实施分液操作,通过蒸发器对有机层进行了浓缩。将所得到的液体进行减压蒸留,获得了对应的三氟甲磺酸酯(410b)(产率88%)。
[0360]
<酯(410c)的合成>
[0361]
将酯(410b)(46.5mmol)和没食子酸甲酯(15mmol),在碳酸钾(46.5mmol)的存在下,在dmac15ml中,在90℃下反应了2小时。分液处理之后,进行管柱纯化,得到了酯(410c)(产率80%)。
[0362]
以下示出所得到的酯(410c)的1h-nmr数据。
[0363]1h-nmr(400mhz,cdcl3)δ(ppm):3.9(s,3h)、4.1-4.5(m,6h)、7.1(s,2h)
[0364]
<羧酸(410d)的合成>
[0365]
将酯(410c)(10mmol)加入到乙醇30ml及水3ml中。向该溶液中添加氢氧化钾(15mmol)并加热了2小时。将该反应液滴加于盐酸水溶液中,加入乙酸乙酯进行分液处理后,用蒸发器浓缩并进行管柱纯化,从而得到了羧酸(410d)(产率79%)。
[0366]
<化合物(a-410)的合成>
[0367]
使羧酸(410d)(4.0mmol)在甲苯20ml与催化剂量的二甲基甲酰胺(dmf)中与亚硫酰氯(6.0mmol)反应而制成酰氯,去除过剩的亚硫酰氯及甲苯之后,向体系中添加了四氢呋喃(thf)8ml、催化剂量的4-二甲基氨基吡啶(dmap)。
[0368]
向其中添加溶于thf8ml的氢醌(2.0mmol),滴加二异丙基乙胺(4.2mmol)与thf3ml的混合液,并在室温下反应了3小时。
[0369]
分液操作后,用蒸发器浓缩并进行管柱纯化,从而得到了化合物(a-410)(产率49%)。
[0370]
以下示出所得到的化合物(a-410)的1h-nmr数据。
[0371]1h-nmr(400mhz,cdcl3)δ(ppm):4.1-4.5(m,12h)、7.1-7.3(s
×
2,4h 4h)
[0372]
通过下述途径合成了化合物(a-501)。
[0373]
[化学式64]
[0374][0375]
<羧酸(501b)的合成>
[0376]
将醇(501a)(20mmol)和琥珀酸酐(20mmol)在四氢呋喃中与催化剂量的三乙基胺一起回流了2小时。用甲苯和1m盐酸水进行分液操作之后,将甲苯进行减压浓缩而得到了501b(产率98%)。
[0377]
<羧酸(501c)的合成>
[0378]
向羧酸(501b)(15mmol)及甲苯40ml的溶液中,加入催化剂量的dmf并使其与亚硫酰氯(18mmol)反应而制成酰氯,去除了过剩的亚硫酰氯及甲苯。
[0379]
接着,向四氢呋喃中,加入3,5-二羟基苯甲酸(5mmol)和501b的酰氯,在冰冷下滴加吡啶(20mmol)之后,在室温下反应了2小时。加入水(5ml)之后,在40℃下进一步搅拌30分钟,然后用乙酸乙酯和1m盐酸水实施分液操作,最后过滤加入甲醇和水而生成的固体,并使其干燥而得到了羧酸(501c)(产率74%)。
[0380]
以下示出所得到的羧酸(501c)的1h-nmr数据。
[0381]1h-nmr(400mhz,cdcl3)δ(ppm):2.7-2.9(m,8h)、4.2-4.5(m,4h)、7.6(s,1h)、7.8(s,2h)
[0382]
<酰氯(501d)的合成>
[0383]
向羧酸(501c)(10mmol)及甲苯30ml的溶液中,加入催化剂量的dmf并使其与亚硫酰氯(14mmol)反应而制成酰氯,去除过剩的亚硫酰氯及甲苯从而得到了酰氯(501d)(产率99%)。
[0384]
<a-501的合成>
[0385]
将酰氯(501d)(9mmol)与催化剂量的dmap设为thf20ml的溶液,向其中添加溶于thf8ml的1,3,5-三羟基苯(2.9mmol),滴加二异丙基乙胺(9.5mmol)与thf5ml的混合液之
后,在室温下反应了3小时。分液操作后,用蒸发器浓缩并进行管柱纯化,从而得到了化合物(a-501)(产率62%)。
[0386]
以下示出所得到的化合物(a-501)的1h-nmr数据。
[0387]1h-nmr(400mhz、cdcl3)δ(ppm):2.7-2.9(m,24h)、4.2-4.5(m,12h)、7.3(s,3h)、7.6(s,3h)、7.8(s,6h)
[0388]
〔含氟化合物的合成〕
[0389]
<化合物(s-2-b)的合成>
[0390]
通过下述途径合成了化合物(s-2-b)。
[0391]
[化学式65]
[0392][0393]
向1l三口烧瓶中,加入了化合物(s-2-a)200g(0.42mol)、四丁基铵硫酸氢盐28.2g(0.08mol)、水13ml及甲苯180ml。接着,在室温下添加溴乙酸叔丁酯96.8g(0.50mol),添加后将内温升温至30℃。在保持内温40℃以下的同时向其中滴加将氢氧化钠66.4g(1.66mol)溶解于水66ml的液体。在内温35℃下搅拌2小时,将反应液冷却至20℃。接着,滴加混合了浓盐酸145ml与水145ml的液体,静置后去除了水层。在有机层中添加食盐51g、水460ml及甲苯180ml,并进行了搅拌。静置后,再次去除水层,并用25%食盐水350ml清洗了有机层。将有机层用无水硫酸镁干燥之后,通过过滤去除干燥剂,并用旋转蒸发器对有机层进行了浓缩。定量地得到了250g无色油状的化合物(s-2-b)。
[0394]
<化合物(s-2-c)的合成>
[0395]
通过下述途径合成了化合物(s-2-c)。
[0396]
[化学式66]
[0397][0398]
在氮气流下,向2l三口烧瓶中,加入red-al122.2g(0.42mol)、甲苯196ml,将内温
冷却至10℃以下。向其中滴加化合物(s-2-b)247g(0.41mol),用甲苯50ml进行了清洗。滴加后,将内温升温至40℃并搅拌30分钟之后,添加了red-al12.0g(0.04mol)。添加后进一步在40℃下搅拌30分钟,将反应液冷却至15℃以下。滴加将罗谢尔盐四水合物132.5g(0.94mol)溶解于水153ml中的液体,并搅拌了30分钟。静置后,去除水层,在有机层中添加将罗谢尔盐四水合物66.3g(0.47mol)溶解于水76ml中的液体,并搅拌了5分钟。静置后,去除水层并用10%食盐水260ml清洗了有机层。将有机层用无水硫酸镁干燥之后,通过过滤去除干燥剂,并用旋转蒸发器对有机层进行了浓缩。得到了217g(产率99%)淡黄色油状的化合物(s-2-c)。
[0399]
<化合物(s-2-d)的合成>
[0400]
通过下述途径合成了化合物(s-2-d)。
[0401]
[化学式67]
[0402][0403]
向2l三口烧瓶中,加入化合物(s-2-c)205g(0.39mol)、乙酸乙酯246ml及甲烷磺酰氯53.6g(0.47mol),并将内温冷却至10℃以下。在保持内温20℃以下的同时向其中滴加了三乙基胺42.8g(0.42mol)。滴加后,将内温升温至25℃并搅拌了2小时。添加混合了己烷246ml、10%食盐水246ml及重曹6.2g的液体,并搅拌了5分钟。静置后,去除水层并用10%食盐水246ml清洗了有机层。将有机层用无水硫酸镁干燥之后,通过过滤去除干燥剂,并用旋转蒸发器对有机层进行了浓缩。得到了233g(产率99%)淡黄色油状的化合物(s-2-d)。
[0404]
<化合物(s-2-e-1)的合成>
[0405]
通过下述途径合成了化合物(s-2-e-1)。
[0406]
[化学式68]
2-e-1)。
[0414]
<化合物(s-2-e-3)的合成>
[0415]
通过下述途径合成了化合物(s-2-e-3)。
[0416]
[化学式70]
[0417][0418]
将原料的3,5-二羟基苯甲酸甲酯变更为3,4,5-三羟基苯甲酸甲酯,且将化合物(s-2-d)与碳酸钾的使用量变更为1.5倍量,除此以外,以与化合物(s-2-e-1)相同的方式进行合成,得到了116g(产率90%)淡黄色油状的化合物(s-2-e-3)。
[0419]
<化合物(s-2-e-4)的合成>
[0420]
通过下述途径合成了化合物(s-2-e-4)。
[0421]
[化学式71]
[0422][0423]
将原料的3,5-二羟基苯甲酸甲酯变更为4-羟基苯甲酸甲酯,且将化合物(s-2-d)
与碳酸钾的使用量变更为一半,除此以外,以与化合物(s-2-e-1)相同的方式进行合成,得到了50.4g(产率92%)淡黄色油状的化合物(s-2-e-4)。
[0424]
<化合物(s-2-f-1)的合成>
[0425]
通过下述途径合成了化合物(s-2-f-1)。
[0426]
[化学式72]
[0427][0428]
向1l三口烧瓶中,加入化合物(s-2-e-1)45.0g(38mmol)、乙醇145ml、氢氧化钾5.3g(95mmol)及水53ml,并在内温70℃下搅拌了1小时。将反应液冷却至20℃后,添加乙酸乙酯240ml、浓盐酸7ml及水241ml,并进行了搅拌。静置后,去除水层并用10%食盐水150ml清洗了有机层。将有机层用无水硫酸镁干燥之后,通过过滤去除干燥剂,并用旋转蒸发器对有机层进行了浓缩。得到了41.1g(产率97%)褐色油状的化合物(s-2-f-1)。
[0429]
<化合物(s-2-f-2)的合成>
[0430]
通过下述途径合成了化合物(s-2-f-2)。
[0431]
[化学式73]
[0432][0433]
将原料的化合物(s-2-e-1)变更为化合物(s-2-e-2),除此以外,以与化合物(s-2-f-1)相同的方式进行合成,得到了40.5g(产率96%)褐色油状的化合物(s-2-f-1)。
[0434]
<化合物(s-2-f-3)的合成>
[0435]
通过下述途径合成了化合物(s-2-f-3)。
[0436]
[化学式74]
[0437][0438]
将原料的化合物(s-2-e-1)变更为化合物(s-2-e-3),除此以外,以与化合物(s-2-f-1)相同的方式进行合成,得到了113.8g(产率85%)褐色油状的化合物(s-2-f-3)。
[0439]
<化合物(s-2-f-4)的合成>
[0440]
通过下述途径合成了化合物(s-2-f-4)。
[0441]
[化学式75]
[0442][0443]
将原料的化合物(s-2-e-1)变更为化合物(s-2-e-4),除此以外,以与化合物(s-2-f-1)相同的方式进行合成,得到了51.5g(产率95%)褐色油状的化合物(s-2-f-4)。
[0444]
<化合物(s-2-g-1)的合成>
[0445]
通过下述途径合成了化合物(s-2-g-1)。
[0446]
[化学式76]
[0447][0448]
向100ml三口烧瓶中,加入化合物(s-2-f-1)9.5g(8.1mmol)、二甲基甲酰胺0.13ml及甲苯23ml,并将内温升温至70℃。向其中添加亚硫酰氯1.2ml(18mmol),并搅拌了1小时。减压蒸馏甲苯,定量地得到了褐色油状的化合物(s-2-g-1)9.7g。
[0449]
<化合物(s-2-g-2)的合成>
[0450]
通过下述途径合成了化合物(s-2-g-2)。
[0451]
[化学式77]
[0452][0453]
将原料的化合物(s-2-f-1)变更为化合物(s-2-f-2),除此以外,以与化合物(s-2-g-1)相同的方式进行合成,定量地得到了褐色油状的化合物(s-2-g-2)9.8g。
[0454]
<化合物(s-2-g-3)的合成>
[0455]
通过下述途径合成了化合物(s-2-g-3)。
[0456]
[化学式78]
[0457][0458]
将原料的化合物(s-2-f-1)变更为化合物(s-2-f-3),除此以外,以与化合物(s-2-g-1)相同的方式进行合成,定量地得到了褐色油状的化合物(s-2-g-3)9.9g。
[0459]
<化合物(s-2-g-4)的合成>
[0460]
通过下述途径合成了化合物(s-2-g-4)。
[0461]
[化学式79]
[0462][0463]
将原料的化合物(s-2-f-1)变更为化合物(s-2-f-4),除此以外,以与化合物(s-2-g-1)相同的方式进行合成,定量地得到了褐色油状的化合物(s-2-g-4)9.6g。
[0464]
[化合物(a-1003)的合成]
[0465]
通过下述途径合成了化合物(a-1003)。
[0466]
[化学式80]
[0467][0468]
向100ml三口烧瓶中,加入化合物(501c)3.0g(2.3mmol)、氢醌117mg(1.0mmol)、n-二甲基氨基吡啶13mg(0.1mmol)、氯仿20ml及edc(1-乙基-3-(3-二甲基氨基丙基)碳二亚胺)盐酸盐0.62g(33mmol),并在25℃下搅拌了1小时。将反应液用硅胶柱层析法进行纯化,得到了1.41g(产率50%)白色固体的化合物(a-1003)。
[0469]
以下示出所得到的化合物(a-1003)的1h-nmr。
[0470]1h-nmr(溶剂:重丙酮)δ(ppm):7.85(d,4h),7.46(s,4h),7.32(t,2h),5.02-4.88(m,8h),3.06-3.03(m,8h),2.94-2.80(m,8h)
[0471]
[化合物(a-1017)的合成]
[0472]
将原料变更为1,5-二羟基萘,除此以外,以与化合物(a-1003)相同的方式进行合成,得到了1.5g(产率52%)白色固体的化合物(a-1017)。
[0473]
以下示出所得到的化合物(a-1017)的1h-nmr。
[0474]1h-nmr(溶剂:重丙酮)δ(ppm):7.98(t,6h),7.69-7.60(m,4h),7.38(t,2h),5.02-4.88(m,8h),3.08-3.05(m,8h),2.95-2.92(m,8h)
[0475]
[化合物(a-1101)的合成]
[0476]
通过下述途径合成了化合物(a-1101)。
[0477]
[化学式81]
[0478][0479]
向2l三口烧瓶中,加入化合物(s-2-g-4)152.7g(0.23mol)、四氢呋喃466ml、乙酸乙酯467ml及氢醌11.5g(0.10mol),并将内温冷却至10℃以下。向其中滴加三乙基胺31.7g(0.31mol),滴加结束后,将内温升温至25℃。在25℃下搅拌2小时,添加甲醇30ml并搅拌了5
分钟。添加乙酸乙酯305ml、1m盐酸水200ml,并进行了搅拌。静置后,去除水层并用10%食盐水305ml清洗了有机层。将有机层用无水硫酸镁干燥之后,通过过滤去除干燥剂,并用旋转蒸发器对有机层进行了浓缩。得到了176.2g褐色油状的粗产物。将粗产物用硅胶柱层析法进行纯化,得到了107.0g(产率75%)白色固体的化合物(a-1101)。
[0480]
以下示出所得到的化合物(a-1101)的1h-nmr。
[0481]1h-nmr(溶剂:重丙酮)δ(ppm):8.17-8.14(m,4h),7.37(s,4h),7.15(d,4h),4.45-4.11(m,8h),4.13-4.11(m,4h)
[0482]
[化合物(a-1102)的合成]
[0483]
将原料的化合物(s-2-g-4)变更为化合物(s-2-g-2),除此以外,以与化合物(a-1101)相同的方式进行合成,得到了164g(产率65%)无色蜡状的化合物(a-1102)。
[0484]
以下示出所得到的化合物(a-1102)的1h-nmr。
[0485]1h-nmr(溶剂:重丙酮)δ(ppm):7.87(dd,2h),7.80(d,2h),7.36(s,4h),7.21(d,2h),4.47-4.44(m,8h),4.40-4.34(m,8h),4.14-4.09(m,8h)
[0486]
[化合物(a-1103)的合成]
[0487]
将原料的化合物(s-2-g-4)变更为化合物(s-2-g-1),除此以外,以与化合物(a-1101)相同的方式进行合成,得到了162g(产率64%)无色蜡状的化合物(a-1103)。
[0488]
以下示出所得到的化合物(a-1103)的1h-nmr。
[0489]1h-nmr(溶剂:重丙酮)δ(ppm):7.40(s,4h),7.37(d,4h),6.88(s,2h),4.44-4.10(m,8h),4.34-4.31(m,8h),4.11-4.09(m,8h)
[0490]
[化合物(a-1104)的合成]
[0491]
将原料的化合物(s-2-g-4)变更为化合物(s-2-g-3),除此以外,以与化合物(a-1101)相同的方式进行合成,得到了160g(产率63%)无色油状的化合物(a-1104)。
[0492]
以下示出所得到的化合物(a-1104)的1h-nmr。
[0493]1h-nmr(溶剂:重丙酮)δ(ppm):7.54(s,4h),7.37(s,4h),4.46-4.33(m,24h),4.14-4.12(m,8h),4.03-4.00(m,4h)
[0494]
[化合物(a-1106)的合成]
[0495]
通过下述途径合成了化合物(a-1106)。
[0496]
[化学式82]
[0497][0498]
将原料的氢醌变更为化合物(s-3-1),除此以外,以与化合物(a-1104)相同的方式进行合成,得到了220.4g(产率59%)无色油状的化合物(a-1106)。
[0499]
以下示出所得到的化合物(a-1106)的1h-nmr。
[0500]1h-nmr(溶剂:重丙酮)δ(ppm):7.93(d,1h),7.65(dd,1h),7.57(d,4h),7.46(d,1h),4.47-4.30(m,26h),4.15-4.13(m,8h),4.03-4.01(m,4h),3.53(t,2h),3.33(t,2h),1.05(t,3h)
[0501]
[化合物(a-1107)的合成]
[0502]
将原料的氢醌变更为四氟氢醌,除此以外,以与化合物(a-1102)相同的方式进行合成,得到了166g(产率64%)无色油状的化合物(a-1107)。
[0503]
以下示出所得到的化合物(a-1107)的1h-nmr。
[0504]1h-nmr(溶剂:重丙酮)δ(ppm):7.94(dd,2h),7.78(s,2h),7.28(d,2h),4.48-4.37(m,16h),4.15-4.10(m,8h)
[0505]
[化合物(a-1108)的合成]
[0506]
将原料的氢醌变更为2,5-二叔丁基对苯二酚,除此以外,以与化合物(a-1102)相同的方式进行合成,得到了177g(产率67%)无色油状的化合物(a-1108)。
[0507]
以下示出所得到的化合物(a-1108)的1h-nmr。
[0508]1h-nmr(溶剂:重丙酮)δ(ppm):7.90(dd,2h),7.77(s,2h),7.27(d,2h),7.20(s,2h),4.48-4.35(m,16h),4.14-4.11(m,8h),1.36(s,18h)
[0509]
[化合物(a-1109)的合成]
[0510]
通过下述途径合成了化合物(a-1109)。
[0511]
[化学式83]
[0512][0513]
将原料的氢醌变更为化合物(s-4-1),除此以外,以与化合物(a-1102)相同的方式进行合成,得到了164g(产率62%)无色油状的化合物(a-1109)。
[0514]
以下示出所得到的化合物(a-1109)的1h-nmr。
[0515]1h-nmr(溶剂:重丙酮)δ(ppm):7.92-7.85(m,2h),7.76(dd,2h),7.37(s,1h),7.22-7.20(m,4h),4.48-4.35(m,16h),4.14-4.10(m,8h),1.95(s,2h),1.42(s,6h),0.82(s,9h)
[0516]
[化合物(a-1115)的合成]
[0517]
将原料的氢醌变更为对苯二胺,除此以外,以与化合物(a-1102)相同的方式进行合成,得到了171g(产率68%)无色固体的化合物(a-1115)。
[0518]
以下示出所得到的化合物(a-1115)的1h-nmr。
[0519]1h-nmr(溶剂:重丙酮)δ(ppm):9.42(s,2h),7.79(s,4h),7.67(d,4h),7.12(d,2h),4.47-4.44(m,8h),4.34-4.31(m,8h),4.11-4.09(m,8h)
[0520]
[化合物(a-1117)的合成]
[0521]
将原料的氢醌变更为对苯二胺,除此以外,以与化合物(a-1104)相同的方式进行合成,得到了216g(产率60%)无色蜡状的化合物(a-1117)。
[0522]
以下示出所得到的化合物(a-1117)的1h-nmr。
[0523]1h-nmr(溶剂:重丙酮)δ(ppm):9.47(s,2h),7.76(s,4h),7.37(s,4h),4.46-4.33(m,20h),4.28-4.26(m,4h),4.13-4.11(m,8h),4.01-3.98(m,4h)
[0524]
[化合物(a-1119)的合成]
[0525]
将原料的氢醌变更为4,4
’‑
二羟基联苯,除此以外,以与化合物(a-1102)相同的方式进行合成,得到了182g(产率71%)无色蜡状的化合物(a-1119)。
[0526]
以下示出所得到的化合物(a-1119)的1h-nmr。
[0527]1h-nmr(溶剂:重丙酮)δ(ppm):7.86(d,2h),7.81-7.76(m,6h),7.38(d,4h),7.22(d,2h),4.48-4.35(m,16h),4.14-4.10(m,8h)
[0528]
[化合物(a-1121)的合成]
[0529]
将原料的氢醌变更为4,4
’‑
二羟基联苯,除此以外,以与化合物(a-1104)相同的方
式进行合成,得到了232g(产率63%)无色蜡状的化合物(a-1121)。
[0530]
以下示出所得到的化合物(a-1121)的1h-nmr。
[0531]1h-nmr(溶剂:重丙酮)δ(ppm):7.82(d,4h),7.55(s,4h),7.38(d,4h),4.47-4.33(m,24h),4.15-4.13(m,8h),4.03-4.01(m,4h)
[0532]
[化合物(a-1131)的合成]
[0533]
将原料的氢醌变更为1,5-二羟基萘,除此以外,以与化合物(a-1102)相同的方式进行合成,得到了188g(产率73%)无色固体的化合物(a-1131)。
[0534]
以下示出所得到的化合物(a-1131)的1h-nmr。
[0535]1h-nmr(溶剂:重丙酮)δ(ppm):8.01(d,2h),8.00-7.87(m,2h),7.62(d,2h),7.51(d,2h),7.28(d,2h),4.49-4.38(m,16h),4.16-4.11(m,8h)
[0536]
[化合物(a-1132)的合成]
[0537]
将原料的氢醌变更为1,5-二羟基萘,除此以外,以与化合物(a-1103)相同的方式进行合成,得到了178g(产率69%)无色固体的化合物(a-1132)。
[0538]
以下示出所得到的化合物(a-1132)的1h-nmr。
[0539]1h-nmr(溶剂:重丙酮)δ(ppm):7.93(d,2h),7.65(t,2h),7.54(d,2h),7.49(s,4h),6.94(s,2h),4.45-4.35(m,16h),4.13-4.11(m,8h)
[0540]
[化合物(a-1133)的合成]
[0541]
将原料的氢醌变更为1,5-二羟基萘,除此以外,以与化合物(a-1104)相同的方式进行合成,得到了235g(产率64%)无色固体的化合物(a-1133)。
[0542]
以下示出所得到的化合物(a-1133)的1h-nmr。
[0543]1h-nmr(溶剂:重丙酮)δ(ppm):7.91(d,2h),7.67(s,4h),7.64(t,2h),7.51(d,2h),4.47-4.37(m,24h),4.16-4.14(m,8h),4.04-4.03(m,4h)
[0544]
[化合物(a-1135)的合成]
[0545]
将原料的氢醌变更为1,5-二氨基萘,除此以外,以与化合物(a-1102)相同的方式进行合成,得到了154g(产率60%)无色固体的化合物(a-1135)。
[0546]
以下示出所得到的化合物(a-1135)的1h-nmr。
[0547]1h-nmr(溶剂:重丙酮)δ(ppm):9.57(s,2h),8.02(d,2h),7.85-7.79(m,6h),7.55(t,2h),7.18(d,2h),4.48-4.45(m,8h),4.38-4.35(m,8h),4.13-4.10(m,8h)
[0548]
[化合物(a-1137)的合成]
[0549]
将原料的氢醌变更为1,5-二氨基萘,除此以外,以与化合物(a-1104)相同的方式进行合成,得到了227g(产率60%)无色固体的化合物(a-1137)。
[0550]
以下示出所得到的化合物(a-1137)的1h-nmr。
[0551]1h-nmr(溶剂:重丙酮)δ(ppm):9.64(s,2h),8.02(d,2h),7.78(d,2h),7.55(s,4h),7.55(t,2h),4.47-4.38(m,24h),4.15-4.14(m,8h),4.03-4.02(m,4h)
[0552]
[化合物(a-1139)的合成]
[0553]
通过下述途径合成了化合物(a-1139)。
[0554]
[化学式84]
[0555][0556]
将原料的氢醌变更为化合物(s-5-1),除此以外,以与化合物(a-1102)相同的方式进行合成,得到了189g(产率68%)无色固体的化合物(a-1139)。
[0557]
以下示出所得到的化合物(a-1139)的1h-nmr。
[0558]1h-nmr(溶剂:重丙酮)δ(ppm):8.36-8.31(m,4h),7.89(d,2h),7.77(s,2h),7.55-7.52(m,4h),7.49-7.22(m,5h),4.48-4.36(m,18h),4.14-4.10(m,8h)
[0559]
[化合物(a-1142)的合成]
[0560]
通过下述途径合成了化合物(a-1142)。
[0561]
[化学式85]
[0562][0563]
将原料的氢醌变更为化合物(s-6-1),且将化合物(s-2-g-4)及三乙基胺的使用量设为2倍,除此以外,以与化合物(a-1101)相同的方式进行合成,得到了175g(产率58%)无色固体的化合物(a-1142)。
[0564]
以下示出所得到的化合物(a-1142)的1h-nmr。
[0565]1h-nmr(溶剂:重丙酮)δ(ppm):8.28-8.25(m,4h),8.07-8.03(m,8h),7.73(d,2h),7.50(s,4h),7.05(d,8h),4.42(s,4h),4.39(s,4h),4.31(t,8h),4.08(s,8h)
[0566]
[化合物(a-1144)的合成]
[0567]
将原料的氢醌变更为间苯三酚,且将化合物(s-2-g-2)及三乙基胺的使用量设为1.5倍,除此以外,以与化合物(a-1102)相同的方式进行合成,得到了195g(产率52%)无色油状的化合物(a-1144)。
[0568]
以下示出所得到的化合物(a-1144)的1h-nmr。
[0569]1h-nmr(溶剂:重丙酮)δ(ppm):7.86(d,3h),7.77(s,3h),7.23(s,3h),7.21(d,3h),4.47-4.34(m,24h),4.13-4.09(m,12h)
[0570]
[化合物(a-1145)的合成]
[0571]
将原料的氢醌变更为间苯三酚,且将化合物(s-2-g-1)及三乙基胺的使用量设为1.5倍,除此以外,以与化合物(a-1103)相同的方式进行合成,得到了209g(产率56%)无色油状的化合物(a-1145)。
[0572]
以下示出所得到的化合物(a-1145)的1h-nmr。
[0573]1h-nmr(溶剂:重丙酮)δ(ppm):7.48(s,6h),7.29(s,3h),6.88(s,3h),4.44(s,6h),4.40(s,6h),4.31(t,12h),4.10(t,12h)
[0574]
[化合物(a-1146)的合成]
[0575]
将原料的氢醌变更为间苯三酚,且将化合物(s-2-g-3)及三乙基胺的使用量设为1.5倍,除此以外,以与化合物(a-1104)相同的方式进行合成,得到了275g(产率51%)无色油状的化合物(a-1146)。
[0576]
以下示出所得到的化合物(a-1146)的1h-nmr。
[0577]1h-nmr(溶剂:重丙酮)δ(ppm):7.55(s,6h),7.23(s,3h),4.48-4.32(m,34h),4.12(t,12h),4.01(t,8h)
[0578]
[化合物(a-1153)的合成]
[0579]
将原料的氢醌变更为1,2-二羟基乙烷,除此以外,以与化合物(a-1104)相同的方式进行合成,得到了178g(产率50%)无色油状的化合物(a-1153)。
[0580]
以下示出所得到的化合物(a-1153)的1h-nmr。
[0581]1h-nmr(溶剂:重丙酮)δ(ppm):7.36(s,4h),4.66(s,4h),4.40(d,12h),4.26(t,12h),4.09(t,8h),3.97(t,4h)
[0582]
[化合物(a-1156)的合成]
[0583]
通过下述途径合成了化合物(a-1156)。
[0584]
[化学式86]
[0585][0586]
<化合物(a-1156b)的合成>
[0587]
向200ml三口烧瓶中加入(a-1156a)20.0g(0.1mol)、乙酸乙酯24ml,并在冰冷下将内温冷却至10℃。向其中滴加甲磺酸氯14.0g(0.12mol),然后滴加三乙基胺11.3g(0.11mol)。滴加结束后,将内温升温至30℃并搅拌了1小时。添加乙酸乙酯24ml、水30ml,并搅拌了5分钟。去除水层,在有机层中添加5%碳酸氢钠水溶液10ml、10%食盐水24ml并进行了搅拌。去除水层,将有机层用无水硫酸镁进行干燥后,通过过滤去除干燥剂,并将滤液进行了减压浓缩。得到了25.8g(产率92%)无色油状的化合物(a-1156b)。
[0588]
<化合物(a-1156c)的合成>
[0589]
在氮气流下,向300ml三口烧瓶中加入了3,4-二羟基苯甲酸甲酯3.0g(18mmol)、n-二甲基乙酰胺74ml。将内温升温至90℃,滴加了化合物(a-1156b)10.1g(37mmol)。滴加结束后,在内温90℃下搅拌2小时,将反应液冷却至室温。向其中滴加乙酸乙酯98ml、1规定浓度盐酸水49ml、浓盐酸5ml,并进行了搅拌。去除水层,并用10%食盐水49ml清洗了有机层。将有机层用无水硫酸镁进行干燥,通过过滤去除干燥剂,并将滤液进行了减压浓缩。得到了8.9g(产率94%)黄色油状的化合物(a-1156c)。
[0590]
<化合物(a-1156d)的合成>
[0591]
向300ml三口烧瓶中加入化合物(a-1156c)8.8g(16mmol)、乙醇63ml、氢氧化钾3.8g(67mmol)、水23ml,并在内温70℃下搅拌了1小时。将反应液冷却至室温,添加乙酸乙酯106ml、浓盐酸6.9ml、水106ml并进行了搅拌。去除水层,将有机层用无水硫酸镁进行干燥后,通过过滤去除干燥剂,然后将滤液进行了减压浓缩。得到了5.6g(产率65%)褐色油状的化合物(a-1156d)。
[0592]
<化合物(a-1156f)的合成>
[0593]
向200ml三口烧瓶中加入化合物(a-1156d)5.3g(10mmol)、n-二甲基甲酰胺0.16ml、甲苯30ml,将内温升温至75℃。向其中滴加亚硫酰氯1.5ml(21mmol),并在75℃下搅拌了1小时。减压蒸馏甲苯,定量地得到了化合物(a-1156e)。向其中添加四氢呋喃52ml、乙酸乙酯52ml、氢醌0.5g(5mmol),并将内温降温至10℃。滴加三乙基胺1.4g(14mmol),滴加后将内温升温至25℃。在25℃下搅拌1小时后,添加水5ml并搅拌了5分钟。向其中添加乙酸乙酯50ml、1规定浓度盐酸水60ml,并进行了搅拌。去除水层,并用10%食盐水50ml清洗了有机层。将有机层用无水硫酸镁进行干燥,通过过滤去除干燥剂,并将滤液进行了减压浓缩。得到了3.6g(产率60%)褐色油状的化合物(a-1156f)。
[0594]
<化合物(a-1156g)的合成>
[0595]
向100ml三口烧瓶中加入化合物(a-1156f)3.1g(2.8mmol)、四氢呋喃31ml、10%钯碳0.3g(0.3mmol),安装了氢球囊。在内温30℃下搅拌2小时,将反应液进行了硅藻土过滤。用四氢呋喃清洗过滤材料,并对滤液进行了减压浓缩。得到了2.1g(产率70%)褐色油状的化合物(a-1156g)。
[0596]
<化合物(a-1156)的合成>
[0597]
向100ml三口烧瓶中加入化合物(a-1156g)1.0g(1.0mmol)、乙腈10ml、三乙基胺0.7g(7mmol)、六氟丙烯3.2g(7mmol),并在25℃下搅拌了3小时。向其中添加1规定浓度盐酸水10ml、乙酸乙酯30ml,并进行了搅拌。去除水层,用10%食盐水30ml清洗了有机层。将有机层用无水硫酸镁进行干燥,通过过滤去除干燥剂,并将滤液进行了减压浓缩。将粗产物用硅胶柱层析法进行纯化,得到了3.3g(产率70%)无色油状的化合物(a-1156)。
[0598]
以下示出所得到的化合物(a-1156)的1h-nmr。
[0599]1h-nmr(溶剂:重丙酮)δ(ppm):7.86(d,2h),7.74(s,2h),7.36(s,4h),7.19(d,2h),4.48-4.45(m,2h),4.32-4.17(m,14h),3.99-3.90(m,16h)
[0600]
[化合物(a-1301)的合成]
[0601]
将原料的氢醌变更为4-羟基联苯,且将化合物(s-2-g-2)及三乙基胺的使用量设为一半,除此以外,以与化合物(a-1102)相同的方式进行合成,得到了97g(产率70%)无色固体的化合物(a-1301)。
[0602]
以下示出所得到的化合物(a-1301)的1h-nmr。
[0603]1h-nmr(溶剂:重丙酮)δ(ppm):7.70(dd,1h),7.75(d,3h),7.70(d,2h),7.50(t,2h),7.40-7.34(m,3h),7.20(d,1h),4.47-4.32(m,8h),4.11(t,4h)
[0604]
[化合物(a-1302)的合成]
[0605]
将原料的氢醌变更为4-羟基联苯,且将化合物(s-2-g-1)及三乙基胺的使用量设为一半,除此以外,以与化合物(a-1103)相同的方式进行合成,得到了97g(产率70%)无色
液体的化合物(a-1302)。
[0606]
以下示出所得到的化合物(a-1302)的1h-nmr。
[0607]1h-nmr(溶剂:重丙酮)δ(ppm):7.77(d,2h),7.70(d,2h),7.49(t,2h),7.41-7.36(m,5h),6.88(t,1h),4.44(s,4h),4.41(s,4h),4.32(t,4h),4.10(t,4h)
[0608]
[化合物(a-1303)的合成]
[0609]
将原料的氢醌变更为4-羟基联苯,且将化合物(s-2-g-3)及三乙基胺的使用量设为一半,除此以外,以与化合物(a-1104)相同的方式进行合成,得到了94g(产率68%)无色液体的化合物(a-1303)。
[0610]
以下示出所得到的化合物(a-1303)的1h-nmr。
[0611]1h-nmr(溶剂:重丙酮)δ(ppm):7.77(d,2h),7.71(d,2h),7.54(s,2h),7.49(t,2h),7.41-7.34(m,3h),4.46-4.33(m,12h),4.13(t,4h),4.02(t,2h)
[0612]
[实施例1~29及比较例1]
[0613]
如下述表1中所记载,使用本发明的化合物作为液晶取向促进剂,形成光学各向异性薄膜,并进行了评价。
[0614]
首先,制备了下述组成的涂布液。另外,将液晶取向促进剂的浓度调整成相对于棒状液晶化合物的合计100质量份成为0.02质量份、0.03质量份、0.10质量份及0.20质量份,从而制备了各涂布液。
[0615][0616]
[化学式87]
[0617][0618]
聚合性棒状液晶化合物m2
[0619]
[化学式88]
[0620][0621]
聚合性棒状液晶化合物m3
[0622]
[化学式89]
[0623][0624]
手性试剂(12)
[0625]
[化学式90]
[0626][0627]
将所制备的涂布液利用微量移液管称取50μl,滴加在带取向膜的玻璃上(se-130)进行了旋涂。
[0628]
接着,在85℃下加热2分钟,并自然冷却1分钟后,在氮气氛下进行紫外线照射(紫外线强度:300mj/m2),由此形成了光学各向异性薄膜。光学各向异性薄膜的膜厚约为4μm。
[0629]
并且,所形成的光学各向异性薄膜显示源自胆甾醇型液晶相的绿色的选择反射性。用shimadzu corporation制uvpc-3000测定了反射波长,其结果中心波长为530nm。
[0630]
用目视及雾度对所制造的各光学各向异性薄膜的取向性进行了评价。利用nippon denshoku industries co.,ltd.制雾度计ndh2000测定了雾度。
[0631]
并且,进行溶解
·
取向促进试验,比较液晶取向促进剂的浓度为0.02质量%、0.03质量%、0.10质量%、0.20质量%的光学各向异性薄膜的雾度值,并以以下的基准对溶解
·
取向促进作用进行了评价。将结果示于下述表1。
[0632]
另外,评价高的表示溶解性良好且取向促进作用也大。评价低的主要表示溶解性低。
[0633]
a:小于0.15
[0634]
b:0.15以上且小于0.35
[0635]
c:0.35以上且小于0.90
[0636]
d:0.90以上
[0637]
[表1]
[0638][0639]
以下示出上述表1中的液晶取向促进剂的结构。
[0640]
化合物(y)[日本特开2002-129162号公报的化合物(30)]
[0641]
[化学式91]
[0642][0643]
[化学式92]
[0644]
[0645]
[0646]
[0647]
[0648]
[0649]
[0650][0651]
如上述表1所示,确认到本发明的化合物的液晶取向作用大,即使浓度变高,相对于溶剂的溶解性也高。并且,确认到本发明的化合物的涂布溶剂的适用范围广,使用适应性高。
[0652]
[实施例30~32]
[0653]
如下述表2中所记载,使用本发明的化合物作为液晶取向促进剂,形成光学各向异性薄膜,并进行了评价。
[0654]
首先,制备了下述组成的涂布液。另外,将液晶取向促进剂的浓度调整成相对于圆盘状液晶化合物的合计100质量份成为0.02质量份、0.03质量份、0.10质量份、0.20质量份及0.30质量份,从而制备了各涂布液。
[0655][0656]
[化学式93]
[0657]
[0658]
聚合性圆盘状液晶化合物m5
[0659]
[化学式94]
[0660][0661]
取向膜表面取向剂1
[0662]
[化学式95]
[0663][0664]
将所制备的涂布液利用微量移液管称取50μl,滴加在带取向膜的玻璃上(pva-103)进行了旋涂。在110℃下加热2分钟,并自然冷却1分钟后,在氮气氛下进行紫外线照射(紫外线强度:500mj/cm2),由此形成了光学各向异性薄膜。光学各向异性薄膜的膜厚约为1.1μm。
[0665]
用目视及雾度对所制造的各光学各向异性薄膜的取向性进行了评价。利用nippon denshoku industries co.,ltd.制雾度计ndh2000测定了雾度。
[0666]
并且,进行溶解
·
取向促进试验,比较液晶取向促进剂的浓度为0.10质量%、0.20质量%、0.30质量%、0.50质量%的光学各向异性薄膜的雾度值,并以以下的基准对溶解
·
取向促进作用进行了评价。将结果示于下述表2。
[0667]
另外,评价高的表示溶解性良好且取向促进作用也大。评价低的主要表示溶解性低。
[0668]
a:小于0.15
[0669]
b:0.15以上且小于0.35
[0670]
c:0.35以上且小于0.90
[0671]
d:0.90以上
[0672]
[表2]
[0673][0674]
以下示出上述表2中的液晶取向促进剂的结构。
[0675]
[化学式96]
[0676][0677]
如上述表2所示,即使在使用圆盘状液晶化合物作为液晶组合物中所含的聚合性液晶化合物的情况下,也确认到本发明的化合物的液晶取向作用大,即使浓度变高,相对于溶剂的溶解性也高。因此,确认到本发明的化合物的涂布溶剂的适用范围广,使用适应性高。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。