1.本发明涉及有机电致发光技术领域,具体而言,涉及一种含七元环的螺芴衍生物、电子传输材料及有机电致发光元件。
背景技术:
2.有机电致发光元件(organic light emitting diodes,oled)具有轻、薄、自发光、低功耗、无背光源、广视角、响应快及可挠性等优点,已经逐步替代液晶显示面板成为新一代的平板显示器,并且在柔性显示方面也有巨大潜力。传统电子传输材料的载流子迁移率(carrier mobility)是空穴传输材料的千分之一,且热稳定性不佳,易导致发光效率滚降快或器件寿命差的问题。据相关文献显示,电子传输材料所占电荷消耗比率达35.9%,仅次于发光层的消耗(39.8%)。因此,开发高载流子迁移率和热稳定性好的电子传输材料是目前oled材料开发的重点之一。
3.目前常用的电子传输材料主要有金属配合物、含氮杂环化合物、全氟化类化合物、有机硅类化合物、有机硼类化合物等。其中,含氮杂环化合物是研究较多的结构,种类繁多,但在制备器件的材料组合中,都或多或少存在着一些缺陷,提高器件效率的同时不能降低工作电压,器件寿命反而降低。所以仍需要继续开发新的电子传输材料,以优化发光器件的材料组合,提升器件的综合性能。
技术实现要素:
4.本发明的主要目的在于提供一种含七元环的螺芴衍生物、电子传输材料及有机电致发光元件,以解决现有技术中的有机电致发光元件难以兼顾发光效率和使用寿命的问题。
5.为了实现上述目的,根据本发明的一个方面,提供了一种含七元环的螺芴衍生物,该螺芴衍生物具有式(a)所示结构:
[0006][0007]
其中,与b直接相连的苯环和b组成的环记为d,d选自取代或非取代的c
10
~c
30
的稠环中的任意一种;ar选自取代或非经取代的c3~c
20
的含氮杂芳基中的任意一种;l选自单键、c6~c
18
的亚芳基、c6~c
18
的亚杂芳基中的任意一种。
[0008]
根据本发明的另一方面,提供了一种电子传输材料,该电子传输材料包括螺芴衍生物,螺芴衍生物包括上述的螺芴衍生物。
[0009]
根据本发明的又一方面,提供了一种有机电致发光元件,包括衬底层、空穴注入层、空穴传输层、发光层、电子传输层和阴极层,该电子传输层由上述的电子传输材料形成。
[0010]
应用本发明的技术方案,本技术的具有上述式(a)所示结构的含七元环的螺芴衍生物,一方面通过螺芴并环结构作为母核使得螺芴衍生物具有较强的刚性,从而极大地提高了螺芴衍生物的热稳定性,进而提高了电子传输材料的高载流子迁移率。另一方面由于引入的ar为缺电子取代基,从而提高了螺芴衍生物的电子传输能力。通过以上两方面的作用,当将上述螺芴衍生物用作有机电致发光元件中的电子传输材料时,能有效降低有机电致发光元件的工作电压,同时提高发光效率和元件寿命。
具体实施方式
[0011]
需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。下面将结合实施例来详细说明本发明。
[0012]
如本技术背景技术所分析的,现有技术中存在有机电致发光元件难以兼顾发光效率和使用寿命的问题,为了解决该问题,本技术提供了一种含七元环的螺芴衍生物、电子传输材料及有机电致发光元件。
[0013]
在本技术的一种典型的实施方式中,提供了一种含七元环的螺芴衍生物,该螺芴衍生物具有式(a)所示结构:
[0014][0015]
其中,与b直接相连的苯环和b组成的环记为d,d选自取代或非取代的c
10
~c
30
的稠环中的任意一种;ar选自取代或非经取代的c3~c
20
的含氮杂芳基中的任意一种;l选自单键、c6~c
18
的亚芳基、c6~c
18
的亚杂芳基中的任意一种。
[0016]
本技术的具有上述式(a)所示结构的含七元环的螺芴衍生物,一方面通过螺芴并环结构作为母核使得螺芴衍生物具有较强的刚性,从而极大地提高了螺芴衍生物的热稳定性,进而提高了电子传输材料的高载流子迁移率。另一方面由于引入的ar为缺电子取代基,从而提高了螺芴衍生物的电子传输能力。通过以上两方面的作用,当将上述螺芴衍生物用作有机电致发光元件中的电子传输材料时,能有效降低有机电致发光元件的工作电压,同时提高发光效率和元件寿命。
[0017]
优选上述d选自取代或非取代的c
10
~c
18
的稠环中的任意一种,优选d选自萘环、菲环、蒽环、苯并噻吩环或苯并呋喃环中的任意一种,可有助于提高螺芴衍生物的刚性,从而提高螺芴衍生物的热稳定性。
[0018]
进一步地,优选上述式(a)选自式(a-1)至式(a-4)中的任意一种:
[0019][0020]
在本技术的一种实施例中,上述ar选自取代或非经取代的c3~c
12
的含氮杂芳基中的任意一种;优选ar选自取代或非经取代的c3~c8的含氮杂芳基中的任意一种。
[0021]
上述ar取代基有利于更进一步地提高螺芴衍生物的缺电子性。
[0022]
在本技术的一种实施例中,上述ar选自
[0023][0023]
中的任意一种;其中,虚线代表连接键;(ar-1)至(ar-10)中的z各自独立地选自h、cn、卤素、取代或非取代的c1~c
10
的直链烷基、取代或非取代的c3~c
10
的支链烷基、取代或非取代的c6~c
18
的芳基、取代或非取代的c6~c
18
的杂芳基中的任意一
种。
[0024]
以上(ar-1)至(ar-10)中的ar取代基主要是吡啶、嘧啶、三嗪、苯并咪唑、喹喔啉、喹唑啉等基团缺电子含氮芳基,优选的取代基有利于进一步地增加这些基团的缺电子性。
[0025]
优选上述z各自独立地选自取代或非取代的c1~c5的直链烷基、取代或非取代的c3~c6的支链烷基、取代或非取代的c6~c
12
的芳基、取代或非取代的c6~c
12
的杂芳基中的任意一种;进一步地,优选z各自独立地选自甲基、乙基、丙基、异丙基、异戊基、苯基、联苯基、萘基、苯并呋喃基、苯并噻吩基中的任意一种;更进一步地,优选z各自独立地选自甲基、苯基、3-联苯基、2-萘基、3-苯并呋喃基中的任意一种,从而有利于进一步地提高ar取代基的缺电子性。
[0026]
优选上述l选自单键、取代或非取代的c6~c
12
的亚芳基、取代或非取代的c6~c
12
的亚杂芳基中的任意一种,优选l选自单键、取代或非取代的亚苯基、取代或非取代的亚联苯基、取代或非取代的亚萘基中的任意一种,进一步地,优选l为单键或亚苯基,从而更有利于ar取代基与母核进行作用,进一步地提高螺芴衍生物的热稳定性。
[0027]
为了得到能够更好地兼顾热稳定性与电子传输性的电子传输材料,优选上述螺芴衍生物选自以下结构中的任意一种:
[0028]
[0029]
[0030]
[0031]
[0032]
[0033]
[0034]
[0035]
[0036][0037]
在本技术的另一种典型的实施方式中,提供了一种电子传输材料,该电子传输材料包括螺芴衍生物,螺芴衍生物包括前述的螺芴衍生物。
[0038]
本技术的包括前述的螺芴衍生物的电子传输材料,具有优良的高载流子迁移率和电子传输能力。
[0039]
在本技术的又一种典型的实施方式中,提供了一种有机电致发光元件,包括衬底层、空穴注入层、空穴传输层、发光层、电子传输层和阴极层,该电子传输层由上述的电子传输材料形成。
[0040]
包括本技术的上述电子传输材料的有机电致发光元件能有效降低有机电致发光元件的工作电压,同时提高发光效率和元件寿命。
[0041]
以下将结合实施例,进一步说明本技术的有益效果。
[0042]
合成实施例
[0043]
1、合成通式
[0044][0045]
其中,mt-1至mt-4中的t对应4种中间体的编号,x为cl或br。
[0046]
2、具体化合物的合成
[0047]
2.1中间体m1-4的合成
[0048][0049]
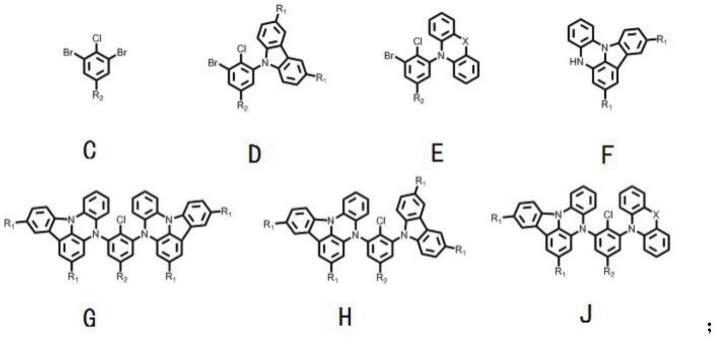
在2000ml的反应瓶中,加入2-萘硼酸(68.80g,400mmol)、2-溴-4-氯-1-碘苯(126.94g,400mmol)、二(三苯基膦)二氯化钯(2.81g,4mmol)、碳酸钾(110.57g,800mmol)、甲苯(1000ml)、乙醇(400ml)和去离子水(400ml),氮气保护下升温至80℃,搅拌3小时;将反应液冷却至室温,加入甲苯(500ml)进行萃取,合并有机相,使用无水硫酸镁进行干燥,并进行硅胶短柱色谱过滤,有机相脱溶;使用正庚烷进行重结晶,得到88.93g中间体m1-1,为类白色固体,收率为70%,纯度为98.8%。
[0050]
在氮气保护下,向干燥的四口反应瓶中加入中间体m1-1(88.93g,280mmol)和thf(560ml),在搅拌下降温至-80℃以下,开始滴加2.5mol/l的正丁基锂的thf溶液(134.4ml),滴加结束后保温1h,取样检测,待锂盐反应完成后,在-80℃以下滴加二苯并环庚酮的thf溶液(58.31g,280mmol),滴加过程中保持温度为-80℃以下,滴加结束后,保温1h,自然升温后搅拌2h;在-0℃以下向其中加入3mol/l的盐酸168ml,搅拌1小时,加乙酸乙酯(400ml)与水(400ml)进行萃取,合并有机相,使用无水硫酸镁进行干燥,过滤,减压除去溶剂;得到中间体m1-2的粗品。所得粗品后直接加入三氟乙酸(400ml),开启搅拌,然后逐渐升温至回流反应2h,反应完成后,直接过滤取得固体,固体用乙醇淋洗(150ml),得到78.07g中间体m1-3,为白色固体粉末,收率为65%,纯度为99.12%。
[0051]
在1000ml的反应瓶中,加入中间体m1-3(77.21g,180mmol)、联硼酸频那醇酯(54.86g,216mmol)和甲苯(750ml),充氮气搅拌15分钟,再加入醋酸钾(52.99g,540mmol),三(二亚苄基茚丙酮)二钯(3.33g),x-phos(3.42g),加热回流3小时。反应结束,进行硅胶短柱色谱过滤,热甲苯淋洗,有机相脱溶;使用乙醇进行热打浆,得到73.08g中间体m1-4,为类白色固体,收率为78%,纯度为99.30%。
[0052]
2.2中间体m2-4的合成
[0053][0054]
在2000ml的反应瓶中,加入1-萘硼酸(68.80g,400mmol)、2-溴-4-氯-1-碘苯(126.94g,400mmol)、二(三苯基膦)二氯化钯(2.81g,4mmol)、碳酸钾(110.57g,800mmol)、甲苯(1000ml)、乙醇(400ml)和去离子水(400ml),氮气保护下升温至80℃,搅拌3小时;将反应液冷却至室温,加入甲苯(500ml)进行萃取,合并有机相,使用无水硫酸镁进行干燥,并进行硅胶短柱色谱过滤,有机相脱溶;使用正庚烷进行重结晶,得到96.55g中间体m2-1,为类白色固体,收率为76%,纯度为98.2%。
[0055]
在氮气保护下,向干燥的四口反应瓶中加入中间体m2-1(95.28g,300mmol)和thf(600ml),在搅拌下降温至-80℃以下,开始滴加2.5mol/l正丁基锂的thf溶液(144ml),滴加结束后保温1h,取样检测,待锂盐反应完成后,在-80℃以下滴加二苯并环庚酮的thf溶液(62.48g,300mmol),滴加过程中保持温度为-80℃以下,滴加结束后,保温1h,自然升温后搅拌2h;在-0℃以下向其中加入3mol/l的盐酸180ml,搅拌1小时,加乙酸乙酯(400ml)与水(400ml)进行萃取,合并有机相,使用无水硫酸镁进行干燥,过滤,减压除去溶剂,得到中间体m2-2的粗品。所得粗品直接加入三氟乙酸(420ml),开启搅拌,然后逐渐升温至回流反应2h,反应完成后,直接过滤取得固体,固体用乙醇淋洗(180ml),得到81.67g中间体m2-3,为白色固体粉末,收率为68%,纯度为99.23%。
[0056]
在1000ml反应瓶中,加入中间体m2-3(81.50g,190mmol)、联硼酸频那醇酯(57.91g,228mmol)和甲苯(840ml),充氮气搅拌15分钟,再加入醋酸钾(55.93g,570mmol),三(二亚苄基茚丙酮)二钯(3.52g),x-phos(3.61g),加热回流3小时。反应结束,进行硅胶短柱色谱过滤,热甲苯淋洗,有机相脱溶;使用乙醇进行热打浆,得到67.46g中间体m2-4,为类白色固体,收率为72%,纯度为99.11%。
[0057]
2.3中间体m3-4的合成
[0058][0059]
在2000ml反应瓶中,加入9-菲硼酸(88.82g,400mmol)、2-溴-4-氯-1-碘苯(126.94g,400mmol)、二(三苯基膦)二氯化钯(2.81g,4mmol)、碳酸钾(110.57g,800mmol)、甲苯(1000ml)、乙醇(400ml)和去离子水(400ml),氮气保护下升温至80℃,搅拌4小时;将反应液冷却至室温,加入甲苯(500ml)进行萃取,合并有机相,使用无水硫酸镁进行干燥,并进行硅胶短柱色谱过滤,有机相脱溶;使用正庚烷进行重结晶,得到116.18g中间体m3-1,为类白色固体,收率为79%,纯度为98.8%。
[0060]
在氮气保护下,向干燥的四口反应瓶中加入中间体m3-1(110.30g,300mmol)和thf(600ml),在搅拌下降温至-80℃以下,开始滴加2.5mol/l正丁基锂的thf溶液(144ml),滴加结束后保温1h,取样检测,待锂盐反应完成后,在-80℃以下滴加二苯并环庚酮的thf溶液(62.48g,300mmol),滴加过程中保持温度为-80℃以下,滴加结束后,保温1h,自然升温后搅拌2h;在-0℃以下向其中加入3mol/l的盐酸180ml,搅拌1小时,加乙酸乙酯(400ml)与水(400ml)进行萃取,合并有机相,使用无水硫酸镁进行干燥,过滤,减压除去溶剂,得到中间体m3-2的粗品。所得粗品直接加入三氟乙酸(420ml),开启搅拌,然后逐渐升温至回流反应2h,反应完成后,直接过滤取得固体,固体用乙醇淋洗(180ml),得到107.78g中间体m3-2,为白色固体粉末,收率为75%,纯度为99.08%。
[0061]
在1000ml反应瓶中,加入中间体m2-2(95.80g,200mmol)、联硼酸频那醇酯(60.58g,240mmol)和甲苯(900ml),充氮气搅拌15分钟,再加入醋酸钾(58.87g,600mmol),三(二亚苄基茚丙酮)二钯(3.71g),x-phos(3.80g),加热回流3小时。反应结束,进行硅胶短柱色谱过滤,热甲苯淋洗,有机相脱溶;使用乙醇进行热打浆,得到82.16g中间体m3-3,为类白色固体,收率为72%,纯度为99.39%。
[0062]
2.4中间体m4-4的合成
[0063][0064]
在2000ml反应瓶中加入二苯并呋喃-4-硼酸(84.80g,400mmol)、2-溴-4-氯-1-碘苯(126.94g,400mmol)、二(三苯基膦)二氯化钯(2.81g,4mmol)、碳酸钾(110.57g,800mmol)、甲苯(1000ml)、乙醇(400ml)和去离子水(400ml),氮气保护下升温至80℃,搅拌4小时;将反应液冷却至室温,加入甲苯(500ml)进行萃取,合并有机相,使用无水硫酸镁进行干燥,并进行硅胶短柱色谱过滤,有机相脱溶;使用正庚烷进行重结晶,得到98.71g中间体m4-1,为类白色固体,收率为69%,纯度为98.2%。
[0065]
在氮气保护下,向干燥的四口反应瓶中加入中间体m4-1(89.40g,250mmol)和thf(500ml),在搅拌下降温至-80℃以下,开始滴加2.5m正丁基锂的thf溶液(120ml),滴加结束后保温1h,取样检测,待锂盐反应完成后,在-80℃以下滴加二苯并环庚酮的thf溶液(52.07g,250mmol),滴加过程中保持温度为-80℃以下,滴加结束后,保温1h,自然升温后搅拌2h;在-0℃以下向其中加入3mol/l的盐酸150ml,搅拌1小时,加乙酸乙酯(320ml)与水(320ml)进行萃取,合并有机相,使用无水硫酸镁进行干燥,过滤,减压除去溶剂,得到中间体m4-2的粗品。所得粗品直接加入三氟乙酸(350ml),开启搅拌,然后逐渐升温至回流反应2h,反应完成后,直接过滤取得固体,固体用乙醇淋洗(150ml),得到85.59g的中间体m4-3,为白色固体粉末,收率为73%,纯度为99.33%。
[0066]
在1000ml反应瓶中,加入中间体m4-3(84.42g,180mmol)、联硼酸频那醇酯(54.52g,216mmol)和甲苯(800ml),充氮气搅拌15分钟,再加入醋酸钾(52.98g,540mmol),三(二亚苄基茚丙酮)二钯(3.34g),x-phos(3.42g),加热回流3小时。反应结束,进行硅胶短柱色谱过滤,热甲苯淋洗,有机相脱溶;使用乙醇进行热打浆,得到77.69g中间体m4-4,为类白色固体,收率为77%,纯度为99.08%。
[0067]
2.5原料m174的合成
[0068][0069]
在500ml反应瓶中加入1-(dibenzo[b,d]furan-3-yl)ethanone(22.05g,0.105mol)、对-溴苯甲醛(18.5g,0.1mol)、200毫升的乙醇,开启搅拌,再加入叔丁醇钠(14.45g,0.15mol)于室温下进行搅拌8小时。待反应完全后,向反应瓶中加入纯水(50ml)搅拌后过滤,滤饼先以去离子水及甲醇进行清洗,再以纯水(30ml)及甲醇(70ml)搅拌30分钟后过滤,上述清洗过滤步骤重复2次后,固体干燥得中间体白色固体29.61g,产率为78.50%,纯度为99.10%。
[0070]
在500ml反应瓶中加入中间体(2.40g,70mmol)、苯甲脒盐酸盐(12.06g,77mmol)与氢氧化钠(4.2g,105mmol)、甲苯(50ml)及乙醇(150ml),开启搅拌并加热至回流。待6小时反应完后,降温至60℃,向反应瓶中加入纯水(50ml)并搅拌5分钟,收集有机层,并真空蒸去有机层的溶剂,得到粗品,粗品再经乙酸乙酯冲洗、过滤及干燥后,获得原料m174的白色固体19.38g,产率为58%。
[0071]
2.6原料m198的合成
[0072][0073]
在反应瓶中,加入4-溴邻苯二胺(20g,0.106mol)、2,3-丁二酮(9.66g,0.112mol)和甲苯(200ml),加热回流3小时。待反应结束,冷却至室温,垫硅胶过滤,滤液真空蒸去溶剂,得到粗品,粗品用正己烷溶解脱色并重结晶,得到18.0g喹喔啉溴代物,为类白色固体粉末,产率为71%,纯度为99.5%。
[0074]
在反应瓶中加入喹喔啉溴代物(16.59g,70mmol)、4-氯苯硼酸(12.04g,77mmol)、四(三苯基膦)钯(1.62g)、碳酸钾(29.02g,210mmol)、甲苯、乙醇和去离子水,氮气保护下升温至80℃,搅拌至反应完毕;将反应液冷却至室温,分液,取有机相,使用无水硫酸镁进行干燥,并进行硅胶短柱色谱过滤,有机相脱溶;重结晶,得到原料m198。
[0075]
2.6化合物a5的合成
[0076][0077]
在500ml反应瓶中,加入中间体m1-4(10.41g,20mmol),2-氯-4,6-二苯基嘧啶(5.33g,20mmol),碳酸钾(8.29g,60mmol),甲苯(90ml),乙醇(30ml),水(30ml),充氮气搅拌15min,再加入醋酸钯(0.067g),x-phos(0.286g),加热回流5小时。反应结束,降温进行过滤得到灰色固体,灰色固体使用氯苯溶解后进行硅胶短柱色谱过滤,滤液真空蒸去溶剂,得到粗品,粗品用氯苯重结晶,抽滤,得到9.49g的化合物a5,为白色固体粉末,产率为76%,纯度99.12%,进行一次升华后纯度为99.79%。
[0078]
参照化合物a5的制备方法,通过采用不同的原料与不同的中间体mt1-4、mt2-4、mt3-4、mt4-4反应合成化合物a25、化合物a49、化合物a66、化合物a88、化合物a101、化合物a129、化合物a142、化合物a174、化合物a189,具体见表1。
[0079]
表1
[0080]
[0081][0082]
3.有机电致发光元件的制备
[0083]
本发明的上述有机化合物特别适用于oled器件中的电子传输层,以下通过具体实施例来详细说明本发明的有机化合物在oled器件中作为电子传输层的应用效果。
[0084]
其中使用到的有机材料的结构式如下:
[0085]
[0086][0087]
采用本发明的含七元环的螺芴并环衍生物作为电子传输层的有机电致发光元件,可依次包括玻璃和透明导电层(ito)衬底层,空穴注入层,空穴传输层,发光层,电子传输层,阴极层。
[0088]
器件实施例1
[0089]
利用sunic sp1710蒸镀机制造oled器件,具体步骤为:将镀有厚度为135nm的ito(氧化铟锡)的玻璃基板(康宁玻璃40mm
×
40mm
×
0.7mm)分别用异丙醇和纯水超声洗涤5分钟,再用紫外线臭氧清洗,之后将玻璃基板传送至真空沉积室中;将掺杂4%hd的空穴传输材料ht1以20nm的厚度真空(约10-7
torr)热沉积在透明ito电极上,形成空穴注入层;在空穴注入层上接着真空沉积120nm厚度的ht1和10nm厚度的ht2作为空穴传输层;在空穴传输层上真空沉积25nm的掺杂4%bd的bh作为发光层;然后真空沉积掺杂50%liq(8-羟基喹啉锂)的化合物a5形成电子传输层,厚度为30nm;最后按顺序沉积2nm厚的金属镱(yb,电子注入层)和掺杂比例为10∶1的镁银合金150nm形成阴极;最后将该器件从沉积室传送至手套箱中,随即用uv可固化环氧树脂及含有吸湿剂的玻璃盖板进行封装。
[0090]
在上述制造步骤中,有机材料、金属镱和金属mg的沉积速率分别保持在0.1nm/s、0.05nm/s和0.2nm/s。
[0091]
该器件结构表示为:ito(135nm)/ht1∶4%hd(20nm)/ht1(120nm)/ht2(10nm)/bh∶4%bd(25nm)/化合物a5∶liq(5∶5,30nm)yb(2nm)/mg∶ag(10∶1,150nm)。
[0092]
器件实施例2至10
[0093]
以与器件实施例1中相同的方式制造有机发光器件,不同之处在于使用下表2中所示的化合物代替器件实施例1中的化合物a5。
[0094]
器件对比例1
[0095]
以与器件实施例1中相同的方式制造有机发光器件,不同之处在于使用化合物eta代替器件实施例1中的化合物a5。
[0096]
器件对比例2
[0097]
以与器件实施例1中相同的方式制造有机发光器件,不同之处在于使用化合物etb代替器件实施例1中的化合物a5。
[0098]
器件的亮度、发光效率、eqe(外量子效率)是由苏州弗式达fs-100ga4测试完成的,器件寿命lt97(初始亮度为4000nits,衰减到3880nits所用的时间,以器件对比例为参照,作归一化处理)是在弗式达fs-mp96测试完成的,所有测量均在室温大气中完成。进一步地,器件在10ma/cm2电流密度下的工作电压(v)、电流效率(c.e.)、外量子效率(eqe)及色坐标(ciex,ciey)的具体性能数据见表2。
[0099]
表2
[0100][0101][0102]
与器件对比例1和2相比,使用本发明的含七元环的螺芴并环衍生物作为电子传输材料的器件,电压有所降低,效率和寿命都得到了明显提升。
[0103]
从以上的描述中,可以看出,本发明上述的实施例实现了如下技术效果:
[0104]
本技术的具有上述式(a)所示结构的含七元环的螺芴衍生物,一方面通过螺芴并环结构作为母核使得螺芴衍生物具有较强的刚性,从而极大地提高了螺芴衍生物的热稳定性,进而提高了电子传输材料的高载流子迁移率。另一方面由于引入的ar为缺电子取代基,从而提高了螺芴衍生物的电子传输能力。通过以上两方面的作用,当将上述螺芴衍生物用作有机电致发光元件中的电子传输材料时,能有效降低有机电致发光元件的工作电压,同时提高发光效率和元件寿命。
[0105]
以上仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人
员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。