1.本发明涉及化合物、造影剂、及化合物的制造方法。
背景技术:
2.使用了碘造影剂的造影x射线检查大大提高x射线图像的诊断能力,成为了在许多诊疗科中占据重要位置的诊断方法。碘造影剂大多自肾脏排泄。因此,作为副作用而引起造影剂肾病这一点成为问题。
3.作为消除上述问题的方法之一,例如,专利文献1中提出了使用非离子性碘造影剂与被肝细胞特异性转运蛋白识别的基团的结合体。该结合体成为转运蛋白的基质并被摄入至肝细胞中,或者,从肝细胞排泄从而使肾排泄的一部分为胆汁排泄,能够实现肾毒性减轻。在专利文献1中,具体而言,公开了下述结合体,其具有碘海醇作为非离子性碘造影剂,并且具有乙氧基苄基或来自熊去氧胆酸的基团作为被肝细胞特异性转运蛋白识别的基团。
4.现有技术文献
5.专利文献
6.专利文献1:日本特开2018-062475号公报
技术实现要素:
7.发明所要解决的课题
8.碘造影剂适宜通过注射至静脉内来施予。将包含专利文献1的结合体的注射剂以高浓度施予至静脉内的情况下,有时药剂的粘稠性变高。因此,要求碘造影剂不仅具有能够实现肾毒性减轻的能力,而且在施予时操作性优异。
9.本发明所要解决的课题是提供能作为碘造影剂使用的新化合物。
10.用于解决课题的手段
11.本技术的发明人为解决上述课题而进行了深入研究,结果发现能成为碘造影剂的新化合物,从而完成了本发明。
12.即,本发明如下所述。
13.1.14.由式(1)表示的化合物或其医药上可允许的盐。
15.[化学式1]
[0016][0017]
[式(1)中,r1~r3各自独立地为:
[0018]
由-nr
xry
(r
x
及ry各自独立地表示氢原子、可具有取代基的c1~c6的烃基、或可具
有取代基的c2~c7的酰基。)表示的氨基;
[0019]
或由-c(=o)nr
zrw
(rz及rw各自独立地表示氢原子、或可具有取代基的c1~c6的烃基。)表示的酰胺基;
[0020]
或者,由式(2)表示的基团,
[0021]
[化学式2]
[0022][0023]
(式(2)中,
[0024]
原子基团(atomic group)为与脱唾液酸糖蛋白受体结合的原子团,
[0025]
连接物(linker)为任意的连接体。)
[0026]
r1~r3中的至少1个为式(2)表示的基团。]
[0027]
[2]
[0028]
如[1]所述的化合物或其医药上可允许的盐,其中,前述原子基团为与脱唾液酸糖蛋白受体结合的糖残基。
[0029]
[3]
[0030]
如[1]或[2]所述的化合物或其医药上可允许的盐,其中,前述原子基团为来自半乳糖、n-乙酰半乳糖胺、n-三氟乙酰半乳糖胺、或半乳糖-n-乙酰葡糖胺的基团。
[0031]
[4]
[0032]
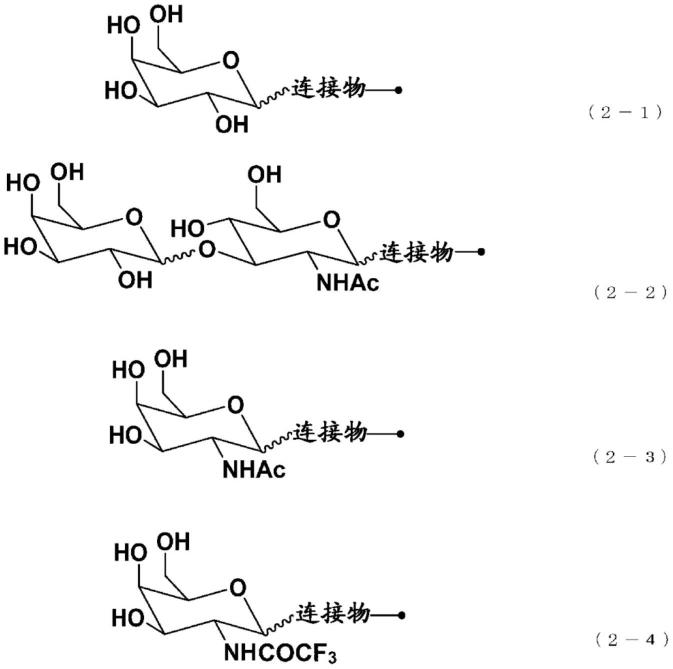
如[1]~[3]中任一项所述的化合物或其医药上可允许的盐,其中,由式(2)表示的基团为由以下的式(2-1)、(2-2)、(2-3)、或(2-4)中的任一者表示的基团。
[0033]
[化学式3]
[0034][0035]
(式(2-1)、(2-2)、(2-3)、及(2-4)中,连接物为任意的连接体。)
[0036]
[5]
[0037]
如[1]~[4]中任一项所述的化合物或其医药上可允许的盐,其中,前述连接体为可具有取代基的烃链,
[0038]
前述烃链的两末端中的一个或两个可以具有杂原子、酰胺键、酯键、羰基键、或芳香族杂环中的任一者。
[0039]
[6]
[0040]
如[5]所述的化合物或其医药上可允许的盐,其中,前述烃链为可具有取代基的亚烷基链,或者为介由选自由杂原子、酰胺键、酯键、羰基键、及芳香族杂环组成的组中的至少一者而结合有可具有取代基的2个以上的亚烷基链的烃链。
[0041]
[7]
[0042]
如[1]~[6]中任一项所述的化合物或其医药上可允许的盐,其中,前述连接体由以下的结构表示,
[0043]
[化学式4]
[0044]
·-l
0-l
x-l
y-l
z-·
[0045]
式中,
[0046]
l0为-o-或-nhc(=o)-,
[0047]
l
x
由以下的结构表示,
[0048]
[化学式5]
[0049][0050]
ly表示单键,或者由以下的结构表示,
[0051]
[化学式6]
[0052][0053]
lz由以下的结构表示,
[0054]
[化学式7]
[0055][0056]
上述式中,
[0057]
r’各自独立地为选自氢原子、可具有取代基的c1~c3烷基、或羟基中的任一者,
[0058]
l1各自独立地为选自醚键(-o-)、硫醚键(-s-)、胺键(-nh-)、酰胺键、酯键、或羰基键中的任一者,
[0059]
l2为酰胺键、或来自芳香族杂环的2价基团,
[0060]
l3为选自-och
2-、-nhch
2-、-c(=o)nh-、-c(=o)nhch
2-、或-nhc(=o)-中的任一者,
[0061]
m1、m2、及m4各自独立地为1以上的整数,
[0062]
m3为0以上的整数,
[0063]
n及n2各自独立地为0以上的整数。
[0064]
[8]
[0065]
如[7]所述的化合物或其医药上可允许的盐,其中,l
x
为以下的结构中的任一者,
[0066]
[化学式8]
[0067][0068]
ly为单键,或者为以下的结构中的任一者,
[0069]
[化学式9]
[0070][0071]
lz为以下的结构中的任一者,
[0072]
[化学式10]
[0073][0074]
上述式中,
[0075]
k1各自独立地为1以上3以下的整数,
[0076]
k2为0或1,
[0077]
m3为0以上的整数,
[0078]
n及n2各自独立地为0以上的整数。
[0079]
[9]
[0080]
如[7]所述的化合物或其医药上可允许的盐,其中,前述连接体由以下的结构表示。
[0081]
[化学式11]
[0082][0083]
(式中,
[0084]
r’各自独立地为选自氢原子、可具有取代基的c1~c3烷基、或羟基中的任一者,
[0085]
l0为-o-或-nhc(=o)-,
[0086]
l1各自独立地为选自醚键(-o-)、硫醚键(-s-)、胺键(-nh-)、酰胺键、酯键、或羰基键中的任一者,
[0087]
m1及m2各自独立地为1以上的整数,
[0088]
n为0以上的整数,
[0089]
n’为0或1。)
[0090]
[10]
[0091]
如[9]所述的化合物或其医药上可允许的盐,其中,前述连接体由以下的结构中的任一者表示。
[0092]
[化学式12]
[0093][0094]
[化学式13]
[0095][0096]
(式中,
[0097]
l0为-o-或-nhc(=o)-,
[0098]
n为0以上的整数,
[0099]
n为0以上的整数,
[0100]
m2为1以上的整数,
[0101]
n’为0或1。)
[0102]
[11]
[0103]
如[1]~[10]中任一项所述的化合物或其医药上可允许的盐,其中,前述氨基由以下的结构中的任一者表示,
[0104]
[化学式14]
[0105][0106]
前述酰胺基由以下的结构中的任一者表示,
[0107]
[化学式15]
[0108][0109]
[12]
[0110]
如[1]~[11]中任一项所述的化合物或其医药上可允许的盐,其中,前述氨基为乙酰基氨基。
[0111]
[13]
[0112]
如[1]~[12]中任一项所述的化合物或其医药上可允许的盐,其中,式(1)中,r1~r3全部为式(2)表示的基团。
[0113]
[14]
[0114]
由以下的结构中的任一者表示的化合物或其医药上可允许的盐。
[0115]
[化学式16]
[0116][0117]
[15]
[0118]
由以下的结构表示的化合物或其医药上可允许的盐。
[0119]
[化学式17]
[0120][0121]
[16]
[0122]
由以下的结构表示的化合物或其医药上可允许的盐。
[0123]
[化学式18]
[0124][0125]
[17]
[0126]
造影剂,其包含[1]~[16]中任一项所述的化合物或其医药上可允许的盐。
[0127]
[18]
[0128]
[1]~[16]中任一项所述的化合物或其医药上可允许的盐的制造方法,其包括使由与脱唾液酸糖蛋白受体结合的原子团部分和连接体部分构成的反应基质、与非离子性碘造影剂部分的反应基质进行反应的工序。
[0129]
发明效果
[0130]
根据本发明,可以提供能作为碘造影剂使用的新化合物。
附图说明
[0131]
[图1]为示出作为实施例的化合物的ryo-1、meg-1、meg-2的、hepg2中的摄入抑制试验的结果的图。
[0132]
[图2]为示出作为实施例的化合物的ryo-1、meg-1、meg-2的、panc-1中的摄入抑制试验的结果的图。
[0133]
[图3]为示出作为实施例的化合物的meg-2、meg-4、ryo-1、及ryo-2的、hepg2中的摄入抑制试验的结果的图。
[0134]
[图4]为示出将作为实施例的化合物的meg-1施予至小鼠时的、施予1小时后的ct图像的图。
具体实施方式
[0135]
以下,对本发明的具体实施方式进行详细说明。需要说明的是,本发明并不限定于以下的实施方式,可以在其要旨的范围内进行各种变形而实施。
[0136]
[化合物]
[0137]
本发明的化合物由式(1)表示。另外,本发明也是由式(1)表示的化合物的医药上可允许的盐。本说明书中,有时将医药上可允许的盐包括在内而简称为化合物。
[0138]
[化学式19]
[0139][0140]
式(1)中,r1~r3各自独立地为:
[0141]
由-nr
xry
(r
x
及ry各自独立地表示氢原子、可具有取代基的c1~c6的烃基或可具有取代基的c2~c7的酰基)表示的氨基;
[0142]
或由-c(=o)nr
zrw
(rz及rw各自独立地表示氢原子、或可具有取代基的c1~c6的烃基)表示的酰胺基;
[0143]
或者,由式(2)表示的基团,
[0144]
[化学式20]
[0145][0146]
(式(2)中,
[0147]
原子基团为与脱唾液酸糖蛋白受体结合的原子团,
[0148]
连接物为任意的连接体。)
[0149]
r1~r3中的至少1个为式(2)表示的基团。
[0150]
本发明的化合物中,r1~r3中的1个为式(2)表示的基团的情况下,r1~r3中的剩余的2个选自上述氨基或上述酰胺基,r1~r3中的2个为式(2)表示的基团的情况下,r1~r3中的剩余的1个选自上述氨基或上述酰胺基。本发明的化合物也包括r1~r3中的3个为式(2)表示的基团的情况。
[0151]
本发明的化合物的r1~r3的各基团中,包含多个式(2)表示的基团、多个上述氨基、或多个上述酰胺基的情况下,这些基团可以相同,也可以不同。
[0152]
作为本发明的化合物,关于r1~r3,就针对r1~r3的各说明中的方式而言,与是否优选无关地,可由各方式形成任意的组合。
[0153]
本发明的化合物可以作为造影剂使用。本发明中,所谓造影剂,是指在图像诊断中为了向拍摄的图像赋予对比度、或者强调特定的组织而施予至患者的医药品或医药组合物。
[0154]
本发明的化合物中的原子基团被肝细胞特异性地表达的脱唾液酸糖蛋白受体识别,由此,该化合物被摄入至肝细胞内。现有的碘性造影剂多数是其全部自肾脏作为尿而被排泄,对肾脏施加负荷,但本发明的化合物能够被摄入至肝脏从而使其作为胆汁排泄。即,本发明的化合物可以将药物代谢途径设为尿和胆汁的双系统,能够实现肾毒性减轻。
[0155]
另外,本发明的化合物在以注射剂的形态施予时,存在即使将该化合物的浓度设为高浓度也能够维持一定的粘度的倾向,操作性优异,能够容易地进行向施予对象的施予。
[0156]
本发明的由式(1)表示的化合物例如可以由以下的式(1-1)、(1-2)、(1-3)、(1-4)、(1-5)、或(1-6)表示。本发明的由式(1)表示的化合物优选具有2个以上的式(2)表示的基团,更优选具有3个式(2)表示的基团。即,作为本发明的化合物,优选由以下的式(1-2)、(1-3)、或(1-5)表示,更优选由以下的式(1-3)表示。根据这样的方式,存在本发明的化合物与
脱唾液酸糖蛋白受体的基质识别部位(例如半乳糖识别部位)的相互作用进一步提高的倾向,存在更高效地导入至肝细胞中的倾向。
[0157]
[化学式21]
[0158][0159]
[化学式22]
[0160][0161]
式(1-1)~(1-6)中的r
x
、ry、rz、rw、原子基团、及连接物与式(1)中的r
x
、ry、rz、rw、原子基团、及连接物的含义相同。-nr
xry
、-c(=o)nr
zrw
、原子基团、或连接物存在有多个的情况下,各自可以相同或不同,但优选相同。
[0162]
式(1)中,由-nr
xry
表示的氨基中的r
x
及ry各自独立地表示氢原子、可具有取代基
的c1~c6的烃基或可具有取代基的c2~c7的酰基。r
x
及ry可以相同或不同,但优选r
x
及ry中的一者表示氢原子,另一者表示可具有取代基的c1~c6的烃基或可具有取代基的c2~c7的酰基。或者,也优选为下述方式:r
x
及ry中的一者表示可具有取代基的c1~c6的烃基,另一者表示可具有取代基的c2~c7的酰基。
[0163]rx
及ry中,可具有取代基的烃基的碳原子数优选为1以上5以下,更优选为1以上4以下。另外,可具有取代基的酰基的碳原子数优选为2以上6以下,更优选为2以上5以下。
[0164]
由-c(=o)nr
zrw
表示的酰胺基中的rz及rw各自独立地表示氢原子、或可具有取代基的c1~c6的烃基。rz及rw可以相同或不同,但优选r
x
及ry中的一者表示氢原子,另一者表示可具有取代基的c1~c6的烃基。
[0165]rz
及rw中,可具有取代基的烃基的碳原子数优选为1以上5以下,更优选为1以上4以下。
[0166]
作为上述c1~c6的烃基,可以为饱和或不饱和的、直链或支链或环状的烃基,例如,可举出碳原子数1以上6以下的烷基、碳原子数2以上6以下的烯基、碳原子数2以上6以下的炔基、碳原子数6的芳基等。上述c1~c6的烃基优选为饱和的、直链或支链的烃基。
[0167]
本说明书中,作为碳原子数1以上6以下的烷基,具体而言,可举出甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、戊基、己基、环丙基、环丁基、环戊基、环己基等。
[0168]
本说明书中,作为碳原子数2以上6以下的烯基,具体而言,可举出烯丙基、甲代烯丙基、1-丁烯-1-基、2-丁烯-1-基、3-丁烯-1-基、1-丁烯-3-基等。
[0169]
本说明书中,作为碳原子数2以上6以下的炔基,具体而言,可举出2-丙炔-1-基、2-丁炔-1-基、3-丁炔-1-基等。
[0170]
本说明书中,作为碳原子数6的芳基,具体而言,可举出苯基等。
[0171]
上述c2~c7的酰基是指由r-co-表示的基团,r例如为碳原子数1以上6以下的烷基、碳原子数2以上6以下的烯基、碳原子数2以上6以下的炔基、碳原子数6的芳基等。作为上述c2~c7的酰基,具体而言,可举出乙酰基、丙烯酰基、苯甲酰基等。
[0172]
作为r
x
、ry、rz及rw中的取代基,没有特别限定,例如,可举出羟基、卤原子、-ora表示的有机氧基、由-n(rb)(rc)表示的氨基等。上述的取代基可以包含于本发明的化合物的任意部分中,而不限于r
x
、ry、rz及rw。
[0173]
作为上述卤原子,可以举出氟原子、氯原子、溴原子、碘原子等。
[0174]
作为ra,例如,可举出碳原子数1以上6以下的烷基、碳原子数2以上6以下的烯基、碳原子数2以上6以下的炔基、碳原子数6的芳基等。
[0175]
作为rb及rc,各自独立地可举出例如氢原子、碳原子数1以上6以下的烷基、碳原子数2以上6以下的烯基、碳原子数2以上6以下的炔基、碳原子数6的芳基等。
[0176]
由-nr
xry
表示的氨基优选为乙酰基氨基(为-nhcor,r为可具有取代基的c1~c6的烃基。可具有取代基的c1~c6的烃基的定义与r
x
及ry同样。)。或者,也优选为下述方式:由-nr
xry
表示的氨基由以下的结构中的任一者表示。
[0177]
[化学式23]
[0178][0179]
由-c(=o)nr
zrw
表示的酰胺基优选由以下的结构中的任一者表示。
[0180]
[化学式24]
[0181][0182]
是指由-nr
xry
表示的氨基及由-c(=o)nr
zrw
表示的酰胺基中的左侧连接键、以及在具体示出的结构中各来自nh或n的连接键、或者来自c=o的连接键为与式(1)中的三碘苯骨架结合的键。
[0183]
本发明的化合物中的原子基团为与脱唾液酸糖蛋白受体结合的原子团,只要在该原子团中具有被脱唾液酸糖蛋白受体识别的结构,就没有特别限定。作为原子基团的被脱唾液酸糖蛋白受体识别的结构,例如,可举出与脱唾液酸糖蛋白受体结合的糖残基、来自免疫球蛋白a(iga)等的肽链的基团等,作为原子基团,可举出来自与脱唾液酸糖蛋白受体结合的糖残基的基团、或来自免疫球蛋白a(iga)等的肽链的基团等。
[0184]
本发明的化合物中的原子基团优选为与脱唾液酸糖蛋白受体结合的糖残基,更优选为来自半乳糖、n-乙酰半乳糖胺、n-三氟乙酰半乳糖胺、或半乳糖-n-乙酰葡糖胺(也称为“半乳糖基-n-乙酰葡糖胺”。)的基团。所谓半乳糖-n-乙酰葡糖胺,是指半乳糖和n-乙酰葡糖胺在半乳糖的1位羟基中与n-乙酰葡糖胺脱水缩合而形成的二糖。
[0185]
本说明书中,所谓“来自半乳糖”、“来自n-乙酰半乳糖胺”、“来自n-三氟乙酰半乳糖胺”、或“来自半乳糖-n-乙酰葡糖胺”,表示也可以为下述方式:为了与连接物结合而对半乳糖、n-乙酰半乳糖胺、n-三氟乙酰半乳糖胺、或半乳糖-n-乙酰葡糖胺的一部分施加了结构改变。作为结构改变的例子,可举出半乳糖、n-乙酰半乳糖胺、n-三氟乙酰半乳糖胺、或半乳糖-n-乙酰葡糖胺的1个羟基被替换为伯氨基(-nh2)而成的结构。
[0186]
半乳糖、n-乙酰半乳糖胺、n-三氟乙酰半乳糖胺、或半乳糖-n-乙酰葡糖胺可以介由该糖中的任意官能团而与连接物进行相互作用。与脱唾液酸糖蛋白受体结合的糖残基与连接物结合的情况下,连接物所结合的碳位置可以为该糖的1位、2位、3位、4位、或6位,优选为1位。
[0187]
对于半乳糖-n-乙酰葡糖胺而言,只要为被脱唾液酸糖蛋白受体识别的糖链,就没有特别限定,半乳糖与n-乙酰葡糖胺的结合可以为β1,3结合、β1,4结合,优选为β1,3结合。
[0188]
本发明的化合物中的由式(2)表示的基团优选为由式(2-1)或式(2-2)表示的基团,更优选为由式(2-1)表示的基团。由式(2)表示的基团也可以为由式(2-3)或(2-4)表示的基团。
[0189]
[化学式25]
[0190][0191]
式(2-1)、(2-2)、(2-3)及(2-4)中的连接物与式(1)中的连接物同义。将来自半乳糖、n-乙酰半乳糖胺、n-三氟乙酰半乳糖胺、或n-乙酰葡糖胺的基团与连接物的结合以波浪线示出,表示其可以为α结合、也可以为β结合。上述的波浪线的结合优选为β结合。
[0192]
本发明的化合物中的连接体是指将与脱唾液酸糖蛋白受体结合的原子团、和三碘苯骨架结合的结构。本发明的化合物具有2个以上的连接体时,该2个以上的连接体可以相同,也可以不同,但优选相同。
[0193]
本发明的化合物中的连接体优选为可具有取代基的烃链,此处,所述烃链的两末端中的一个或两个可以具有杂原子、酰胺键、酯键、羰基键、或芳香族杂环中的任一者。即,本发明的化合物中的连接体可以为在两末端中的一个或两个具有杂原子、酰胺键、酯键、羰基键、及芳香族杂环中的任一者的、可具有取代基的烃链。上述烃链可以包含不饱和键,可以为直链或支链或环状的烃基。另外,上述烃链也可以为直链及/或支链的烃、与环状的烃的组合。作为上述烃链,例如,可举出亚烷基链、亚烯基链、亚炔基链、及亚芳基、以及它们的组合等。上述烃链的主链中可以包含除碳原子及氢原子以外的原子(例如,氧原子、氮原子)。另外,上述烃链的主链中可以具有羰基结构。
[0194]
连接体可以在其两末端中的任意末端具有杂原子、酰胺键、酯键、羰基键、或芳香族杂环,优选在两末端中的任意末端均具有杂原子、酰胺键、酯键、羰基键、或芳香族杂环。即,本发明的化合物中,优选与脱唾液酸糖蛋白受体结合的原子团或三碘苯骨架、和连接体的结合部分各自独立地为杂原子、酰胺键、酯键、羰基键、或芳香族杂环。
[0195]
上述的烃链更优选为可具有取代基的亚烷基链,或者为介由选自由杂原子、酰胺键、酯键、羰基键、及芳香族杂环组成的组中的至少一者而结合有可具有取代基的2个以上的亚烷基链的烃链。此处,亚烷基链可以为直链,或者也可以具有支链。构成上述亚烷基链
的主链的元素可以包含除碳原子以外的原子,但优选上述亚烷基链的主链仅由碳原子构成。
[0196]
以将与脱唾液酸糖蛋白受体结合的原子团、和三碘苯骨架之间最短地连接的亚烷基链作为主链时,构成该主链的碳原子数优选为1以上10以下,更优选为1以上8以下,进一步优选为1以上6以下。上述的碳原子数在上述的范围内更优选为2以上,例如也可以为2以上6以下。
[0197]
通过使上述碳原子数在上述的范围内,从而存在下述倾向:本发明的化合物变得容易与脱唾液酸糖蛋白受体的基质识别部位(例如半乳糖识别部位)进行相互作用,被高效地导入至肝细胞中。特别是在本发明的化合物在式(1)中具有2个以上(或3个)的式(2)表示的基团时,通过使上述碳原子数在上述的范围内,从而存在本发明的化合物与脱唾液酸糖蛋白受体的基质识别部位(例如半乳糖识别部位)的相互作用进一步提高的倾向,存在更高效地导入至肝细胞中的倾向。此处的碳原子数不包括后述的芳香族杂环、酰胺键、酯键、及羰基键等亚烷基链中可包含的官能团的碳。
[0198]
作为杂原子,例如,可举出氧原子、硫原子、氮原子等。这些杂原子优选以醚键(-o-)、硫醚键(-s-)、胺键(-nh-)的形式被包含。杂原子可以包含在亚烷基链中,也可以配置在亚烷基链的末端、即同与脱唾液酸糖蛋白受体结合的原子团、或三碘苯骨架结合的位置。
[0199]
酰胺键、酯键、及羰基键分别由-nh-co-、-co-o-、及-co-表示。酰胺键、酯键、及羰基键可以包含在亚烷基链中,也可以配置在亚烷基链的末端、即同与脱唾液酸糖蛋白受体结合的原子团、或三碘苯骨架结合的位置。
[0200]
作为芳香族杂环,例如可举出三唑环,特别优选为1,2,3-三唑。芳香族杂环可以包含在亚烷基链中,也可以配置在亚烷基链的末端、即同与脱唾液酸糖蛋白受体结合的原子团、或三碘苯骨架结合的位置。
[0201]
可以在1个亚烷基链中包含1个或2个以上的芳香族杂环、杂原子、酰胺键、酯键、及羰基键。1个烃链中包含的芳香族杂环、杂原子、酰胺键、酯键、及羰基键的总计优选为2以上,更优选为2以上6以下,进一步优选为2以上4以下。
[0202]
酰胺键及酯键的方向没有特别限定,为酰胺键时,可以为-nh-co-,也可以为-co-nh-,为酯键时,可以为-co-o-,也可以为-o-co-。
[0203]
芳香族杂环的结合的位置及方向没有特别限定,例如在亚烷基链中包含1,2,3-三唑的情况下,例如可以结合在1位及4位。另外,1,2,3-三唑结合在1位及4位的情况下,1位可以位于与脱唾液酸糖蛋白受体结合的原子团侧,4位也可以位于与脱唾液酸糖蛋白受体结合的原子团侧。
[0204]
对于本发明的化合物中的连接体而言,以将与脱唾液酸糖蛋白受体结合的原子团、和三碘苯骨架之间最短地连接的亚烷基链作为主链时,构成该主链的原子数优选为1以上15以下,更优选为1以上13以下,进一步优选为1以上12以下。构成该主链的原子数在上述的范围内优选为2以上、3以上、或4以上,也可以为5以上。另外,该原子数在上述的范围内可以为11以下。通过使上述原子数在上述的范围内,从而存在下述倾向:本发明的化合物变得容易与脱唾液酸糖蛋白受体的基质识别部位(例如半乳糖识别部位)进行相互作用,被高效地导入至肝细胞中。上述的原子数可以设定为将上述的上限值及下限值任意组合而得到的范围内。
[0205]
作为上述的烃链及亚烷基链可具有的取代基,可举出与r
x
、ry、rz及rw中的取代基同样的取代基,优选选自羟基或氨基(-nh2),更优选为羟基。另外,作为该取代基,可以为被羟基取代的烷基,也可以为被羟基和经羟基取代的烷基取代的侧链。
[0206]
作为本发明的化合物中的连接体的优选方式,可举出由以下的结构表示的连接体。
[0207]
[化学式26]
[0208]
·-l
0-l
x-l
y-l
z-·
[0209]
此处,式中,l0为-o-或-nhc(=o)-,l
x
由以下的结构表示,
[0210]
[化学式27]
[0211][0212]
ly表示单键,或者由以下的结构表示(优选为单键。),
[0213]
[化学式28]
[0214][0215]
lz由以下的结构表示。
[0216]
[化学式29]
[0217][0218]
上述式中,r’各自独立地为选自氢原子、可具有取代基的c1~c3烷基、或羟基中的任一者;l1各自独立地为选自醚键(-o-)、硫醚键(-s-)、胺键(-nh-)、酰胺键、酯键、或羰基键中的任一者;l2为酰胺键、或来自芳香族杂环的2价基团;l3为选自-och
2-、-nhch
2-、-c(=o)nh-、-c(=o)nhch
2-、或-nhc(=o)-中的任一者。
[0219]
m1、m2、及m4各自独立地为1以上的整数;m3为0以上的整数;n及n2各自独立地为0以上的整数。
[0220]
l0表示连接体和与脱唾液酸糖蛋白受体结合的原子团的结合部分。l0优选为-o-(醚键)。需要说明的是,l0为-nhc(=o)-时,与脱唾液酸糖蛋白受体结合的原子团和连接物如以下的顺序:(与脱唾液酸糖蛋白受体结合的原子团)-nhc(=o)-连接物这样结合。
[0221]
l
x
、ly、及lz中,作为r’中的可具有取代基的c1~c3烷基,可举出甲基、乙基、正丙基、异丙基等。作为上述的c1~c3烷基可具有的取代基,可举出与r
x
、ry、rz及rw中的取代基同样的取代基,但优选选自羟基或氨基(-nh2)。
[0222]
l1中,酰胺键及酯键的方向没有特别限定,为酰胺键时,可以为-nh-co-,也可以为-co-nh-,为酯键时,可以为-co-o-,也可以为-o-co-。l2中的酰胺键也同样。
[0223]
l2中,所谓来自芳香族杂环的2价基团,是指从芳香族杂环除去2个氢原子而得到的2价基团。作为这样的2价基团,没有特别限定,例如可举出将1,2,3-三唑的1位及4位的氢原子除去而得到的2价基团。在该情况下,来自1,2,3-三唑的2价基团的方向没有限定,1位可以与l
x
结合,4位也可以与l
x
结合。
[0224]
作为l1的优选方式,例如可举出醚键及酰胺键。作为l3的优选方式,例如可举出-c(=o)nhch
2-、及-nhc(=o)-。
[0225]
m1、m2、及m4各自独立地为1以上的整数,优选为1以上5以下的整数,更优选为1以上4以下、或1以上3以下的整数。m1、m2、及m4各自独立地可以为2以上的整数。m2可以为1以上10以下、1以上8以下、或1以上6以下。
[0226]
m3为0以上的整数,可以为1以上5以下的整数,也可以为1以上4以下、或1以上3以下的整数。
[0227]
n及n2各自独立地为0以上的整数,可以为5以下,优选为4以下,更优选为3以下。n及n2之和优选为0以上4以下,更优选为1以上3以下。
[0228]
上述的连接体中,l
x
优选为以下的结构中的任一者,
[0229]
[化学式30]
[0230][0231]
ly优选为单键,或者为以下的结构中的任一者,
[0232]
[化学式31]
[0233][0234]
lz优选为以下的结构中的任一者。
[0235]
[化学式32]
[0236][0237]
上述式中,k1各自独立地为1以上3以下的整数;k2为0或1;m3为0以上的整数;n及n2各自独立地为0以上的整数。
[0238]
k1各自独立地为1以上3以下的整数,n为2以上时,多个k2可以相同,也可以不同。
[0239]
m3、n及n2的优选的数值范围与上述同样。
[0240]
作为本发明的化合物中的连接体的其他优选方式,可举出由以下的结构表示的连接体。
[0241]
[化学式33]
[0242][0243]
式中,r’各自独立地为选自氢原子、可具有取代基的c1~c3烷基、羟基中的任一者,
[0244]
l0为-o-或-nhc(=o)-,
[0245]
l1各自独立地表示单键,或者为选自醚键(-o-)、硫醚键(-s-)、胺键(-nh-)、酰胺键、酯键、羰基键中的任一者,
[0246]
m1及m2各自独立地为1以上的整数,
[0247]
n为0以上的整数,
[0248]
n’为0或1。
[0249]
作为可具有取代基的c1~c3烷基的c1~c3烷基,可举出甲基、乙基、正丙基、异丙基等。作为可具有取代基的c1~c3烷基的取代基,可以举出与r
x
、ry、rz及rw中的取代基同样的取代基。
[0250]
l0的定义与上文同样,优选为-o-(醚键)。
[0251]
m1及m2各自独立地优选为1以上5以下的整数,更优选为1以上4以下的整数,进一步优选为1以上3以下的整数。
[0252]
m2也可以为1以上10以下、1以上8以下、或1以上6以下。
[0253]
n只要为0以上的整数即可,没有特别限定,但优选为0以上5以下,更优选为0以上4以下,进一步优选为1以上3以下。
[0254]
n为0时,上述的连接体结构由下式表示。
[0255]
[化学式34]
[0256][0257]
n’为0或1。n’为0时,上述式的连接体由下式表示。n’优选为1。
[0258]
[化学式35]
[0259][0260]
本发明的化合物中的连接体进一步优选由以下的结构中的任一者表示。
[0261]
[化学式36]
[0262][0263]
[化学式37]
[0264][0265]
式中,l0为-o-或-nhc(=o)-;n为0以上的整数;n为0以上的整数;m2为1以上的整数;n’为0或1。
[0266]
l0的定义与上文同样,优选为-o-(醚键)。
[0267]
n优选为0以上5以下,更优选为0以上4以下,进一步优选为1以上3以下。
[0268]
n优选为0或者为1以上5以下的整数,更优选为0或者为1以上4以下的整数,进一步优选为0或者为1以上3以下的整数。
[0269]
m2可以为1以上10以下、1以上8以下、或1以上6以下的整数,优选为1以上5以下的整数,更优选为1以上4以下的整数,进一步优选为1以上3以下的整数。
[0270]
n或n为0时,m2优选为1以上10以下的整数。
[0271]
n或n为1以上的整数时,m2优选为1以上6以下的整数。
[0272]
n’优选为1。
[0273]
作为本发明的化合物,可以为式(1)中针对由-nr
xry
表示的氨基、由-c(=o)nr
zrw
表示的酰胺基、及由式(2)表示的基团分别说明的优选方式的任意组合。另外,对于针对各基团说明的优选方式而言也同样,可以为任意的组合。即,虽然没有特别限定,但作为一例而示出时,可以就由-nr
xry
表示的氨基、由-c(=o)nr
zrw
表示的酰胺基、及由式(2)表示的基团而言仅由式(2)表示的基团为特定的优选方式,也可以就由-nr
xry
表示的氨基、由-c(=o)nr
zrw
表示的酰胺基、及由式(2)表示的基团而言、由-nr
xry
表示的氨基及由-c(=o)nr
zrw
表示的酰胺基为针对各自所说明的特定优选方式。
[0274]
作为本发明的化合物,由下述任意式表示的化合物是优选的。
[0275]
[化学式38]
[0276][0277]
[化学式39]
[0278][0279]
作为本发明的化合物,由下述任意式表示的化合物是更优选的。
[0280]
[化学式40]
[0281][0282]
作为本发明的化合物,由下述任意式表示的化合物是进一步优选的。
[0283]
[化学式41]
[0284][0285]
[化学式42]
[0286][0287]
上文中作为本发明的化合物的优选方式而示出的化合物中的连接体部分(即,根据由上述任意式表示的化合物和式(2-1)~(2-4)理解出的相当于连接物的结构)的各结构成为式(1)表示的化合物中优选的连接物结构。
[0288]
即,作为式(1)中的连接物的任意的连接体可以为由上述任意式表示的化合物中的连接物,作为式(2-1)~(2-4)中的连接物的任意的连接体也可以为由上述任意式表示的化合物中的连接物。
[0289]
另外,作为“作为本发明的化合物中的连接体的优选方式,可举出由以下的结构表示的连接体”而说明的连接体可以为由上述任意式表示的化合物中的连接物。
[0290]
作为本发明的化合物的医药上可允许的盐,例如,可举出盐酸盐、硫酸盐、磷酸盐等无机酸盐、乙酸盐、丙酸盐、酒石酸盐、富马酸盐、马来酸盐、苹果酸盐、柠檬酸盐、甲磺酸盐、对甲苯磺酸盐、三氟乙酸盐等有机酸盐等。
[0291]
作为本发明的化合物的医药上可允许的盐,例如,可举出钠盐、钾盐、钙盐等碱金属或碱土金属等。
[0292]
[化合物的制造方法]
[0293]
本发明的化合物可以通过使用有机合成方法来制造。本发明的化合物由与脱唾液酸糖蛋白受体结合的原子团部分、连接体部分、及非离子性碘造影剂部分构成。可以使相当于上述与脱唾液酸糖蛋白受体结合的原子团部分、连接体部分、及非离子性碘造影剂部分的反应基质分别进行反应并连接,由此制造本发明的化合物。
[0294]
本发明的化合物例如可以通过下述方法来制造:使由与脱唾液酸糖蛋白受体结合的原子团部分和连接体部分构成的反应基质、与非离子性碘造影剂部分的反应基质进行反应的方法;向非离子性碘造影剂部分的反应基质中导入连接体部分之后,使其与相当于与脱唾液酸糖蛋白受体结合的原子团部分的反应基质进行反应的方法;等等。
[0295]
作为本发明的化合物的制造方法,使由与脱唾液酸糖蛋白受体结合的原子团部分和连接体部分构成的反应基质与非离子性碘造影剂部分的反应基质进行反应的方法是优选的,由式(1)表示的化合物的制造方法例如可以由以下的合成路径表示。本发明之一为由式(1)表示的化合物的制造方法,其包括使由与脱唾液酸糖蛋白受体结合的原子团部分和连接体部分构成的反应基质、与非离子性碘造影剂部分的反应基质进行反应的工序。非离子性碘造影剂部分的反应基质优选为具有三碘苯骨架的化合物。
[0296]
[化学式43]
[0297][0298]
合成路径中,x1、和x2中的至少一者包含反应性基团,x1的反应性基团与x2的反应性基团进行反应而形成化学键。反应性基团以外的x2优选为-nr
xry
或-c(=o)nr
zrw
(r
x
、ry、rz及rw的定义与上文同样。)。
[0299]
作为此处形成的化学键,没有特别限定,例如,可举出碳-碳键(可以为饱和键,也可以为不饱和键)、醚键(-o-)、硫醚键(-s-)、胺键(-nh-)、酰胺键、酯键、羰基键及芳香族杂环等。作为这些键的形成反应,应用已知的有机合成反应即可。
[0300]
形成碳-碳键的情况下,作为反应,没有特别限定,例如,可举出维蒂希(wittig)反应、格氏(grignard)反应、铃木-宫浦偶联反应、根岸偶联反应等。x1及x2为通过这些反应来进行反应的有机基团即可。
[0301]
在维蒂希反应的情况下,例如,优选x1及x2中的一者为醛基或酮基,另一者为磷叶立德(phosphorus ylide)基。在格氏反应的情况下,例如,优选x1及x2中的一者为醛基或酮基,另一者为形成格氏反应剂的基团(-mgx)。在铃木-宫浦偶联反应的情况下,例如,优选x1及x2中的一者为硼酸或硼酸酯基,另一者为卤素基团或三氟甲磺酸酯(triflate)基。在根岸偶联反应的情况下,例如,优选x1及x2中的一者为形成有机锌的基团(-znx),另一者为卤素基团。
[0302]
形成醚键的情况下,例如,优选x1及x2中的一者为羟基,另一者为卤素基团。
[0303]
形成硫醚键的情况下,例如,优选x1及x2中的一者为硫醇基,另一者为卤素基团。
[0304]
形成胺键的情况下,例如,优选x1及x2中的一者为氨基,另一者为卤素基团。
[0305]
形成酰胺键的情况下,例如,优选x1及x2中的一者为氨基,另一者为羧酸基、酰卤化物基(-co-x)、或酸酐基。
[0306]
形成酯键的情况下,例如,优选x1及x2中的一者为羟基,另一者为羧酸基或酰卤化物基(-co-x)。
[0307]
形成羰基键的情况下,例如,优选x1及x2中的一者为weinreb酰胺基(-co-n(ome)2),另一者为形成格氏反应剂的基团(-mgx)或形成有机锂的基团(-li)。
[0308]
形成芳香族杂环的情况下,可以使用以huisgen环化反应为代表的点击反应。例如,在x1及x2中的一者为叠氮基(-n3)、另一者为末端炔的情况下,能够形成1,2,3-三唑环。
[0309]
这些所形成的键之中,优选为酰胺键。此时,优选x1为氨基,x2为羧酸基、酰卤化物基(-co-x)、或酸酐基,更优选x1为氨基,x2为酰卤化物基(-co-x)。
[0310]
由与脱唾液酸糖蛋白受体结合的原子团部分和连接体部分构成的反应基质s1(以下,也称为中间体s1)、及非离子性碘造影剂部分的反应基质s2(以下,也称为中间体s2)可以适宜地通过有机合成方法来合成。
[0311]
例如,中间体s1可以通过向成为原子基团的基团中导入连接体部、接着导入反应性基团x1从而制备。例示原子基团为糖残基结构的情况来进行表示时,如以下的合成路径
那样。具体而言,可以通过下述方式来制备:将五-o-乙酰基-d-吡喃半乳糖或七-o-乙酰基-d-吡喃半乳糖基-n-乙酰葡糖胺等糖或其衍生物作为起始物,导入连接体部,接着导入反应性基团x1。在中间体s1的合成中,可以适宜地包括官能团的基于保护基的保护及/或脱保护的工序等对中间体s1的合成而言必要的转化工序。
[0312]
[化学式44]
[0313][0314]
在将与脱唾液酸糖蛋白受体结合的原子团部分和连接体部分结合之前,可以将与脱唾液酸糖蛋白受体结合的原子团部分的官能团预先转化为其他的取代基。例示原子基团为糖残基结构的情况来进行表示时,可以将糖部分的羟基预先转化为其他的取代基。作为这样的取代基,可举出伯氨基、酰基氨基等。
[0315]
如上述那样,通过将与脱唾液酸糖蛋白受体结合的原子团部分的官能团预先转化为其他的取代基,能够利用任意的键使与脱唾液酸糖蛋白受体结合的原子团部分和连接体结合。例如,原子基团为糖残基结构的情况下,可以如下所述地将糖部分的羟基替换为氨基,由此,能够利用酰胺键使糖部分和连接体结合。
[0316]
[化学式45]
[0317][0318]
连接体的导入可以阶段性地进行。即,可以通过2阶段以上的反应将连接体结构延长,从而形成最终的连接体结构。
[0319]
中间体s2例如可以通过下述方式来制备:如以下的合成路径所示地,将具有由r
s1
~r
s3
表示的任意取代基的芳香族化合物作为起始物,导入碘基,接着导入反应性基团x2,从而进行向s2的衍生。在中间体s2的合成中,可以适宜地包括官能团的基于保护基的保护及/或脱保护的工序等对中间体s2的合成而言必要的转化工序。
[0320]
[化学式46]
[0321][0322]
对于r
s1
~r
s3
而言,优选根据与脱唾液酸糖蛋白受体结合的原子团的导入数而使取代基为不同的取代基。
[0323]
在本发明的化合物的合成中,可以适宜地在反应溶剂、催化剂、添加剂等的存在下进行。另外,反应温度、反应压力、反应时间等反应条件也可以适宜地调整。进而,对所得到的产物进行适宜的后处理之后,可以取得本发明的化合物。作为后处理的具体方法,可以举出萃取处理及/或结晶析出、重结晶、色谱法等已知的纯化。
[0324]
[造影剂]
[0325]
本发明的化合物可以作为造影剂使用。因此,本发明的一个实施方式为包含本发明的化合物的造影剂。
[0326]
应用本发明的造影剂的成像技术设想到了使用现有的碘性造影剂进行的造影x射线检查中的几乎全部检查。即,包括从头颈部至胸部、上下腹部、四肢的造影ct检查、以及脑、心脏、大动脉、腹部等的使用血管导管的检查、治疗等。另外,本造影剂被排泄至胆汁及尿,因此,能够期待向排泄性尿路造影、排泄性胆管造影检查等的应用。
[0327]
本发明的化合物可以作为造影剂而以已知的医药组合物的形态使用。上述医药组合物可以利用以往已知的方法来制造。
[0328]
本发明的化合物可以使用一种,也可以以两种以上的混合物的形式使用。
[0329]
本发明的造影剂中,可以含有药剂学上可允许的添加剂。作为添加剂,没有特别限定,可举出稳定剂、赋形剂、粘合剂、崩解剂、润滑剂、抗氧化剂、矫味剂、着色剂、香料等。
[0330]
本发明的造影剂可被制剂化以适合包括静脉内、皮内、皮下、经口(例如,也包括吸入等)、经皮及经粘膜的施予的、治疗上适当的施予途径,但可利用操作性优异的性质而优选进行静脉内注射。
[0331]
本发明的造影剂的施予量可根据成为施予对象的患者的状态、施予方法等而适宜确定。
[0332]
本发明还进一步提供包含由式(1)表示的化合物或其医药上可允许的盐的医药组合物。所述医药组合物可以还包含药剂学上可允许的添加剂。医药组合物也可以作为造影剂使用。
[0333]
本发明还提供造影x射线检查方法,其使用包含由式(1)表示的化合物或其医药上可允许的盐的医药组合物。
[0334]
本发明还提供用于造影x射线检查的由式(1)表示的化合物或其医药上可允许的盐。
[0335]
实施例
[0336]
以下,利用实施例来详细说明本实施方式,但本发明并不限定于以下的实施例。
[0337]
[实施例1]meg-1的合成
[0338]
为了合成meg-1,首先进行中间体i及中间体ii的合成。
[0339]
《中间体i的合成》
[0340]
中间体i按照以下的合成路径来合成。
[0341]
[化学式47]
[0342][0343]
将市售的泛影酸(东京化成工业株式会社制,2.01g,3.27mmol)溶解于亚硫酰氯(20.0ml),使用油浴,于120℃进行3小时回流。将亚硫酰氯蒸馏除去之后,用正己烷清洗,以黄白色的粉末的形态得到中间体i(1.39g,67%)。
[0344]
中间体i的1h-nmr如下所述。
[0345]1h-nmr(500mhz,dmso-d6)δ:10.13(s,1h),10.04(s,1h),2.03(s,6h)
[0346]
《中间体ii的合成》
[0347]
中间体ii按照以下的合成路径来合成。
[0348]
[化学式48]
[0349][0350]
(步骤1)
[0351]
步骤1及后续的步骤2的反应条件参照日本专利第4293735号。
[0352]
具体而言,首先,在氩下将市售的五-o-乙酰基-β-d-吡喃半乳糖(东京化成工业株式会社制,3.89g,9.97mmol)和2-溴乙醇(0.71ml,9.45mmol)溶解于脱水二氯甲烷(41.0ml)。接下来,在冰冷却条件下缓慢地滴加硼酸(4.11ml,32.1mmol),搅拌1小时后,在暗处搅拌一夜。搅拌后,反应溶液变化为橙色。向反应溶液中加入水而使反应停止,进行浓缩。反应后,加入适量的乙酸乙酯,使用水、饱和碳酸钠水溶液、饱和食盐水进行分液。使用硫化钠进行干燥后,进行过滤,并进行浓缩。其后,使用硅胶柱色谱法(正己烷:乙酸乙酯=3:2)进行纯化,以无色透明的糖状物质的形态得到溴体(3.03g,67%)。
[0353]
溴体的1h-nmr如下所述。
[0354]1h-nmr(500mhz,cdcl3)δ:5.40(dd,j=3.5,1.0hz,1h),5.24(dd,j=10.9,8.0hz,1h),5.03(dd,j=10.3,3.4hz,1h),4.54(d,j=8.0hz,1h),4.17-4.21(m,2h),4.12(dd,j=11.2,6.6hz,1h),3.92(td,j=6.6,1.1hz,1h),3.80-3.85(m,1h),3.46-3.51(m,2h),2.16(s,3h),2.09(s,3h),2.06(s,3h),1.99(s,3h)
[0355]
(步骤2)
[0356]
在氩下将由步骤1得到的溴体(3.02g,6.66mmol)和叠氮化钠(1.00g,15.4mmol)溶解于脱水二甲基甲酰胺(35.0ml),于80℃搅拌一夜。反应后,加入适量的乙酸乙酯,使用水、饱和碳酸钠水溶液、饱和食盐水进行分液。使用硫化钠进行干燥后,进行过滤,并进行浓缩。
其后,使用硅胶柱色谱法(正己烷:乙酸乙酯=3:2)进行纯化,以无色透明的油状物质的形态得到叠氮体(2.37g,86%)。
[0357]
叠氮体的1h-nmr如下所述。
[0358]1h-nmr(500mhz,cdcl3)δ:5.41(dd,j=3.5,1.5hz 1h),5.27(dd,j=10.5,8.0hz 1h),5.04(dd,j=10.0,3.5hz 1h),4.56(d,j=8.0hz 1h),4.11-4.21(m,2h),4.03-4.07(m,1h),3.91(td,j=7.0,1.0hz 1h),3.68-3.72(m,1h),3.49-3.54(m,1h),3.29-3.33(m,1h),2.16(s,3h),2.07(s,3h),2.06(s,3h),1.99(s,3h)
[0359]
(步骤3)
[0360]
步骤3及后续的步骤4的反应条件参照journal of medicinal chemistry,2005,48,645-652。
[0361]
具体而言,在氩下将由步骤2得到的叠氮体(1.75g,4.21mmol)溶解于脱水甲醇(40.0ml)。加入甲醇钠甲醇溶液(meoh中的1m)直至ph达到9。其后,于室温下搅拌直至反应结束(tlc:iso-proh-水=7:3,rf=0.810),使用离子交换树脂(12.0g,amberlite h )进行中和。对溶液进行过滤后,用蒸发器进行浓缩,以黄色或褐色的油状物质的形态得到脱乙酰基体(1.02g,97%)。
[0362]
脱乙酰基体的1h-nmr如下所述。
[0363]1h-nmr(500mhz,d2o)δ:4.29(d,j=8.0hz,1h),3.77-3.78(m,1h),3.64-3.71(m,1h),3.53-3.62(m,4h),3.50(dd,j=10.0,3.7hz,1h),3.36-3.41(m,3h)
[0364]
(步骤4)
[0365]
将由步骤3得到的脱乙酰基体(52.9mg,0.212mmol)和钯碳催化剂(10%pd-c,6.70mg)加入至脱水甲醇(4.50ml)中,进行氢取代。于室温搅拌20小时后,进行过滤,使用蒸发器进行浓缩,由此,以黄色或褐色的固体的形态得到中间体ii(43.3mg,92%)。
[0366]1h-nmr(500mhz,d2o)δ:4.26(d,j=7.4hz,1h),3.76-3.81(m,1h),3.63(d,j=4.0hz,1h),3.53-3.61(m,4h),3.50(dd,j=10.0,3.7hz,1h),3.38(dd,j=9.7,8.0hz,1h),2.70-2.73(m,2h)
[0367]
《meg-1的合成》
[0368]
meg-1按照以下的合成路径来合成。
[0369]
[化学式49]
[0370][0371]
具体而言,首先,将中间体ii(29.1mg,0.130mmol)和中间体i(82.2mg,0.130mmol)溶解于脱水dmf(4.00ml)。接着,缓慢地滴加三乙胺(19.9μml),于室温下搅拌一夜。使用蒸发器将溶剂浓缩,使用硅胶柱色谱法(氯仿:甲醇=4:1)进行纯化,以黄色或褐色的固体的形态得到粗制meg-1(38.6mg,36%)。其后,使用尺寸排阻色谱法(sec)进一步纯化,以无色
透明的晶体的形态得到meg-1。
[0372]
meg-1的1h-nmr如下所述。
[0373]1h-nmr(500mhz,d2o)δ:4.31-4.35(m,1h),3.80-3.99(m,1h),3.78-3.88(m,1h),3.74(s,1h),3.46-3.66(m,6h),3.36-3.40(m,1h),2.11(s,6h)
[0374]
[实施例2]meg-2的合成
[0375]
依据实施例1的meg-1的合成方法,按照以下的合成路径来合成meg-2。首先,进行中间体ii’的合成。
[0376]
《中间体ii’的合成》
[0377]
中间体ii’按照以下的合成路径来合成。
[0378]
[化学式50]
[0379][0380]
(步骤5)
[0381]
步骤5及后续的步骤6的反应条件参照chinese journal of chemistry,2006,24,1058-1061。
[0382]
具体而言,首先,在氩下将市售的五-o-乙酰基-β-d-吡喃半乳糖(东京化成工业株式会社制,3.91g,10.0mmol)和2-(2-氯乙氧基)乙醇(1.70ml,16.0mmol)溶解于脱水二氯甲烷(41.0ml)。接着,在冰冷却条件下缓慢地滴加硼酸(3.90ml,30.4mmol),搅拌1小时后,在暗处搅拌一夜。搅拌后,反应溶液变化为橙色。向反应溶液中加入水而使反应停止,进行浓缩。反应后,加入适量的乙酸乙酯,使用水、饱和碳酸钠水溶液、饱和食盐水进行分液。使用硫化钠进行干燥后,进行过滤,并进行浓缩。其后,使用硅胶柱色谱法(正己烷:乙酸乙酯=3:2)进行纯化,以无色透明的油状物质的形态得到氯体。
[0383]
氯体的1h-nmr如下所述。
[0384]1h-nmr(500mhz,cdcl3)δ:5.39(s,1h),5.19-5.23(m,1h),5.02(td,j=6.7,3.4hz,1h),4.59(dd,j=7.7,2.0hz,1h),4.09-4.20(m,2h),3.91-3.99(m,2h),3.72-3.78(m,3h),3.61-3.69(m,4h),2.15(s,3h),2.07(s,3h),2.05(s,3h),1.99(s,3h)
[0385]
(步骤6)
[0386]
在氩下将由步骤5得到的氯体(1.84g,4.06mmol)和叠氮化钠(0.612g,9.41mmol)溶解于脱水二甲基甲酰胺(35.0ml),于80℃搅拌一夜。反应后,加入适量的乙酸乙酯,使用水、饱和碳酸钠水溶液、饱和食盐水进行分液。使用硫化钠进行干燥后,进行过滤,并进行浓缩。其后,使用硅胶柱色谱法(正己烷:乙酸乙酯=3:2)进行纯化,以透明的黄色油状物质的形态得到叠氮体(1.75g,94%)。
[0387]
叠氮体的1h-nmr如下所述。
3.69(m,11h),3.32-3.37(m,1h),2.10(s,6h)
[0406]
[实施例3]ryo-1的合成
[0407]
为了合成ryo-1,首先进行中间体iii的合成。
[0408]
《中间体iii的合成》
[0409]
中间体iii按照以下的合成路径来合成。对于中间体iii而言,参照jinyong fan et al,journal of materials science:materials in medicine,2010,21,319-327来进行。
[0410]
[化学式52]
[0411][0412]
具体而言,首先,使作为起始物的内酯及过剩的乙二胺的无水二甲基亚砜(dmso)溶液回流2小时。通过加入丙酮而使作为产物的中间体iii沉淀,对所得到的黄色沉淀物进行减压干燥。所得到的中间体iii的质谱分析的结果如下所述。
[0413]c14h29
n2o
11
的lcms(esi)m/z[m h]
计算值:401;实际值:401.
[0414]
需要说明的是,对于上述内酯而言,参照alexandra.m.b et al,european polymer journal,2012,48,963-973,按照以下的合成路径来合成。
[0415]
[化学式53]
[0416][0417]
具体而言,首先,于40℃对包含4-o-β-吡喃半乳糖基-d-葡糖酸(fujifilm wako pure chemical corporation制)18.0g(50mmol)的脱盐水(375ml)的水溶液进行1小时加热。其后,在减压下将水除去,用甲醇(375ml)对残余物进行清洗。接着,使甲醇蒸发,重复4次该操作,进一步使用2-丙醇进行2次该操作。以白色的固体(14.19g,83%)的形态得到目标内酯。
[0418]
《ryo-1的合成》
[0419]
依据实施例1的meg-1的合成方法,按照以下的合成路径来合成ryo-1。
[0420]
[化学式54]
[0421][0422]
于室温向将中间体i溶解于dmf(3ml)而得到的溶液中添加中间体iii(100mg,0.25mmol)。接下来,于50℃将反应混合物搅拌一夜。其后,直接使用硅胶柱色谱法(氯仿:甲醇:水=30:20:4;v/v)对反应混合物进行分离,得到白色固体(rf=0.24;氯仿:甲醇:水=30:20:4;v/v)。接着,使用尺寸排阻再循环hplc(洗脱液:水,流速:3ml/mins)对产物进行分离,于60℃使包含产物的组分干燥,以白色固体的形态得到ryo-1。
[0423]
ryo-1的1h-nmr如下所述。
[0424]1h-nmr(500mhz,d2o)δ:8.04(s,1h),4.38(d,j=7.4hz,1h),4.25(d,j=1.7hz,1h),4.05(s,1h),3.33-3.83(m,14h),2.08(d,j=2.3hz,6h)
[0425]
[实施例4]meg-3的合成
[0426]
为了合成meg-3,首先进行中间体iv的合成。
[0427]
《中间体iv的合成》
[0428]
中间体iv按照以下的合成路径来合成。
[0429]
[化学式55]
[0430][0431]
(步骤9)
[0432]
步骤9的反应条件参照中国专利102503814。
[0433]
具体而言,于60℃将三碘均三甲苯(1.0g,2.0mmol)的吡啶(30ml)与水(10ml)的溶液搅拌10分钟。需要说明的是,三碘均三甲苯参照美国专利6310243来合成。接下来,于90℃经5小时每次少量地添加高锰酸钾(12g,75.9mmol)。搅拌24小时后,在热的状态下对反应混合物进行过滤,用5%氢氧化钾水溶液进行清洗。在减压下于60℃将滤液浓缩。向残余物中加入水,对混合物进行过滤,将不溶的白色固体除去。加入5n的盐酸水溶液而将ph调节为1,利用乙酸乙酯对滤液进行3次萃取。将有机相蒸馏除去,以白色固体的形态得到羧酸(0.82g,70%)。
[0434]
(步骤10)
[0435]
步骤10的反应条件参照美国专利6310243号。
[0436]
具体而言,将由步骤9得到的羧酸(200mg,0.339mmol)装入茄形瓶中,添加亚硫酰氯(1ml)、及dmf(1滴),于120℃回流2.5小时。于50℃在减压下进行蒸馏除去后,溶解于甲苯
(10ml)中,于50℃搅拌2小时。其后,通过过滤将固体除去后,进行浓缩,用少量的己烷洗涤,以白色粉末的形态得到中间体iv(92.6mg,43%)。
[0437]
《meg-3的合成》
[0438]
按照以下的合成路径来合成meg-3。
[0439]
[化学式56]
[0440][0441]
具体而言,首先,将中间体ii(56.3mg,0.252mmol)和中间体iv(50.1mg,0.0781mmol)溶解于脱水dmf(5.00ml)。接着,缓慢地滴加三乙胺(30.0μml),于室温下搅拌一夜。使用蒸发器将溶剂浓缩后,使用尺寸排阻色谱法(sec)进行3次纯化,以褐色透明的晶体的形态得到meg-3。
[0442]
meg-3的1h-nmr如下所述。
[0443]1h-nmr(500mhz,d2o)δ:4.36-4.36(m,1h),3.96-4.01(m,1h),3.81-3.86(m,1h),3.76-3.79(m,1h),3.50-3.60(m,6h),3.39-3.42(m,1h)
[0444]
[实施例5]meg-4的合成
[0445]
依据实施例4的meg-3的合成方法,按照以下的合成路径来合成meg-4。
[0446]
[化学式57]
[0447][0448]
具体而言,将中间体ii’(25.1mg,0.0935mmol)和中间体iv(20.0mg,0.0312mmol)溶解于脱水dmf(3.30ml)。接着,缓慢地滴加三乙胺(13.0μml),于室温下搅拌一夜。使用蒸发器将溶剂浓缩后,使用尺寸排阻色谱法(sec)进行纯化,以混合物的形态得到meg-4。
[0449]
meg-4的1h-nmr及质谱分析的结果如下所述。
[0450]1h-nmr(500mhz,d2o)δ:4.24-4.33(dd,j=7.4,5.2hz,1h),3.87-3.94(m,3h),3.79-3.80(m,3h),3.51-3.71(m,33h),3.34-3.46(m,3h);c
63h84
i3n3o
36
的hrms(esi)m/z[m na]
计算值:1358.0592;实际值1358.0599.
[0451]
[实施例6]kbt的合成
[0452]
为了合成kbt,首先进行中间体iii’的合成。
[0453]
《中间体iii’的合成》
[0454]
中间体iii’按照以下的合成路径来合成。
[0455]
[化学式58]
[0456][0457]
(步骤11)
[0458]
将三碘均三甲苯(19.5g,39mmol)添加至冰乙酸(200ml)、乙酸酐(400ml)、及浓硫酸(40ml)的混合物中。经3小时每次少量地加入高锰酸钾(24.6g,156mmol)。搅拌16小时后,将溶剂蒸馏除去,加入水(200ml)。利用二氯甲烷(250ml)对悬浮液进行萃取,用水对有机相进行清洗,用硫酸镁进行干燥后,进行蒸馏除去。利用硅胶色谱法(洗脱液:氯仿)对固体的残余物进行纯化。得到乙酰基体(1,3,5-三碘-2,4,6-三乙酰氧基甲基苯)8.2g(收率31%)。
[0459]
乙酰基体的1h-nmr如下所述。
[0460]1h-nmr(cdcl3)δ:5.70(s,1h),2.11(s,2h)
[0461]
(步骤12)
[0462]
使由步骤11得到的乙酰基体(7.56g,11.25mmol)悬浮于甲醇(120ml),加入k2co3(0.26g,1.9mmol)。在周围的温度下将混合物搅拌16小时。其后,用2m的盐酸水溶液进行中和后,将有机溶剂蒸馏除去。使残余物悬浮于水,通过过滤而收集白色的固体,依次用水、甲醇、及醚进行清洗,由此得到醇(1,3,5-三碘-2,4,6-三羟基甲基苯)6.0g(收率94%)。
[0463]
醇的1h-nmr如下所述。
[0464]1h-nmr(dmso-d6)δ:5.11-5.07(s,6h),3.32(s,3h)
[0465]
(步骤13)
[0466]
使由步骤12得到的醇(1.0g,1.83mmol)悬浮于亚硫酰氯(50ml)。加入3滴dmf,将混合物加热回流3小时。在减压下除去亚硫酰氯。使固体的残余物悬浮于甲苯(20ml),将溶液注入冰中。用甲苯对产物进行萃取,将有机相用水清洗3次,用饱和食盐水清洗1次。利用硫酸钠进行干燥后,进行过滤、浓缩,由此得到氯体5.9g(1,3,5-三(氯甲基)-2,4,6-三碘苯)(收率97%)。
[0467]
氯体的1h-nmr如下所述。
[0468]1h-nmr(cdcl3)δ:5.29(s,6h)
[0469]
(步骤14)
[0470]
在氩气氛下将叠氮化钠(65mg,1mmol)加入由步骤13得到的氯体(0.1g,0.17mmol)的dmso溶液中。于60℃将溶液搅拌1小时后,用水使反应停止。将产物用乙酸乙酯萃取3次,将有机相用饱和食盐水清洗。用无水硫酸钠干燥后,进行过滤,在减压下进行浓缩,由此以白色的固体的形态得到叠氮化物(1,3,5-三(叠氮基甲基)-2,4,6-三碘苯)(0.1017g,收率为98%)。
[0471]
叠氮化物的1h-nmr如下所述。
[0472]1h-nmr(cdcl3)δ:5.20(s,6h).
[0473]
(步骤15)
[0474]
在氩气氛下,于0℃将三苯基膦(0.79g,3mmol)加入由步骤14得到的叠氮化物(0.53g,0.85mmol)的thf(5ml)溶液中。将反应混合物搅拌10分钟。于0℃向该溶液中加入水(0.8ml)之后,在周围温度下将混合物搅拌一夜。用2n的盐酸水溶液对产物进行萃取,将水溶液用醚清洗1次,由此将三苯基膦除去,用乙酸乙酯清洗4次,由此将三苯基氧化膦除去。向水层中加入5n的氢氧化钾水溶液直至ph变为约14,然后,将产物用氯仿萃取3次。在减压下将有机层浓缩,以白色固体的形态得到中间体iii’。
[0475]
中间体iii’的1h-nmr如下所述。
[0476]1h-nmr(cdcl3)δ:4.47(s,6h)
[0477]
《kbt的合成》
[0478]
按照以下的合成路径来合成kbt。
[0479]
[化学式59]
[0480][0481]
使中间体iii’(10mg,0.018mmol)、与《中间体iii的合成》中作为原料使用的内酯(27mg,0.079mmol)的乙腈(3ml)混合物回流120小时。进行冷却后,将氯仿和水添加至混合物中,用氯仿进行萃取。在减压下将有机层浓缩,得到白色的晶体。通过使用水作为洗脱液的尺寸排阻色谱法进行纯化,得到kbt。
[0482]
kbt的1h-nmr如下所述。
[0483]1h-nmr(500mhz,d2o)δ:4.85(s,6h),4.40(d,j=7.4hz,3h),4.23-4.28(m,3h),4.03(br s,3h),3.86-3.39(m,33h)
[0484]
[实施例7]ryo-2的合成
[0485]
按照以下的合成路径来合成ryo-2。
[0486]
[化学式60]
[0487][0488]
具体而言,于室温向将中间体iv(50mg,0.78mmol)溶解于dmf(3ml)而得到的溶液中添加与实施例3同样地合成的中间体iii(109mg,0.27mmol)及dipea(二异丙基乙基胺)(48μl,0.27mmol)。接下来,于50℃将反应混合物搅拌一夜。其后,直接使用硅胶柱色谱法(甲醇:水=1:1;v/v)对反应混合物进行分离,得到白色固体。接着,使用尺寸排阻再循环hplc(洗脱液:水,流速:5ml/mins)对产物进行分离,于60℃使包含产物的组分干燥,以白色固体的形态得到ryo-2。
[0489]
ryo-2的1h-nmr如下所述。
[0490]1h-nmr(500mhz,d2o)δ:8.04(s,3h),4.38(d,j=7.4hz,3h),4.25(s,3h),4.05(s,3h),3.81(dd,j=6.6,4.3hz,3h),3.67-3.75(m,9h),3.32-3.62(m,33h)
[0491]
[实施例8]pk-1及pk-2的合成
[0492]
为了合成pk-1,首先进行中间体v’的合成。另外,用于合成pk-2的中间体v可以如下所述地合成。
[0493]
《中间体v’及v的合成》
[0494]
中间体v’按照以下的合成路径来合成。中间体v可以按照以下的合成路径来合成。
[0495]
[化学式61]
[0496]
[0497]
(步骤16)
[0498]
于室温向碳酸氢钠(742mg,8.83mmol)与37%甲醛水溶液(27ml)的混合溶液中缓慢地滴加丙二酸二乙酯(15ml,99.4mmol),然后搅拌4小时。加入饱和食盐水而使反应停止,将产物用乙醚萃取4次。将合并的有机层用硫酸钠干燥后,进行过滤,在减压下将溶剂蒸馏除去。以无色油状物的形态得到2,2-双(羟基甲基)丙二酸二乙酯(diethyl 2,2-bis(hydroxymethyl)malonate)(21,3g,98%)(rf=0.31,乙酸乙酯:正己烷=1:1;v/v)。
[0499]
2,2-双(羟基甲基)丙二酸二乙酯的1h-nmr如下所述。
[0500]1h-nmr(500mhz,cdcl3)δ:4.15-4.26(m,4h),4.06(m,j=12.0hz,4h),3.13-3.41(m,2h),1.18-1.24(m,6h)
[0501]
(步骤17)
[0502]
在氩气氛下,将2,2-双(羟基甲基)丙二酸二乙酯(5.00g,22.9mmol)与超脱水丙酮(15ml)混合,加入丙酮缩二甲醇(3.62ml,29.5mmol),接着加入浓硫酸(0.1ml)。于室温将反应混合溶液搅拌24小时后,加入饱和碳酸氢钠水溶液,搅拌15分钟。对该混合溶液进行过滤,将滤液用丙酮萃取2次,在减压下将溶剂蒸馏除去。向残余物中加入饱和碳酸氢钠水溶液,将产物用乙醚萃取,用饱和食盐水洗涤。将该萃取液用硫酸钠干燥,进行过滤、浓缩,利用中性硅胶柱色谱法对残余物进行纯化,以无色油状物的形态得到2,2-二甲基-1,3-二氧杂环己烷-5,5-二甲酸二乙酯(diethyl2,2-dimethyl-1,3-dioxane-5,5-dicarboxylate)(4.25g,71%)(rf=0.9,乙酸乙酯:正己烷=1:1;v/v)。
[0503]
2,2-二甲基-1,3-二氧杂环己烷-5,5-二甲酸二乙酯的1h-nmr如下所述。
[0504]1h-nmr(500mhz,cdcl3)δ:4.25(s,4h),4.20(q,j=7.1hz,4h),1.38(s,6h),1.23(t,j=7.2hz,6h)
[0505]
(步骤18)
[0506]
于180℃使2,2-二甲基-1,3-二氧杂环己烷-5,5-二甲酸二乙酯(391mg,1.5mmol)与氯化钠(106mg,1.81mmol)、水(2、3滴)、dmso(3ml)的混合溶液回流20小时。放置冷却至室温后,加入饱和食盐水,将产物用乙醚萃取4次,用饱和食盐水清洗2次。用硫酸钠使有机层干燥,进行过滤,在减压下进行浓缩。对残余物进行蒸馏(6torr,153℃),以淡黄色油状物的形态得到2,2-二甲基-5-乙氧甲酰基-1,3-二氧杂环己烷(2,2-dimethyl-5-carboethoxy-1,3-dioxane)(181mg,64%)。
[0507]
2,2-二甲基-5-乙氧甲酰基-1,3-二氧杂环己烷的1h-nmr如下所述。
[0508]1h-nmr(500mhz,cdcl3)δ:4.17(q,j=21.8hz,2h),4.04(d,j=20.0hz,4h),2.80(m,j=28.1hz,1h),1.45(s,3h),1.42(s,3h),1.27(d,j=14.3hz,3h)
[0509]
(步骤19-步骤20)
[0510]
步骤19可以通过利用thf溶剂中的氢氧化锂使2,2-二甲基-5-乙氧甲酰基-1,3-二氧杂环己烷水解来实施。步骤20可以通过于室温在二氯甲烷溶剂中使由步骤19得到的产物与草酰氯反应来实施。
[0511]
(步骤21)
[0512]
在氩气氛下,向氢化锂铝(88.3mg,2,33mmol)的thf(2ml)溶液中滴加2,2-二甲基-5-乙氧甲酰基-1,3-二氧杂环己烷(207mg,1.10mmol)的thf(2ml)溶液,然后,将混合溶液加热回流20小时。放置冷却后,加入6m氢氧化钠水溶液(10ml),将产物用乙酸乙酯萃取4次。用
硫酸钠使该有机层干燥后,进行过滤,在减压下进行浓缩,以淡黄色油状物的形态得到2,2-二甲基-1,3-二氧杂环己烷-5-甲醇(2,2-dimethyl-1,3-dioxane-5-methanol)(80mg,50%)。
[0513]
2,2-二甲基-1,3-二氧杂环己烷-5-甲醇的1h-nmr如下所述。
[0514]1h nmr(500mhz,cdcl3)δ:4.03(dd,j=12.0,4.0hz,2h),3.77-3.80(m,4h),1.83(m,1h),1.66(t,j=5.1hz,1h),1.45(s,3h),1.41(s,3h)
[0515]
(步骤22)
[0516]
将2,2-二甲基-1,3-二氧杂环己烷-5-甲醇(59mg,0.40mmol)与二氯甲烷(2ml)、及三乙胺(0.1ml)混合,冷却至0℃。加入对甲苯磺酰氯(102mg,0.7mmol),于室温将反应混合液搅拌1小时。其后,用饱和氯化铵盐将反应溶液洗涤2次,用饱和食盐水洗涤1次,用二氯甲烷对产物进行萃取。用硫酸钠使有机层干燥,进行浓缩,由此以淡褐色油状物的形态得到中间体v’(l=对甲苯磺酰基)(82mg,90%)。
[0517]
中间体v’(l=对甲苯磺酰基)的1h-nmr如下所述。
[0518]1h-nmr(500mhz,cdcl3)δ:7.79-7.81(d,j=8.0hz,2h),7.36(d,j=8.0hz,2h),4.17(d,j=6.9hz,2h),3.97(dd,j=12.3,3.7hz,2h),3.68(dd,j=12.0,4.6hz,2h),2.45(s,3h),1.95(m,j=22.3hz,1h),1.40(s,3h),1.30(s,3h)
[0519]
《中间体vi’及vi的合成》
[0520]
接下来,由中间体v’按照以下的合成路径来合成中间体vi’。另外,由中间体v按照以下的合成路径来合成中间体vi。
[0521]
[化学式62]
[0522][0523]
[化学式63]
[0524][0525]
具体而言,中间体vi’的合成可以通过用盐酸使市售的泛影酸水解后、使其与中间
体v’反应来实施。中间体vi的合成可以通过用碱对市售的泛影酸进行处理后、使其与中间体v反应来实施。
[0526]
《pk-1及pk-2的合成》
[0527]
可以使用由实施例1中的中间体ii的合成方法的步骤2得到的叠氮体、和中间体vi’或vi,来合成下述的pk-1及pk-2。
[0528]
[化学式64]
[0529][0530]
具体而言,可以按照以下的合成路径来合成pk-1。利用实施例1中的中间体ii的合成方法的步骤4的方法对由中间体ii的合成方法的步骤2得到的叠氮体进行氨基化。使用dcc或edc等肽键形成试剂使所得到的氨基体与中间体vi’偶联之后,通过碱处理及酸处理进行脱保护,由此得到pk-1。
[0531]
[化学式65]
[0532][0533]
可以利用同样的方法,按照以下的合成路径由中间体vi合成pk-2。
[0534]
[化学式66]
[0535][0536]
[实施例9]kog-1的合成
[0537]
为了合成kog-1,首先进行中间体ii”的合成。
[0538]
《中间体ii”的合成》
[0539]
中间体ii”按照以下的合成路径来合成。
[0540]
[化学式67]
[0541][0542]
(步骤23)
[0543]
在氩气氛下,使市售的五-o-乙酰基-β-d-吡喃半乳糖(东京化成工业株式会社制,5.04g,12.9mmol)和苄基胺(2.8ml,25.6mmol)溶解于脱水thf(50ml)。于室温搅拌23小时后,在减压下将溶剂蒸馏除去,加入乙酸乙酯。将该有机层用2.0m盐酸洗涤,用硫酸钠干燥,过滤后,在减压下将溶剂蒸馏除去。使用硅胶色谱法(正己烷:乙酸乙酯=1:1)对残余物进行纯化,以无色油状物的形态得到2,3,4,6-四-o-乙酰基-β-d-吡喃半乳糖(4.5g,定量性的)(rf=0.36,正己烷:乙酸乙酯=1:1)。
[0544]
2,3,4,6-四-o-乙酰基-β-d-吡喃半乳糖的1h-nmr如下所述。
[0545]1h-nmr(500mhz,cdcl3)δ:5.52(t,j=3.4hz,1h),5.48(t,j=1.7hz,1h),5.42(dd,j=10.9,3.4hz,1h),5.16(dd,j=10.9,3.4hz,1h),4.48(t,j=6.6hz,1h),4.07-4.16(m,2h),3.48(br,1h),2.16(s,3h),2.11(s,3h),2.06(s,3h),2.00(s,3h)
[0546]
(步骤24)
[0547]
在氩气氛下,使2,3,4,6-四-o-乙酰基-β-d-吡喃半乳糖(3.87g,11.1mmol)和三氯乙腈(5.50ml,55.4mmol)溶解于脱水二氯甲烷(35ml)。在冰冷却条件下,滴加1,8-二氮杂双
环[5.4.0]-7-十一碳烯(1,8-diazabicyclo[5.4.0]undec-7-ene)(dbu,0.34ml,2.23mmol),搅拌2小时。其后,在减压下将溶剂蒸馏除去,加入乙酸乙酯。利用水和饱和食盐水对该有机层进行清洗,用无水硫酸钠干燥后,进行过滤,在减压下进行浓缩,由此得到粗制1-(2,2,2-三氯乙烷亚氨酸酯)-2,3,4,6-四乙酸酯-β-d-吡喃半乳糖苷(1-(2,2,2-trichloroethanimidate)-2,3,4,6-tetraacetate-β-d-galactopyranoside)(rf=0.40,正己烷:乙酸乙酯=2:1)。
[0548]
1-(2,2,2-三氯乙烷亚氨酸酯)-2,3,4,6-四乙酸酯-β-d-吡喃半乳糖苷的1h-nmr如下所述。
[0549]1h-nmr(400mhz,cdcl3)δ:8.67(s,1h),6.60(d,j=3.5hz,1h),5.56(d,j=2.0hz,1h),5.35-5.45(m,2h),4.44(t,j=6.5hz,1h),4.06-4.19(m,2h),2.17(s,3h),2.02-2.05(m,9h)
[0550]
(步骤25)
[0551]
在氩气氛下,使粗制1-(2,2,2-三氯乙烷亚氨酸酯)-2,3,4,6-四乙酸酯-β-d-吡喃半乳糖苷和2-[2-(2-氯乙氧基)乙氧基]乙醇(2-[2-(2-chloroethoxy)ethoxy]ethanol)(4.6ml,32.2mmol)溶解于脱水二氯甲烷(90ml)。在冰冷却条件下滴加三氟化硼乙醚(boron trifluoride diethyl etherate)(10.6ml,86.2mmol),在冰冷却的状态下搅拌1小时后,将光遮断并于室温搅拌一夜。在反应后加入水,在减压下将溶剂蒸馏除去。向其中加入乙酸乙酯,依次利用水、饱和碳酸氢钠溶液、及饱和食盐水进行清洗,用无水硫酸钠干燥后,进行过滤。在减压下将滤液浓缩,使用硅胶柱色谱法(正己烷:乙酸乙酯=4:1)进行纯化,由此,以黄色油状物的形态得到粗制(2r,3s,4s,5r,6r)-2-(乙酰氧基甲基)-6-(2-(2-(2-氯乙氧基)乙氧基)乙氧基)四氢-2h-吡喃-3,4,5-三基三乙酸酯((2r,3s,4s,5r,6r)-2-(acetoxymethyl)-6-(2-(2-(2-chloroethoxy)ethoxy)ethoxy)tetrahydro-2h-pyran-3,4,5-triyl triacetate)。
[0552]
所得到的氯体的1h-nmr及质谱分析的结果如下所述。
[0553]1h-nmr(500mhz,cdcl3)δ:5.38(q,j=1.4hz,1h),5.20(dd,j=10.9,8.0hz,1h),5.01(dd,j=10.3,3.4hz,1h),4.56(d,j=8.0hz,1h),4.19-4.07(m,3h),3.78-3.61(m,12h),2.14(s,3h),2.05(d,j=6.9hz,6h),1.98(s,3h);c
20h31cl3
nao
12
的lrms(esi)m/z[m na]
计算值:521;实际值521.
[0554]
(步骤26)
[0555]
在氩气氛下,使所得到的氯体和叠氮化钠(709mg,10.9mmol)溶解于脱水dmso(32ml),于80℃搅拌一夜。反应后,加入乙酸乙酯,依次利用水、饱和碳酸氢钠水溶液、及饱和食盐水对有机层进行清洗。用无水硫酸钠对有机层进行干燥后,进行过滤,在减压下进行浓缩。使用硅胶柱色谱法(正己烷:乙酸乙酯=2:3)对残余物进行纯化,由此,以黄色油状物的形态得到(2r,3s,4s,5r,6r)-2-(乙酰氧基甲基)-6-(2-(2-(2-叠氮基乙氧基)乙氧基)乙氧基)四氢-2h-吡喃-3,4,5-三基三乙酸酯((2r,3s,4s,5r,6r)-2-(acetoxymethyl)-6-(2-(2-(2-azidoethoxy)ethoxy)ethoxy)tetrahydro-2h-pyran-3,4,5-triyl triacetate)(94mg,在3个工序中为44%)。
[0556]
所得到的叠氮体的1h-nmr及质谱分析的结果如下所述。
[0557]1h-nmr(500mhz,cdcl3)δ:5.46(d,j=2.9hz,1h),5.37(dd,j=10.9,3.4hz,1h),
5.17(d,j=3.4hz,1h),5.13(dd,j=10.9,3.4hz,1h),4.33-4.17(m,2h),4.16-4.04(m,2h),3.72-3.61(m,9h),3.40(t,j=4.9hz,2h),2.14(s,3h),2.08(s,3h),2.04(s,3h),1.99(s,3h);c
20h31
n3nao
12
的lrms(esi)m/z[m na]
计算值:528;实际值528.
[0558]
(步骤27)
[0559]
将所得到的叠氮体(330mg,0.65mmol)、10%钯碳(30mg)及脱水甲醇(8ml)混合,在氢气氛下,于室温搅拌24小时。在反应后进行过滤,在减压下将溶剂蒸馏除去,以褐色油状物的形态得到作为(2r,3s,4s,5r,6r)-2-(乙酰氧基甲基)-6-(2-(2-(2-氨基乙氧基)乙氧基)乙氧基)四氢-2h-吡喃-3,4,5-三基三乙酸酯((2r,3s,4s,5r,6r)-2-(acetoxymethyl)-6-(2-(2-(2-aminoethoxy)ethoxy)ethoxy)tetrahydro-2h-pyran-3,4,5-triyl triacetate)(170mg,54%)的中间体ii”。
[0560]
中间体ii”的1h-nmr如下所述。
[0561]1h-nmr(500mhz,cdcl3)δ:5.39(d,j=3.4hz,1h),5.21(dd,j=10.3,8.0hz,1h),5.02(dd,j=10.3,3.4hz,1h),4.58(d,j=8.0hz,1h),4.20-4.10(m,2h),3.99-3.90(m,2h),3.79-3.73(m,1h),3.69-3.59(m,8h),3.52(t,j=5.2hz,2h),2.88(t,j=5.2hz,2h),2.15(s,3h),2.06(d,j=6.3hz,6h),1.99(s,3h)
[0562]
《kog-1的合成》
[0563]
可以按照以下的合成路径而由中间体vii合成kog-1。需要说明的是,中间体vii是由中间体ii”及中间体iv合成的。
[0564]
[化学式68]
[0565][0566]
(步骤28)
[0567]
在氩气氛下,使中间体ii”(800mg,1.68mmol)和中间体iv(308mg,0.48mmol)溶解
于脱水dmf(10ml),然后缓慢地滴加三乙胺(810μl)。于室温将该反应混合液搅拌23小时后,在减压下将溶剂蒸馏除去。使用硅胶柱色谱法(氯仿:甲醇=9:1)对残余物进行纯化后,使用尺寸排阻色谱法(氯仿)进一步纯化,由此得到作为kog-1的前体的中间体vii。
[0568]
中间体vii的质谱分析的结果如下所述。
[0569]c69h96
n3nao
39
的lrms(esi)m/z[m na]
计算值:1994;实际值1994.
[0570]
(步骤29)
[0571]
使中间体vii溶解于脱水甲醇,加入1m甲醇甲醇盐(methanol methoxide)/甲醇溶液直至ph变为9,于室温搅拌3小时。加入离子交换树脂(amberlite h )来进行中和,进行过滤,将滤液浓缩,由此能够得到kog-1。
[0572]
[实施例10]meg-4的合成
[0573]
按照与实施例5不同的以下的合成路径来合成meg-4。
[0574]
[化学式69]
[0575][0576]
(步骤30)
[0577]
将由实施例1中的中间体ii的合成方法的步骤2得到的叠氮体即2-(2-叠氮基乙氧基)乙基-2,3,4,6-四乙酸酯-β-d-吡喃半乳糖苷(2-(2-azidoethoxy)ethyl-2,3,4,6-tetraacetate-β-d-galactopyranoside)(33.7mg,0.731mmol)、10%钯碳(3.7mg)及脱水甲醇/异丙醇(1.0ml/1.5ml)混合,在氢气氛下,于室温搅拌2小时。在反应后进行过滤,在减压下将溶剂蒸馏除去,以褐色油状物的形态得到粗制(2r,3s,4s,5r,6r)-2-(乙酰氧基甲基)-6-(2-(2-氨基乙氧基)乙氧基)四氢-2h-吡喃-3,4,5-三基三乙酸酯((2r,3s,4s,5r,6r)-2-(acetoxymethyl)-6-(2-(2-aminoethoxy)ethoxy)tetrahydro-2h-pyran-3,4,5-triyl triacetate)(31.6mg,99%)(rf=0.13,氯仿:甲醇=9:1)。
[0578]
所得到的胺的1h-nmr如下所述。
[0579]1h-nmr(500mhz,cdcl3)δ:5.40(d,j=2.9hz,1h),5.18(dd,j=10.3,8.0hz,1h),5.08(dd,j=10.9,3.4hz,1h),4.60(d,j=8.0hz,1h),4.21(q,j=5.9hz,1h),4.13(dd,j=11.5,6.9hz,1h),3.97-4.02(m,2h),3.68-3.76(m,5h),3.17(s,2h),2.18(s,3h),2.10(s,3h),2.06(s,3h),1.99(s,3h)
[0580]
(步骤31)
[0581]
在氩气氛下,使所得到的胺(由1.90mmol的叠氮体合成的产物)和中间体iv
(302mg,0.471mmol)溶解于脱水dmf(10ml),然后缓慢地滴加三乙胺(660μl)。于室温将该反应混合液搅拌45小时后,在减压下将溶剂蒸馏除去。使用硅胶柱色谱法(氯仿:甲醇=100:1~1:9)对残余物进行纯化后,使用尺寸排阻色谱法(氯仿)进一步纯化,由此,以黄色油状物的形态得到作为meg-4的前体的中间体viii(42.2mg,5%)。
[0582]
中间体viii的1h-nmr及质谱分析的结果如下所述。
[0583]1h-nmr(500mhz,cdcl3)δ:5.38(d,j=3.2hz,3h),5.14-5.18(m,3h),4.98-5.01(m,3h),4.50-4.53(m,3h),4.11-4.15(m,6h),3.97(s,6h),3.63-3.75(m,21h),2.15(s,9h),2.02-2.05(m,18h),1.98(s,9h);c
63h84
i3n3o
36
的hrms(esi)m/z[m na]
计算值:1862.1694;实际值1862.1686.
[0584]
(步骤32)
[0585]
在氩气氛下,使中间体viii(5.2mg,2.83μmol)溶解于脱水甲醇(1.0ml),加入1m甲醇甲醇盐/甲醇溶液直至ph变为9,然后,于室温搅拌3小时。其后,向反应溶液中加入离子交换树脂(amberlite h )而进行中和,进行过滤,将滤液浓缩,由此得到meg-4。
[0586]
[试验例1]化合物的摄入抑制试验
[0587]
对于实施例中制作的ryo-1、meg-1、meg-2,使用肝癌细胞株hepg2,依据j gastroenterol 1998,33,855-859来进行介由肝细胞特异性地表达的脱唾液酸糖蛋白受体的摄入抑制试验。具体而言,采用以下的方法。
[0588]
首先,于37℃使其与将血清类粘蛋白(orosomucoid)(sigma)固相化而得到的神经氨酸苷酶(neuraminidase)(sigma)进行12小时反应,由此进行脱唾液酸化。使用氯胺-t法,将该经脱唾液酸化的血清类粘蛋白(asor)用放射性碘(125i)标记,制作i-asor。
[0589]
将所得到的i-asor调整为6μg/ml的pbs溶液,于4℃使其在6个孔上与利用经冰冷却的mem(gibco)进行了清洗的hepg2细胞反应30分钟。对细胞进行充分清洗后,使用0.1n的naoh将细胞溶解,对摄入至细胞内的放射线量a进行测定(对照样品)。通过除i-asor之外还以100μg/ml添加meg-1、meg-2、或ryo-1来进行同样的测定,从而对添加有各化合物时的放射线量a进行测定。
[0590]
接下来,为了排除i-asor对细胞的非特异性结合的影响,如下所述地对i-asor的非特异性结合的背景进行测定。即,将i-asor(6μg/ml)与100倍的浓度(600μg/ml)的未标记asor混合,使其与hepg2同样地进行反应,然后,对所得到的细胞进行清洗、溶解,对放射线量b进行测定。作为背景的放射线量b在对照样品、及添加有各化合物的样品的全部中共同使用。
[0591]
接下来,在各样品中,计算a-b,由此算出特异性地摄入至hepg2内的i-asor。实验一式三份地进行,算出标准偏差。
[0592]
确认到在hepg2中,ryo-1、meg-1、meg-2均竞争性地抑制配体的摄入。将hepg2中的摄入抑制试验的结果示于图1。需要说明的是,图1中,标注为“6μg/ml sp”的样品为对照样品。
[0593]
使用不具有脱唾液酸糖蛋白受体的胰腺癌细胞株panc-1进行了同样的实验。具体而言,采用以下的方法。
[0594]
将与上文同样地操作而用放射性碘(125i)标记得到的i-asor调整为6μg/ml的pbs溶液,于4℃使其在6个孔上与利用经冰冷却的mem(gibco)进行了清洗的panc1细胞反应30
分钟。对细胞进行充分清洗后,使用0.1n的naoh将细胞溶解,对摄入至细胞内的放射线量a进行测定(对照样品)。通过除i-asor之外还以100μg/ml添加meg-1、meg-2、或ryo-1来进行同样的测定,从而对添加有各化合物时的放射线量a进行测定。
[0595]
接下来,为了排除i-asor对细胞的非特异性结合的影响,如下所述地对i-asor的非特异性结合的背景进行测定。即,将i-asor(6μg/ml)与100倍的浓度(600μg/ml)的未标记asor混合,使其与panc-1同样地进行反应,然后,对所得到的细胞进行清洗、溶解,对放射线量b进行测定。作为背景的放射线量b在对照样品、及添加有各化合物的样品的全部中共同使用。
[0596]
接下来,在各样品中,计算a-b,由此算出特异性地摄入至panc-1内的i-asor。实验一式三份地进行,算出标准偏差。
[0597]
在panc-1中,ryo-1、meg-1、meg-2均未抑制配体的摄入。将panc-1中的摄入抑制试验的结果示于图2。
[0598]
由以上结果确认到,本发明的化合物可被肝细胞特异性地摄入。
[0599]
[试验例2]介由脱唾液酸糖蛋白受体的摄入活性在化合物间的比较
[0600]
使用与上述试验例1同样的方法,对于meg-2、meg-4、ryo-1、及ryo-2,比较了介由脱唾液酸糖蛋白受体的摄入活性。
[0601]
制备包含与试验例1同样地得到的0.4μg/ml的i-asor、和1.0mm/ml的meg-2、meg-4、ryo-1、或ryo-2的pbs溶液,于37℃使其在6个孔上与利用经冰冷却的mem(gibco)进行了清洗的hepg2反应30分钟。对细胞进行充分清洗后,使用0.1n的naoh将细胞溶解,对摄入至hepg2内的放射线量a进行测定。
[0602]
作为阳性对照,制备包含0.4μg/ml的i-asor、和1000μg/ml的未标记asor的pbs溶液,实施于4℃使其在6个孔上与利用经冰冷却的mem(gibco)进行了清洗的hepg2反应30分钟的实验。
[0603]
为了排除i-asor对细胞的非特异性结合的影响,如下所述地对i-asor的非特异性结合的背景进行测定。即,将i-asor(0.4μg/ml)与100倍的浓度(40μg/ml)的未标记asor混合,使其与hepg2同样地进行反应,然后,对所得到的细胞进行清洗、溶解,对放射线量b进行测定。作为背景的放射线量b在对照样品、阳性对照样品、及添加有各化合物的样品的全部中共同使用。
[0604]
接下来,在各样品中,计算a-b,由此算出特异性地摄入至hepg2内的i-asor。实验一式三份地进行,算出标准偏差。
[0605]
meg-2、meg-4、ryo-1、及ryo-2均竞争性地抑制hepg2中的配体的摄入。就这样的抑制能力而言,确认到meg-4比meg-2更高,另外,ryo-2比ryo-1更高。将结果示于图3。由此暗示了具有较多的与脱唾液酸糖蛋白受体结合的原子团的化合物的介由脱唾液酸糖蛋白受体的摄入活性高。
[0606]
[试验例3]造影实验
[0607]
通过吸入异氟烷而将自日本clea株式会社购入的小鼠(icr雄性9周龄)麻醉。一边利用delpetμct100应用软件(delbio公司)监视呼吸状态,一边缓慢地从尾静脉注射施予meg-1。meg-1的施予量设定为52mg/只动物。
[0608]
在施予前、施予中、施予15分钟后及施予1小时后拍摄ct图像。对于从胸至下腹部
的图像数据,以45μm的像素尺寸(pixel size)进行重建,进行向dicom数据的转换。进而,利用vivoquant软件(invicro公司)对图像进行分析,结果确认到:呈现在施予前观察不到的高吸收的液体经时性地出现在肠道内及膀胱内。
[0609]
将施予1小时后的图像示于图4。
[0610]
代替meg-1而使用meg-2、meg-4、或ryo-2进行了同样的试验,结果,在任意情况下均确认到:呈现在施予前观察不到的高吸收的液体经时性地出现在肠道内及膀胱内。
[0611]
由以图4为代表的上述结果确认到,具有由脱唾液酸糖蛋白受体识别的部位的化合物具有胆汁排泄功能,通过双系统的排泄途径而被排泄。即,上述化合物能够减轻副作用,并且能够通过使肝细胞功能可视化来进行肝脏的诊断。
[0612]
产业上的可利用性
[0613]
根据本发明,在图像诊断等中具有产业上的可利用性。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。