1.本发明涉及萘并[1,2-d]噻唑化合物技术领域,更具体的涉及2-酰氨基萘并[1,2-d]噻唑的合成方法。
背景技术:
[0002]
有机硫化合物由于具有特殊的结构和潜在的药理活性,被广泛应用于医药、农药、染料、有机导电材料以及液晶显示材料等领域。为此,引起人们对其进行广泛关注和不懈研究。经典构建c-s键的方法是以卤代芳烃或芳基硼酸为原料,在过渡金属的催化下与二硫化物或硫醇进行交叉偶联获得。脱氢交联法是构建c-s键最直接、有效的策略之一,它具有原子经济性高,合成步骤少等优点。然而,与构建c-n、c-c或c-o键的反应相比,构建c-s键的合成策略相对较少。这可能是由于含硫化合物容易被过度氧化所致。因此,在温和的条件下发展更经济、高效和实用的构建c-s键合成策略是非常必要的。
[0003]
萘并噻唑及其衍生物是许多医药中重要的含硫结构单元,具有抗癌、抗白血病、抗肿瘤等生物学和药物学活性。因此,开发高效、绿色的合成策略以获得萘并噻唑及其衍生物仍然是有机化学和药物化学领域最具吸引力的研究方向之一。萘并[1,2-d]噻唑是重要的萘并噻唑衍生物之一,目前合成萘并[1,2-d]噻唑的方法主要有:(1)以1,4-萘醌和硫脲为原料合成。如:ulrich,p.等以2-取代-1,4-萘醌和硫脲的衍生物为原料,在盐酸的作用下反应48h,成功得到萘并[1,2-d]噻唑的衍生物,但它们的产率为9~68%。(ulrich,p.;cerami,a.j.med.chem.1982,25,654.)(2)以1-萘胺和硫为主要原料合成。如:zhang l.f.等以1-萘胺、硫和甲苯(对二甲苯)为原料,在250~275℃条件下反应1.5~3.5h,成功获得2-芳基萘并[1,2-d]噻唑。产率为41~50%。(zhang,l.-f.;ni,z.-h.;li,d.-y.;qin,z.-h.;wei,x.-y.chinese chem.lett.2012,23,281.)jonaghani m.z.等则以1-萘胺、硫和2-甲基喹啉为起始原料,首先在对甲苯磺酸的作用下得到硫代酰胺衍生物,再依次与氢氧化钠和铁氰化钾作用得到2-(2-喹啉基)萘并[1,2-d]噻唑,最后与浓硫酸反应2.5h,得到一种性能良好的hg
2
荧光探针2-(2-喹啉基)萘并[1,2-d]噻唑-8-磺酸。(jonaghani,m.z.;boeini,h.z.spectrochim.acta a 2017,178,66.)尽管以上方法都获得了成功,但是这些方法还存在一些缺点,如反应时间长,有毒的环合试剂,反应条件苛刻等。为此,开发一种简单而新颖的合成萘并[1,2-d]噻唑衍生物的方法很有必要和十分迫切。
技术实现要素:
[0004]
针对以上问题,本发明提供了一种2-酰氨基萘并[1,2-d]噻唑的合成方法。
[0005]
本发明的第一个目的是提供2-酰氨基萘并[1,2-d]噻唑的合成方法,按照以下步骤制备:
[0006]
将3-(1-萘基)-1-取代酰基硫脲溶于醋酸或醇类溶剂,加入醋酸锰催化剂,发生自由基关环反应,后处理得到2-酰氨基萘并[1,2-d]噻唑;
[0007]
其合成路线如下所示:
[0008][0009]
其中,r1为c1~c
10
的直链烷基或c5~c6的环烷基中的一种,r2为h或卤素。
[0010]
优选的,c1~c
10
的直链烷基为ch3、(ch2)2ch3、(ch2)3ch3、(ch2)9ch3、(ch2)5ch3中的一种。
[0011]
优选的,c5~c6的环烷基为中的一种。
[0012]
优选的,卤素为br。
[0013]
优选的,3-(1-萘基)-1-取代酰基硫脲和醋酸锰的摩尔比为0.79-1.30:1.98-3.30。
[0014]
优选的,醇类溶剂为乙醇或甲醇。
[0015]
优选的,3-(1-萘基)-1-取代酰基硫脲与溶剂的比例为0.79-1.30mmol:15ml。
[0016]
优选的,反应温度为90-100℃,反应功率为600-800w,反应时间为25-30min。
[0017]
优选的,后处理步骤是:将反应结束后得到的反应液冷却到室温,加入去离子水,得到灰白色固体,用二氯甲烷进行萃取,将有机相合并后、干燥、过滤、浓缩、提纯。
[0018]
更优选的,r1为ch3,r2为h时,3-(1-萘基)-1-取代酰基硫脲为3-(1-萘基)-1-乙酰基硫脲,3-(1-萘基)-1-乙酰基硫脲、醋酸锰和溶剂的比例为1.24-1.32mmol:3.10-3.30mmol:15ml。
[0019]
更优选的,r1为(ch2)2ch3,r2为h时,3-(1-萘基)-1-取代酰基硫脲为3-(1-萘基)-1-丁酰基硫脲,3-(1-萘基)-1-丁酰基硫脲、醋酸锰和溶剂的比例为1.10-1.20mmol:2.75-2.83mmol:15ml。
[0020]
更优选的,r1为(ch2)3ch3,r2为h时,3-(1-萘基)-1-取代酰基硫脲为3-(1-萘基)-1-戊酰基硫脲,3-(1-萘基)-1-戊酰基硫脲、醋酸锰和溶剂的比例为1.05-1.15mmol:2.63-2.71mmol:15ml。
[0021]
更优选的,r1为(ch2)9ch3,r2为h时,3-(1-萘基)-1-取代酰基硫脲为3-(1-萘基)-1-十一烷酰基硫脲,3-(1-萘基)-1-十一烷酰基硫脲、醋酸锰和溶剂的比例为0.81-0.93mmol:2.03-2.11mmol:15ml。
[0022]
更优选的,r1为(ch2)2ch3,r2为br时,3-(1-萘基)-1-取代酰基硫脲为3-[1-(4-溴萘基)]-1-丁酰基硫脲,3-[1-(4-溴萘基)]-1-丁酰基硫脲、醋酸锰和溶剂的比例为0.85-0.97mmol:2.13-2.21mmol:15ml。
[0023]
更优选的,r1为(ch2)5ch3,r2为br时,3-(1-萘基)-1-取代酰基硫脲为3-[1-(4-溴萘基)]-1-庚酰基硫脲、醋酸锰和溶剂的比例为0.79-0.82mmol:1.98-2.03mmol:15ml。
[0024]
更优选的,r1为r2为h时,3-(1-萘基)-1-取代酰基硫脲为3-(1-萘基)-1-环戊基甲酰基硫脲,3-(1-萘基)-1-环戊基甲酰基硫脲、醋酸锰和溶剂的比例为1.01-1.07mmol:2.53-2.67mmol:15ml。
[0025]
更优选的,r1为r2为h时,3-(1-萘基)-1-取代酰基硫脲为3-(1-萘基)-1-环己基甲酰基硫脲,3-(1-萘基)-1-环己基甲酰基硫脲、醋酸锰和溶剂的比例为0.96-1.04mmol:2.40-2.53mmol:15ml。
[0026]
与现有技术相比,本发明具有以下有益效果:
[0027]
本发明将3-(1-萘基)-1-取代酰基硫脲和醋酸锰(iii)溶入有机溶剂(醋酸、乙醇或甲醇)中,设定微波功率和温度,在微波辐射下反应一段时间后,得到2-酰氨基萘并[1,2-d]噻唑。然后应用该方法成功获得其它7种2-取代酰氨基萘并[1,2-d]噻唑类化合物。该发明不仅具有条件温和、操作简单,反应速率快以及底物适用范围广等优点,而且还能够有效提高原子的利用效率。
[0028]
本发明采用一步合成法通过原料自身发生自由基的关环得到的化合物;所用的醋酸锰为低毒性催化剂,醋酸、乙醇或甲醇为溶剂,原料易得且毒性小,从而减少了对环境的污染,在制备过程中通过微波促进反应,反应时间很短。本发明所制备的2-酰氨基萘并[1,2-d]噻唑可作为光学材料使用,如作为检测金属离子的荧光探针。
具体实施方式
[0029]
下面对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0030]
以下各实施例中所述实验方法,如无特殊说明,均为常规方法;所涉及试剂和材料,如无特殊说明,均可在市场上购买到。
[0031]
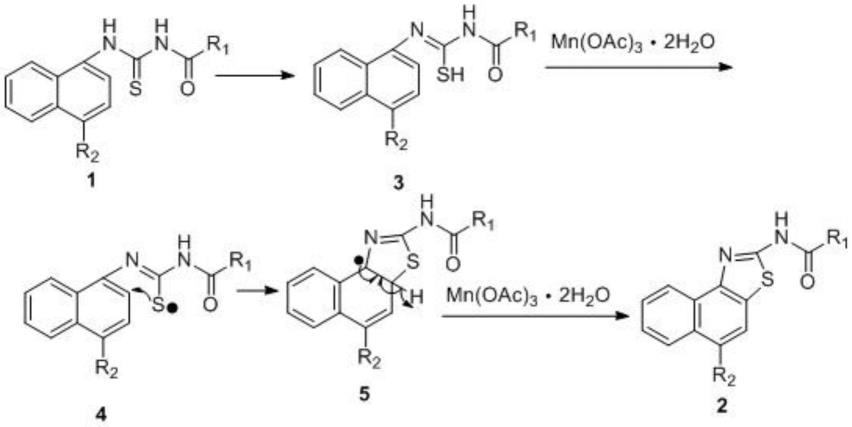
本发明在制备时,首先,酰基硫脲1中的硫羰基可由酮式转为硫代烯醇(式3),接着mn(oac)3·
2h2o将式3中的巯基氧化成硫自由基(式4),三价锰变成二价锰,生成的硫自由基进攻萘环β位碳完成c-s键的构建,然后噻唑啉环(式5)上的c-h键会被mn(oac)3·
2h2o氧化生成碳自由基,进而与噻唑啉环5位碳自由基偶合即可获得产物(式2)。对应机理图如下所示:
[0032]
[0033]
实施例1
[0034]
本实施例制备的一种2-乙酰氨基萘并[1,2-d]噻唑,其结构式如下:
[0035][0036]
具体制备工艺流程如下所示,
[0037][0038]
具体制备过程为:将0.30g,即1.24mmol的3-(1-萘基)-1-乙酰基硫脲、0.83g、即3.10mmol的醋酸锰和15ml冰醋酸依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度100℃,功率700w,加热时间25min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=2:3],得2-乙酰氨基萘并[1,2-d]噻唑260mg,产率87%。
[0039]
本实施例制备的2-乙酰氨基萘并[1,2-d]噻唑:260mg,白色固体,产率87%。m.p.280~282℃;1h nmr(400mhz,cdcl3):δ9.74(s,1h),8.57(d,j=8.30hz,1h),7.95(d,j=8.10hz,1h),7.86(d,j=8.70hz,1h),7.77(d,j=8.70hz,1h),7.65-7.61(m,1h),7.58-7.55(m,1h),2.29(s,3h).
13
c nmr(101mhz,cdcl3):δ169.7,158.5,144.7,132.3,128.7,127.6,127.3,127.0,126.2,124.2,123.2,120.0,23.2。
[0040]
实施例2
[0041]
将1.32mmol的3-(1-萘基)-1-乙酰基硫脲、3.30mmol的醋酸锰和15ml乙醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度95℃,功率800w,加热时间27min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=2:3],得2-乙酰氨基萘并[1,2-d]噻唑。
[0042]
实施例3
[0043]
将1.27mmol的3-(1-萘基)-1-乙酰基硫脲、3.20mmol的醋酸锰和15ml甲醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度90℃,功率600w,加热时间30min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=2:3],得
2-乙酰氨基萘并[1,2-d]噻唑。
[0044]
实施例4
[0045]
本实施例制备的一种2-丁酰氨基萘并[1,2-d]噻唑,其结构式如下:
[0046][0047]
具体制备过程为:将0.30g,即1.10mmol的3-(1-萘基)-1-丁酰基硫脲、0.74g、即2.75mmol的醋酸锰和15ml冰醋酸依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度100℃,功率700w,加热时间25min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-丁酰氨基萘并[1,2-d]噻唑256mg,产率86%。
[0048]
本实施例制备的2-丁酰氨基萘并[1,2-d]噻唑:256mg,白色固体,产率86%。m.p.139~140℃;1h nmr(400mhz,cdcl3):δ10.75(s,1h),8.59(d,j=8.10hz,1h),7.96(d,j=7.90hz,1h),7.88(d,j=8.70hz,1h),7.78(d,j=8.70hz,1h),7.66-7.60(m,1h),7.59-7.53(m,1h),2.29(t,j=7.50hz,2h),1.70-1.58(m,2h),0.75(t,j=7.40hz,3h).
13
c nmr(101mhz,cdcl3):δ171.9,159.1,144.0,132.3,128.5,127.9,127.1,126.7,126.0,124.7,123.0,119.0,38.2,18.5,13.3。
[0049]
实施例5
[0050]
将1.20mmol的3-(1-萘基)-1-丁酰基硫脲、2.83mmol的醋酸锰和15ml乙醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度95℃,功率600w,加热时间30min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-丁酰氨基萘并[1,2-d]噻唑。
[0051]
实施例6
[0052]
将1.15mmol的3-(1-萘基)-1-丁酰基硫脲、2.80mmol的醋酸锰和15ml甲醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度90℃,功率800w,加热时间27min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-丁酰氨基萘并[1,2-d]噻唑。
[0053]
实施例7
[0054]
本实施例制备的一种2-戊酰氨基萘并[1,2-d]噻唑,其结构式如下:
[0055][0056]
具体制备过程为:将0.30g,即1.05mmol的3-(1-萘基)-1-戊酰基硫脲、0.70g、即2.63mmol的醋酸锰和15ml冰醋酸依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度100℃,功率700w,加热时间25min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-戊酰氨基萘并[1,2-d]噻唑262mg,产率88%。
[0057]
本实施例制备的2-戊酰氨基萘并[1,2-d]噻唑:262mg,白色固体,产率88%。m.p.80~82℃;1h nmr(400mhz,cdcl3):δ10.35(s,1h),8.59(d,j=8.20hz,1h),7.96(d,j=8.00hz,1h),7.88(d,j=8.70hz,1h),7.78(d,j=8.70hz,1h),7.68-7.52(m,2h),2.36(t,j=7.60hz,2h),1.70-1.60(m,2h),1.25-1.12(m,2h),0.78(t,j=7.30hz,3h).
13
c nmr(101mhz,cdcl3):δ171.5,158.2,144.2,132.3,128.4,127.9,127.3,126.7,126.0,124.6,123.0,119.0,36.2,27.0,22.1,13.5。
[0058]
实施例8
[0059]
将1.15mmol的3-(1-萘基)-1-戊酰基硫脲、2.71mmol的醋酸锰和15ml乙醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度95℃,功率600w,加热时间30min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-戊酰氨基萘并[1,2-d]噻唑。
[0060]
实施例9
[0061]
将2.10mmol的3-(1-萘基)-1-戊酰基硫脲、2.68mmol的醋酸锰和15ml甲醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度90℃,功率800w,加热时间27min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-戊酰氨基萘并[1,2-d]噻唑。
[0062]
实施例10
[0063]
本实施例制备的一种2-十一烷酰氨基萘并[1,2-d]噻唑,其结构式如下:
[0064][0065]
具体制备过程为:将0.30g,即0.81mmol的3-(1-萘基)-1-十一烷酰基硫脲、0.54g、即2.03mmol的醋酸锰和15ml冰醋酸依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度100℃,功率700w,加热时间25min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-十一烷酰氨基萘并[1,2-d]噻唑268mg,产率90%。
[0066]
本实施例制备的2-十一烷酰氨基萘并[1,2-d]噻唑:268mg,白色固体,产率90%。m.p.76~77℃;1h nmr(400mhz,cdcl3):δ10.80(s,1h),8.59(d,j=8.10hz,1h),7.96(d,j=8.00hz,1h),7.88(d,j=8.70hz,1h),7.78(d,j=8.70hz,1h),7.66-7.53(m,2h),2.29(t,j=7.40hz,2h),1.78-1.66(m,2h),1.34-0.99(m,14h),0.88(t,j=7.00hz,3h).
13
c nmr(101mhz,cdcl3):δ172.0,159.2,144.0,132.4,128.5,127.9,127.1,126.7,126.0,124.7,123.0,119.0,36.4,31.9,29.5,29.4,29.3,29.0,28.9,25.0,22.7,14.2。
[0067]
实施例11
[0068]
将0.93mmol的3-(1-萘基)-1-十一烷酰基硫脲、2.11mmol的醋酸锰和15ml甲醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度95℃,功率600w,加热时间30min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-十一烷酰氨基萘并[1,2-d]噻唑。
[0069]
实施例12
[0070]
将0.87mmol的3-(1-萘基)-1-十一烷酰基硫脲、2.09mmol的醋酸锰和15ml乙醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度90℃,功率800w,加热时间27min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-十一烷酰氨基萘并[1,2-d]噻唑。
[0071]
实施例13
[0072]
本实施例制备的一种2-丁酰氨基-5-溴萘并[1,2-d]噻唑,其结构式如下:
[0073][0074]
具体制备过程为:将0.30g,即0.85mmol的3-[1-(4-溴萘基)]-1-丁酰基硫脲、0.57g、即2.13mmol的醋酸锰和15ml冰醋酸依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度100℃,功率700w,加热时间25min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-丁酰氨基-5-溴萘并[1,2-d]噻唑224mg,产率75%。
[0075]
本实施例制备的2-丁酰氨基-5-溴萘并[1,2-d]噻唑:224mg,白色固体,产率75%。m.p.200~202℃;1h nmr(400mhz,dmso):δ12.63(s,1h),8.57(d,j=7.30hz,1h),8.52(s,1h),8.23(d,j=5.70hz,1h),7.77-7.45(m,2h),2.53(t,j=7.20hz,2h),1.71-1.63(m,2h),0.94(t,j=7.40hz,3h).
13
c nmr(101mhz,dmso):δ172.7,159.2,144.9,130.0,128.1,128.0,127.9,127.8,127.5,123.9,116.5,37.4,18.6,14.0。
[0076]
实施例14
[0077]
将0.97mmol的3-[1-(4-溴萘基)]-1-丁酰基硫脲、2.21mmol的醋酸锰和15ml甲醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度95℃,功率600w,加热时间30min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-丁酰氨基-5-溴萘并[1,2-d]噻唑。
[0078]
实施例15
[0079]
将0.91mmol的3-[1-(4-溴萘基)]-1-丁酰基硫脲、2.17mmol的醋酸锰和15ml乙醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度90℃,功率800w,加热时间27min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-丁酰氨基-5-溴萘并[1,2-d]噻唑。
[0080]
实施例16
[0081]
本实施例制备的一种2-庚酰氨基-5-溴萘并[1,2-d]噻唑,其结构式如下:
[0082][0083]
具体制备过程为:将0.30g,即0.79mmol的3-[1-(4-溴萘基)]-1-庚酰基硫脲、0.53g、即1,98mmol的醋酸锰和15ml冰醋酸依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度100℃,功率700w,加热时间25min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-庚酰氨基-5-溴萘并[1,2-d]噻唑256mg,产率86%。
[0084]
本实施例制备的2-庚酰氨基-5-溴萘并[1,2-d]噻唑:256mg,白色固体,产率86%。m.p.136~138℃;1h nmr(400mhz,cdcl3):δ9.79(s,1h),8.63-8.56(m,1h),8.38-8.30(m,1h),8.18(s,1h),7.71-7.62(m,2h),2.45-2.37(m,2h),1.71-1.65(m,2h),1.30-1.13(m,6h),0.85(t,j=7.00hz,3h).
13
c nmr(101mhz,cdcl3):δ171.5,158.4,144.1,130.5,128.1,128.0,127.4,127.2,123.5,122.6,118.3,36.5,31.2,28.9,24.9,22.4,14.0。
[0085]
实施例17
[0086]
将0.82mmol的3-[1-(4-溴萘基)]-1-庚酰基硫脲、2.03mmol的醋酸锰和15ml甲醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度90℃,功率600w,加热时间30min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-庚酰氨基-5-溴萘并[1,2-d]噻唑。
[0087]
实施例18
[0088]
将0.80mmol的3-[1-(4-溴萘基)]-1-庚酰基硫脲、2.00mmol的醋酸锰和15ml乙醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度95℃,功率800w,加热时间27min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-庚酰氨基-5-溴萘并[1,2-d]噻唑。
[0089]
实施例19
[0090]
本实施例制备的一种2-环戊基甲酰氨基萘并[1,2-d]噻唑,其结构式如下:
[0091][0092]
具体制备过程为:将0.30g,即1.01mmol的3-(1-萘基)-1-环戊基甲酰基硫脲、0.68g、即2.53mmol的醋酸锰和15ml冰醋酸依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度100℃,功率700w,加热时间25min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-环戊基甲酰氨基萘并[1,2-d]噻唑240mg,产率81%。
[0093]
本实施例制备的2-环戊基甲酰氨基萘并[1,2-d]噻唑:240mg,白色固体,产率81%。m.p.170~172℃;1h nmr(400mhz,cdcl3):δ10.06(s,1h),8.59(d,j=8.20hz,1h),7.95(d,j=8.00hz,1h),7.87(d,j=8.70hz,1h),7.77(d,j=8.70hz,1h),7.66-7.60(m,1h),7.59-7.53(m,1h),2.73-2.67(m,1h),1.95-1.77(m,4h),1.74-1.65(m,2h),1.45-1.44(m,2h).
13
c nmr(101mhz,cdcl3):δ175.1,158.8,144.1,132.3,128.4,127.9,127.2,126.7,126.0,124.6,123.0,119.0,45.5,30.3,25.8.
[0094]
实施例20
[0095]
将1.07mmol的3-(1-萘基)-1-环戊基甲酰基硫脲、2.67mmol的醋酸锰和15ml甲醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度95℃,功率600w,加热时间30min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-环戊基甲酰氨基萘并[1,2-d]噻唑。
[0096]
实施例21
[0097]
将1.03mmol的3-(1-萘基)-1-环戊基甲酰基硫脲、2.6mmol的醋酸锰和15ml乙醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度90℃,功率800w,加热时间27min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-环戊基甲酰氨基萘并[1,2-d]噻唑。
[0098]
实施例22
[0099]
本实施例制备的一种2-环己基甲酰氨基萘并[1,2-d]噻唑,其结构式如下:
[0100][0101]
具体制备过程为:将0.30g,即0.96mmol的3-(1-萘基)-1-环己基甲酰基硫脲、0.64g、即2.40mmol的醋酸锰和15ml冰醋酸依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度100℃,功率700w,加热时间25min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-环己基甲酰氨基萘并[1,2-d]噻唑250mg,产率84%。
[0102]
本实施例制备的2-环己基甲酰氨基萘并[1,2-d]噻唑:250mg,白色固体,产率84%。m.p.154~156℃;1h nmr(400mhz,cdcl3):δ10.36(s,1h),8.63(d,j=8.00hz,1h),7.97(d,j=8.00hz,1h),7.88(d,j=8.70hz,1h),7.78(d,j=8.70hz,1h),7.66-7.62(m,1h),7.59-7.55(m,1h),2.24-2.12(m,1h),1.72-1.19(m,7h),1.15-0.59(m,3h).
13
c nmr(101mhz,cdcl3):δ174.9,159.3,144.0,132.3,128.5,127.9,127.2,126.7,126.1,124.7,123.0,119.1,44.9,29.1,25.2,24.8。
[0103]
实施例23
[0104]
将1.04mmol的3-(1-萘基)-1-环己基甲酰基硫脲、2.53mmol的醋酸锰和15ml乙醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度95℃,功率600w,加热时间30min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-环己基甲酰氨基萘并[1,2-d]噻唑。
[0105]
实施例24
[0106]
将0.99mmol的3-(1-萘基)-1-环己基甲酰基硫脲、2.47mmol的醋酸锰和15ml甲醇依次加入到50ml圆底烧瓶中后,放入微波合成仪,设定加热温度90℃,功率800w,加热时间27min,开启磁力搅拌。反应结束后,将反应液冷却至室温,加入15ml去离子水,有大量灰白色固体出现,然后用二氯甲烷(15ml
×
3)萃取,合并有机相,用无水硫酸钠干燥,过滤,滤液浓缩后得灰白色固体,然后用硅胶过柱提纯[洗脱剂为二氯甲烷/石油醚(60-90℃),v/v=1:1],得2-环己基甲酰氨基萘并[1,2-d]噻唑。
[0107]
尽管已描述了本发明的优选实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例作出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。
[0108]
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精
神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
