用于治疗癌症的usp7抑制剂
技术领域
1.本发明涉及通过抑制成纤维细胞中的usp7活性来治疗癌症。根据本发明的癌症治疗可以通过例如,抑制肿瘤微环境中的细胞外基质重塑、抑制vegf、抑制血管生成或转移、调节免疫系统或其组合。
背景技术:
2.在从健康组织到癌症,然后再到转移的转变过程中,肿瘤微环境(tme)引发了关键变化,该变化允许原发性肿瘤形成和生长,以及从原发性肿瘤发展到侵袭性和转移性疾病。
3.影响tme中支持这些转变的特定因素和分子机制为提高癌症治疗策略的效力和范围提供了新的途径。
4.usp7因其在致癌途径的调节中起着关键作用且因此靶向usp7以抑制癌细胞增殖的潜力而成为一种潜在的治疗靶标。
5.先前未描述过usp7抑制成纤维细胞或肿瘤微环境的影响。
技术实现要素:
6.本文首次证明usp7在影响肿瘤微环境中起关键作用。本文证明的usp7抑制作用包括促进细胞外基质(ecm)的重塑,从而促进肿瘤侵袭和转移。usp7还影响全身和肿瘤微环境(tme)中的血管生成和vegf水平。
7.值得注意的是,本文证明usp7抑制,特别是在tme的成纤维细胞室中,导致细胞侵袭和血管生成显著减少。usp7抑制还导致肿瘤免疫环境的调节(例如,通过促进cd8 t细胞的浸润)。
8.显著地,体内usp7抑制抑制体外不受usp7抑制剂直接抑制影响的细胞模型中的肿瘤生长。因此,本文提供了一种通过抑制成纤维细胞中的usp7治疗癌症的新发现的方法,该方法独立于先前确定的usp7在直接驱动癌细胞中的肿瘤发生中的作用。
9.因此,在一个方面,提供了一种通过向有此需要的受试者施用usp7抑制剂来治疗癌症的方法,其中usp7活性在非癌细胞中受到抑制。优选地,在tme的非癌细胞中抑制usp7活性。
10.在一个方面,提供了一种通过抑制成纤维细胞中的usp7活性来治疗癌症的方法,该方法包括向有此需要的受试者施用包含usp7抑制剂的组合物。
11.在某些实施方案中,施用包含usp7抑制剂的组合物通过抑制癌症相关成纤维细胞(caf)中的usp7活性来治疗癌症。
12.本文证明,抑制成纤维细胞中的usp7减少成纤维细胞的致瘤作用,例如通过减少细胞侵袭;减少mmp分泌;减少基底膜降解;以及降低全身和tme中的vegf水平中的一种或多种。
13.因此,在某些实施方案中,施用包含usp7抑制剂的组合物通过降低受试者血清中的vegf水平来治疗癌症。在某些实施方案中,施用包含usp7抑制剂的组合物通过降低肿瘤
微环境中的vegf水平来治疗癌症。
14.在某些实施方案中,施用包含usp7抑制剂的组合物通过抑制癌症相关成纤维细胞(caf)产生vegf来治疗癌症。
15.在另一个方面,提供了一种通过调节肿瘤免疫环境治疗癌症的方法,该方法包括向有此需要的受试者施用包含usp7抑制剂的组合物,其中施用usp7抑制剂调节了肿瘤免疫环境。
16.在另一个方面,提供了一种通过增加肿瘤浸润淋巴细胞(til)治疗癌症的方法,该方法包括向有此需要的受试者施用包含usp7抑制剂的组合物,其中施用usp7抑制剂增加til的数量,优选cd8 til的数量。
17.在另一个方面,提供了一种通过降低tme中的treg细胞相对于cd8 t细胞的比例来治疗癌症的方法,该方法包括向有此需要的受试者施用包含usp7抑制剂的组合物,其中施用usp7抑制剂降低了tme中的treg细胞相对于cd8 t细胞的比例。
18.在另一个方面,提供了一种通过减少tme中的巨噬细胞数量来治疗癌症的方法,该方法包括向有此需要的受试者施用包含usp7抑制剂的组合物,其中施用usp7抑制剂减少了tme中的巨噬细胞的数量。
19.在另一个方面,提供了一种通过向有此需要的受试者施用组合疗法治疗癌症的方法,该组合疗法包括包含usp7抑制剂的组合物和包含免疫检查点抑制剂的组合物。
20.在另一个方面,提供了一种用于治疗癌症的方法中的usp7抑制剂,该方法包括向有此需要的受试者施用组合疗法,该组合疗法包括包含usp7抑制剂的组合物和包含免疫检查点抑制剂的组合物。
21.在另一个方面,提供了一种用于治疗癌症的方法中的免疫检查点抑制剂,该方法包括向有此需要的受试者施用组合疗法,该组合疗法包括包含usp7抑制剂的组合物和包含免疫检查点抑制剂的组合物。
22.在另一个方面,提供了一种用于治疗癌症的方法中的组合疗法,该方法包括向有此需要的受试者施用组合疗法,该组合疗法包括包含usp7抑制剂的组合物和包含免疫检查点抑制剂的组合物。
23.在某些实施方案中,施用包含usp7抑制剂的组合物通过抑制癌症相关成纤维细胞的细胞外基质(ecm)重塑来治疗癌症。在此类实施方案中,ecm是肿瘤微环境的ecm。
24.在某些实施方案中,施用包含usp7抑制剂的组合物通过抑制基底膜的降解、任选地抑制管状基底膜的降解来治疗癌症。
25.在某些实施方案中,施用包含usp7抑制剂的组合物通过抑制血管生成(任选地新血管生成)来治疗癌症。在此类实施方案中,肿瘤微环境中的血管生成受到抑制。
26.在某些实施方案中,施用包含usp7抑制剂的组合物通过调节肿瘤免疫环境(例如,通过促进cd8 t细胞的浸润)来治疗癌症。
27.在某些实施方案中,以达到抑制肿瘤生长的剂量施用组合物。
28.在某些实施方案中,以达到抑制肿瘤侵袭的剂量施用组合物。
29.在某些实施方案中,以达到抑制肿瘤转移的剂量施用组合物。
30.在某些实施方案中,以实现肿瘤免疫环境调节的剂量施用组合物。
31.在某些实施方案中,通过该方法治疗的癌症由癌细胞形成,并且癌细胞对usp7抑
制剂具有抗性。在某些实施方案中,通过该方法治疗的癌症由癌细胞形成,并且癌细胞对体外usp7抑制剂具有抗性。
32.在另一个方面,提供了一种用于根据本发明的治疗癌症的方法中的usp7抑制剂。
33.本文提供的每个方面或实施方案可以与任何其他方面(多个)或实施方案(多个)组合,除非指出相反的情况。特别地,指示为优选或有利的任何特征可以与任何其他一个或多个特征或最有利地指示为优选或有利的那些特征组合。
附图说明
34.图1:usp7抑制降低成纤维细胞中svegf水平:原代真皮成纤维细胞和pbmc的共培养物。用溶媒(dmso)和不同浓度的ad-04处理原代人真皮成纤维细胞与激活的pbmc的共培养物48小时。使用elisa测量生物标志物水平,并以对数转化率表示。
35.图2:usp7抑制降低成纤维细胞中svegf水平:原代真皮成纤维细胞、pbmc和癌细胞的共培养物。用溶媒(dmso)和不同浓度的ad-04处理原代人成纤维细胞与非小肺癌或结直肠癌细胞的共培养物。使用elisa测量生物标志物水平,并以对数转化率表示。
36.图3:usp7抑制降低成纤维细胞中分泌的vegf水平:癌细胞、激活的pbmc和原代真皮或肺成纤维细胞的共培养物。与原代人肺(wi-38和imr-90)或真皮(hdf)成纤维细胞和用抗cd3/cd28抗体刺激的pbmc共培养的ht29细胞用指定浓度的ad-04和ent-ad-04处理48小时。通过elisa测量释放到细胞培养基中的vegf水平。
37.图4:usp7抑制剂不调节癌细胞中的分泌型vegf水平:在原代成纤维细胞中观察到的usp7抑制剂对vegf分泌的调节在肿瘤细胞中不发生。用指定浓度的ad-04和ent-ad-04处理ht29、h1299、lncap和mcf-7细胞48小时。通过elisa测量释放到细胞培养基中的svegf水平。
38.图5:usp7抑制降低激活的成纤维细胞中的分泌型vegf水平。在共培养物中观察到的usp7介导的vegf分泌减少可完全归因于原代成纤维细胞中的分泌型vegf的调节。用cd3/cd28刺激的pbmc、fgf-2或tgf-β激活wi-38、imr-90和hdf,并用指定浓度的ad-04和ent-ad-04处理48小时。通过elisa测量释放到细胞培养基中的vegf水平。
39.图6:usp7抑制降低了癌症相关成纤维细胞中的分泌型vegf水平。癌症相关成纤维细胞分泌型vegf,因此不需要进一步激活以观察usp7介导的vegf分泌减少。原发性结直肠腺癌相关成纤维细胞用指定浓度的ad-04和ent-ad-04处理48小时。通过elisa测量释放到细胞培养基中的vegf水平。
40.图7:usp7抑制介导的vegf分泌减少依赖于功能性p53途径。只有在具有功能和野生型p53途径的细胞中,usp7抑制才能下调vegf分泌。具有不稳定p53基因的sv40永生化成纤维细胞wi-38-va13用溶媒(dmso)、指定浓度的ad-04和ent-ad-04处理48小时,然后通过elisa检测vegf。
41.图8:除了调节分泌的vegf外,usp7抑制还降低了原代成纤维细胞裂解物中的细胞内vegf水平,但不降低癌细胞或永生化成纤维细胞中的vegf水平。用溶媒(dmso)和ad-04处理细胞48小时,裂解并通过elisa测量细胞内vegf。
42.图9:usp7抑制降低低氧(缺氧)条件下成纤维细胞中的vegf水平。在与生长因子(fgf或tgfβ)或激活的pbmc孵育时观察到的usp7介导的vegf减少所需的成纤维细胞的必要
激活也可以通过低氧重现。成纤维细胞在低氧室内用指定浓度的ad-04和ent-ad-04处理48小时。收集上清液并通过elisa检测vegf。
43.图10:usp7抑制降低原代人成纤维细胞中hif-1α半衰期。已知分泌型vegf受hif-1α转录因子的控制:usp7抑制剂依赖性vegf分泌减少是通过调节hif-1α稳定性介导的。低氧培养的原代成纤维细胞用放线菌酮(100μg/ml)预处理,然后暴露于ad-04(1μm)或溶媒(dmso)。在指定的时间点(0至60分钟)裂解细胞,并通过hif-1α的免疫印迹探测分析样品。hif-1α半衰期通过光密度测定分析定量。β-肌动蛋白作为标准化对照包括在内。这些数据是来自至少两个独立实验的代表性数据。
44.图11:ad-04抑制usp7增加成纤维细胞中k48连接的hif-1α多泛素化。在低氧条件下用溶媒(dmso)、ad-04(1μm)或mg132(10μm)处理原代人成纤维细胞。使用tube
tm
琼脂糖(tube1是k63/k48特异性的,而tube2是k63特异性的)下拉k63/k58(上图)或k63(下图),然后使用抗hif-1α抗体进行免疫印迹分析。这些数据是来自至少两个独立实验的代表性数据。
45.图12:hif-1α网络途径分级。正如所料,与非激活(常氧)成纤维细胞样品相比,来源于低氧激活的成纤维细胞的样品中hif1a mrna表达网络是最显著富集的途径(nci自然数据库)。
46.图13:低氧诱导vegfa mrna表达,经usp7抑制剂ad-04处理后以基因水平下调。通过rnaseq在原代人成纤维细胞中评估usp7抑制剂对vegfa表达水平的影响。在常氧或低氧条件下,在dmso溶媒或usp7抑制剂ad-04存在下,通过rnaseq分析监测人原代成纤维细胞培养物中的vegfa水平。
47.图14:在经ad-04处理的低氧激活的成纤维细胞中,hif1a mrna表达网络(kegg定义)正在发生变化。usp7抑制剂处理原代人成纤维细胞可在6小时(黑条)和24小时(灰条)调节hif1a表达网络中的已知基因。
48.图15:usp7抑制剂在低氧激活的成纤维细胞中诱导差异vegfa选择性剪接。除了调节vegfa分泌外,usp7抑制剂处理的原代人成纤维细胞显示vegfa选择性剪接的选择性模式。在24小时时,短(191aa-左图)抗血管生成vegf-165的mrna表达上调,且长(371aa-右图)促血管生成的vegf-65亚型的mrna表达强烈下调。(黑色(左箱线图)-dmso;灰色(右箱线图)-ad-04)。
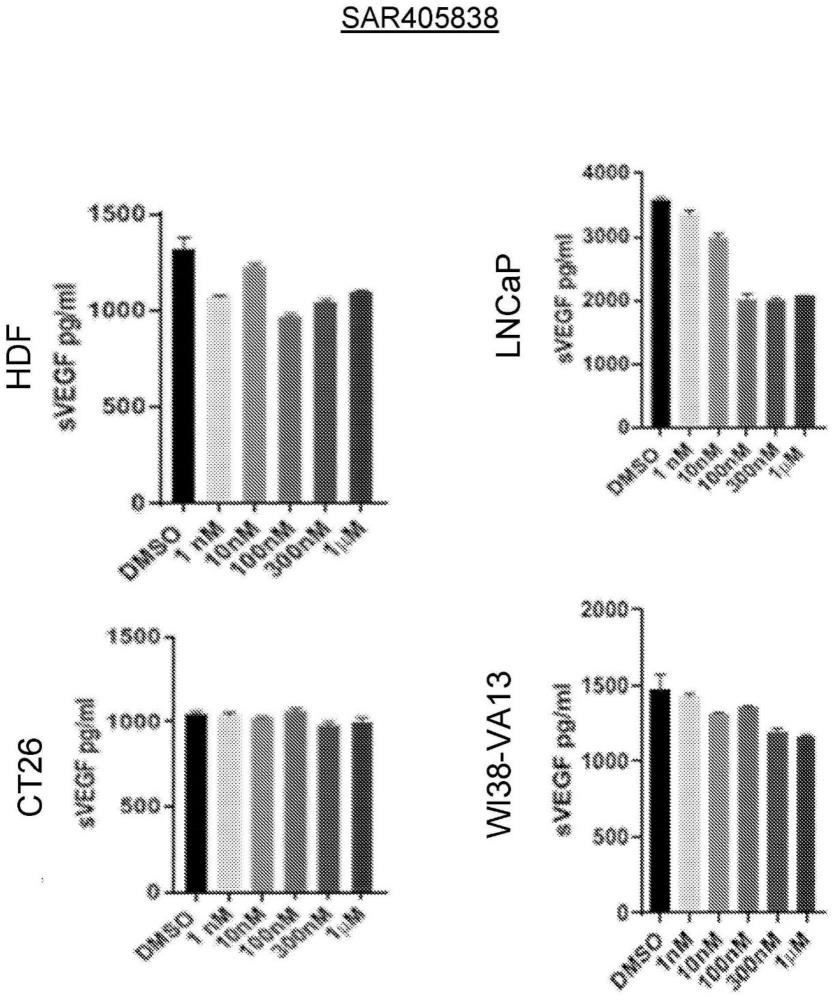
49.图16:mdm-2拮抗剂不影响常氧条件下激活的成纤维细胞、癌细胞或永生化成纤维细胞中的svegf水平。usp7抑制剂对原代人成纤维细胞中vegf分泌的调节不是通过mdm-2拮抗剂剂量下的mdm2蛋白泛素化介导的,该剂量不影响常氧条件下的增殖。细胞用溶媒(dmso)、ad-04、其非活性对映体(ent-ad-04)、nutlin-3a、rg7112或sar405838以指定浓度在常氧条件下处理48小时。收集细胞培养上清液,通过elisa测量svegf。
50.图17:mdm-2拮抗剂不影响低氧条件下激活的成纤维细胞、癌细胞或永生化成纤维细胞中的svegf水平。usp7抑制剂对原代人成纤维细胞中vegf分泌的调节不是通过mdm-2拮抗剂剂量下的mdm2蛋白泛素化介导的,该剂量不影响低氧条件下的增殖。细胞用溶媒(dmso)、ad-04、其非活性对映体(ent-ad-04)、nutlin-3a、rg7112或sar405838以指定浓度在低氧条件下处理48小时。收集细胞培养上清液,通过elisa测量svegf。
51.图18:与mdm2拮抗剂相比,usp7抑制剂ad-04不影响原代成纤维细胞、永生化成纤
维细胞或癌细胞的增殖。usp7介导的vegf分泌减少的功能性影响不是对成纤维细胞或癌细胞增殖的影响。在常氧(图18a和18a)或低氧(图18c)条件下用溶媒(dmso)和逐渐增加浓度的ad-04、其非活性对映体(ent-ad-04)、nutlin-3a、rg7112或sar405838处理癌细胞、原代和永生化成纤维细胞。在72小时后,通过细胞滴度glo测量细胞活力。将结果标准化为对照非处理组(平均值
±
sd,n=3)。
52.图19:ad-04在原代人成纤维细胞中显示出强大的usp7靶接合。为了确保usp7抑制剂的作用通过人原代成纤维细胞中的直接usp7调节来介导,我们使用基于活性的探针竞争分析来监测usp7细胞靶接合。hdf和imr-90用溶媒(dmso)或增加浓度的ad-04抑制剂处理2小时。在孵育后,裂解细胞并用泛素-丙炔胺(ub-pa)活性探针孵育。然后使用抗usp7抗体通过免疫印迹分析样品。ec
50
值通过光密度测定分析确定。使用imagej软件对谱带强度进行量化,其中上谱带(usp7-ub)和下谱带(usp7)计算为相应dmso对照的百分比(-/ ub-pa),然后将值归一化为每个浓度的上谱带和上谱带之和。
53.图20:ad-04显示了癌细胞中的有效usp7靶接合。为了确保usp7抑制剂的作用通过癌细胞中的直接usp7调节来介导,我们使用基于活性的探针竞争分析来监测usp7细胞靶接合。ht-29和ct-26细胞用溶媒(dmso)或增加浓度的ad-04抑制剂处理2小时。在孵育后,裂解细胞并用泛素-丙炔胺(ub-pa)探针孵育。然后使用抗usp7抗体通过免疫印迹分析样品。ec
50
值通过光密度测定分析确定。使用imagej软件对谱带强度进行量化,其中上谱带(usp7-ub)和下谱带(usp7)计算为相应dmso对照的百分比(-/ ub-pa),然后将值归一化为每个浓度的上谱带和上谱带之和。
54.图21:原代真皮成纤维细胞中usp7 crispr/cas9敲除导致svegf降低。选择性usp7抑制剂ad-04对原代人成纤维细胞中vegf分泌的影响的特异性通过选择性usp7 crispr敲除得到证实。使用抗usp7抗体和β-肌动蛋白作为负载对照的免疫印迹分析证实了usp7敲除的效率(左)。将usp7敲除的hdf在低氧条件下孵育48小时,并通过elisa在细胞培养上清液中测量svegf水平。
55.图22:usp7抑制不会调节癌细胞、永生化成纤维细胞、激活的成纤维细胞和内皮细胞的增殖。细胞用溶媒(dmso)、ad-04或ent-ad-04处理96小时。在增殖分析期间,使用延时显微镜每2小时拍摄照片。图表用于显示随时间的细胞融汇。
56.图23:usp7抑制不调节癌细胞、永生化成纤维细胞、激活的成纤维细胞和内皮细胞的迁移。在刮伤愈合分析中,将细胞接种在包被孔上,损伤并用溶媒(dmso)、ad-04或ent-ad-04处理。在4天的分析期间,使用延时显微镜每2小时拍摄伤口闭合的照片。图表显示了伤口闭合的百分比随时间的变化。
57.图24:usp7抑制(ad-04)抑制人原代成纤维细胞的细胞侵袭,但不抑制内皮细胞或癌细胞。刮伤细胞侵袭分析用于测量指示的细胞通过基底膜侵袭的能力。使用延时显微镜在4天内每2小时拍摄照片,伤口闭合随时间的变化如图表所示。
58.图25:usp7抑制(ad-04)降低侵袭原代人成纤维细胞中的mmp-7分泌。刮伤细胞侵袭分析用于测量原代人成纤维细胞通过基底膜侵袭的能力。在96小时后收集侵袭分析的细胞培养上清液,并使用测量mmp-7水平。
59.图26:usp7抑制(ad-04)抑制成纤维细胞和内皮细胞共培养物的小管形成,但不抑
制预形成的小管。在小管形成之前(顶部)或之后(底部),用溶媒(dmso)和指定浓度的ad-04、ent-ad-04和avastin处理hdf和huvec共培养物,然后进行cd31染色。显示了在第14天拍摄的代表性照片。通过测量小管长度(μm)来量化小管形成,如图所示。
60.图27:usp7抑制(ad-04)降低了新形成的小管中hdf和huvec共培养物中的mmp-2分泌水平。在小管形成之前(左)或之后(右),用溶媒(dmso)和指定浓度的ad-04、ent-ad-04和avastin处理hdf和huvec共培养物。收集来自小管形成的细胞培养上清液,并使用测量mmp-2水平。
61.图28:usp7抑制对体外huvec小管形成没有直接影响。将huvec接种在生长因子减少的包被孔上,然后用溶媒(dmso)和指定浓度的ad-04、ent-ad-04或avastin处理。在小管发生后6h后拍摄小管形成图像。
62.图29:usp7在体外不抑制ct-26癌细胞系增殖。ct-26同基因模型是体内模型,用于监测化合物对tme的影响。细胞用溶媒(dmso)或增加浓度的ad-04、其非活性对映体(ent-ad-04)、nutlin-3a或舒尼替尼处理。在72小时后,通过测量细胞活力。
63.图30:usp7对体内肿瘤微环境的影响。usp7抑制剂ad-04在体内抑制同基因异种移植物中的ct-26细胞系的生长。每周测量两次用溶媒对照、ad-04或索拉非尼处理的ct-26异种移植物植入的免疫活性同基因小鼠的肿瘤体积,直到肿瘤植入后第15天。ad-04产生了53%的肿瘤生长抑制,与阳性对照索拉非尼达到的水平非常相似,为58%(左图),并且在研究期间耐受性良好(右图)。n=13;误差条表示s.e.m.;*,p《0.05;**p《0.01(双因素方差分析)。各组荷瘤小鼠无明显的体重变化(右)。
64.图31:usp7抑制调节体内分泌型vegf水平。给携带ct-26的同基因小鼠服用usp7抑制剂ad-04导致循环vegf血清水平降低。在肿瘤移植后第15天收集用溶媒、ad-04或索拉非尼处理的ct-26荷瘤小鼠的血清样品,并通过elisa测量循环血清vegf水平。将显示的所有统计差异与对照处理进行比较;****p《0.0001,***p《0.001;**p《0.01;*p《0.05;ns:不显著(单因素方差分析/dunnet事后检验)。
65.图32:usp7抑制调节肿瘤微环境免疫室。usp7抑制增加了细胞毒性淋巴细胞的募集,并减少了ct-26肿瘤中的巨噬细胞数量。在用usp7抑制剂给药10天的ct-26同基因模型中,肿瘤浸润免疫细胞的流式细胞术分析。
66.图33:adc-159(一种新型口服usp7抑制剂)降低低氧条件下成纤维细胞中的vegf水平。新型口服usp7抑制剂adc-159在低氧室内48小时内降低由低氧水平激活的原代人成纤维细胞的vegf分泌。收集上清液并通过elisa检测vegf。
67.图34:adc-160、adc-198、adc-199和adc-556usp7抑制剂降低低氧条件下成纤维细胞中的vegf水平。成纤维细胞在低氧室内用指定浓度的usp7抑制剂处理48小时。收集上清液并通过elisa检测vegf。
68.图35:adc-159显示对来自成纤维细胞和内皮细胞共培养物的新形成小管的抑制作用。在小管形成之前(顶部)或之后(底部),用溶媒(dmso)或指定浓度的adc-159和avastin处理hdf和huvec共培养物,然后进行抗cd31染色。显示了在小管发生后第14天拍摄的代表性照片。通过测量小管长度(μm)来量化小管形成。
69.图36:adc-159在体内防止ct-26同基因小鼠模型中的肿瘤血管成熟。在用溶媒和
adc-159处理15天后制备ffpe肿瘤块,并用内皮细胞标志物cd31(绿色)、周细胞标志物ng2(红色)和dapi(蓝色;n=5个块/组)共同染色。成熟血管具有cd31和ng2染色阳性的细小分支(a,左图),当用usp7抑制剂处理时,所述细小分支消失(b,右图)。
70.图37:adc-159在体内ct-26同基因小鼠模型中减少肿瘤面积并增加坏死面积。每周测量两次用溶媒对照和100mg/kg剂量下的adc-159处理的ct-26异种移植物植入的免疫活性同基因小鼠的肿瘤体积,直到肿瘤植入后第15天。制备ffpe肿瘤块,然后使用halo软件量化肿瘤和坏死。
71.图38:usp7抑制联合免疫检查点抑制剂(ici)对体内肿瘤微环境的影响。usp7抑制剂adc-159在体内抑制同基因异种移植物中的ct-26细胞系的生长,并与ici(a)协同作用。每周测量两次用溶媒对照、adc-159、抗-pd-l1、抗-ctla-4或组合处理的ct-26异种移植物植入的免疫活性同基因小鼠的肿瘤体积,直到肿瘤植入后第13天。adc-159产生23%的肿瘤生长抑制(tgi),与抗-pdl-1(22%)达到的水平非常相似。adc-159和抗-pdl-的组合进一步减少了肿瘤生长(tgi=43%)。用抗-ctla-4处理导致43%的tgi,这与adc-159和抗-ctra-4组合产生的tgi相似(上图)。处理耐受性良好,未检测到明显的体重变化(b)。n=15;误差条表示s.e.m.;**p《0.01。***p《0.001,(双因素方差分析)。
72.图39:当与抗pd-l1和抗ctla-4免疫检查点抑制剂组合时,usp7抑制导致存活率增加。(a)在使用ct-26小鼠结直肠癌细胞的小鼠同种异体移植研究中,kaplan-meier存活曲线显示,经adc-159和ici处理的动物存活时间延长。(b)当usp7抑制剂adc-159与ici组合时,个体生长曲线显示抗肿瘤反应改善。n=10只动物/组。
73.图40:usp7抑制剂adc-159在体内调节肿瘤微环境免疫室。usp7抑制增加了细胞毒性淋巴细胞的募集,如用单独的usp7抑制剂adc-159或与ici联合给药13天的ct-26同基因模型中肿瘤浸润免疫细胞的流式细胞术分析所示。
具体实施方式
74.本文首次证明usp7在影响肿瘤微环境中起着关键作用,而肿瘤微环境不是由癌症细胞本身介导的。例如,usp7被证明介导了成纤维细胞对肿瘤微环境(tme)的影响。本文证明usp7影响成纤维细胞介导的tme细胞外基质(ecm)的重塑,并促进vegf的成纤维细胞表达。因此,抑制成纤维细胞中的usp7,尤其是癌症相关成纤维细胞(caf),能够通过多种不同的效应限制肿瘤生长和侵袭。
75.本文证明在tme的成纤维细胞室中的usp7抑制导致细胞侵袭和血管生成两者显著减少。tme中成纤维细胞的usp7抑制也导致肿瘤免疫环境调节,从而促进cd8 t细胞的浸润。
76.成纤维细胞中的usp7抑制作为有效的癌症治疗的效果是显著且有利的,因为即使当癌细胞对usp7抑制剂的直接抑制具有抗性时,治疗也是有效的。
77.因此,本文提供了一种通过抑制非癌细胞中的usp7治疗癌症的新发现的方法,该方法独立于先前确定的usp7在直接驱动癌细胞中肿瘤发生中的作用。令人惊讶的是,在成纤维细胞中抑制usp7显示出强的抗肿瘤作用,该强的抗肿瘤作用不依赖于usp7抑制剂对癌细胞本身的任何作用。
78.因此,在一个方面,提供了一种通过施用usp7抑制剂来治疗癌症的方法,其中usp7活性在非癌细胞中受到抑制。优选地,在tme的非癌细胞中抑制usp7活性。
79.在另一个方面,提供了一种通过抑制成纤维细胞中的usp7活性来治疗癌症的方法,该方法包括向有此需要的受试者施用包含usp7抑制剂的组合物。
80.癌症相关成纤维细胞(或肿瘤相关成纤维细胞)是存在于肿瘤微环境中的成纤维细胞。通常,caf来源于正常周围组织中的正常成纤维细胞,但也可以来源于周细胞、平滑肌细胞、纤维细胞或间充质干细胞(msc)。caf的标志物包括平滑肌肌动蛋白(αsma)、波形蛋白、血小板源生长因子受体α(pdgfr-α)、血小板源生长因子受体β(pdgfr-β)、成纤维细胞特异性蛋白1(fsp-1)和成纤维细胞激活蛋白(fap)。
81.通常,caf表现出激活的成纤维细胞表型,例如,与正常成纤维细胞相比,显示出升高水平的成纤维细胞激活蛋白(fap)。caf分泌生长因子,如vegf和fgf,其例如通过促进血管生成来促进肿瘤生长。caf还通过重塑肿瘤微环境(tme)中的细胞外基质(ecm)来促进肿瘤生长、侵袭和转移。例如,caf能够重塑ecm以包含更多生存信号,如igf-1和igf-2,从而促进周围癌细胞的生存。caf还可产生降解ecm组分(如基底膜)的酶(如基质金属蛋白酶mmp)。肿瘤细胞可以通过迁移穿过降解的膜,在某些情况下通过附着迁移的caf来侵入组织。肿瘤细胞通过ecm的迁移可导致转移。
82.在某些实施方案中,施用包含usp7抑制剂的组合物通过抑制癌症相关成纤维细胞(caf)中的usp7活性来治疗癌症。
83.本文证明,抑制成纤维细胞中的usp7减少成纤维细胞的致瘤作用,例如通过减少细胞侵袭、减少mmp分泌、减少基底膜降解、以及降低全身和tme vegf水平。因此,施用usp7抑制剂可以通过抑制一种或多种(例如两种或三种或全部)的这些致瘤效应来治疗癌症。
84.因此,在本文提供的方法的某些实施方案中,施用包含usp7抑制剂的组合物通过抑制癌症相关成纤维细胞的细胞外基质(ecm)重塑来治疗癌症。在此类实施方案中,ecm是肿瘤微环境的ecm。
85.在某些实施方案中,施用包含usp7抑制剂的组合物通过抑制基底膜的降解来治疗癌症。
86.在某些实施方案中,施用包含usp7抑制剂的组合物抑制选自弹性蛋白、胶原(例如iv型胶原)和层粘连蛋白的一种或多种ecm组分的降解。在某些实施方案中,施用包含usp7抑制剂的组合物抑制iv型胶原的降解。
87.在某些优选实施方案中,施用usp7抑制剂抑制成纤维细胞(任选caf)分泌mmp2和/或mmp7。在某些优选实施方案中,施用usp7抑制剂抑制成纤维细胞(任选caf)分泌mmp7。在某些优选实施方案中,施用usp7抑制剂抑制成纤维细胞(任选caf)分泌mmp2。在某些优选实施方案中,施用usp7抑制剂抑制成纤维细胞(任选caf)分泌mmp2和mmp7。
88.在实施例中证明,抑制激活的成纤维细胞中的usp7抑制了成纤维细胞通过基底膜的侵袭。这可能是由于usp7抑制后成纤维细胞对基质金属蛋白酶的表达减少所致。抑制成纤维细胞侵袭(例如,caf侵袭)是特别有利的,因为肿瘤细胞可以自身附着于侵袭性成纤维细胞,因此减少成纤维细胞侵袭可以减少癌细胞侵袭。
89.在某些实施方案中,施用包含usp7抑制剂的组合物抑制成纤维细胞通过基底膜的侵袭。在某些实施方案中,施用包含usp7抑制剂的组合物抑制caf通过基底膜的侵袭。
90.通过抑制成纤维细胞分泌mmp2、mmp7或两者,在成纤维细胞中抑制usp7特别有利,因为除了降解诸如基底膜的ecm组分外,已知mmp2和mmp7有助于上皮细胞向间充质细胞转
化(emt),例如通过触发tgf-β激活。
91.emt是上皮细胞失去粘附并成为间充质干细胞的过程,其是肿瘤转移的已知因素,特别是在上皮细胞衍生的癌症中。因此,通过减少成纤维细胞产生mmp2和mmp7,抑制usp7能够调节emt,这进一步减少肿瘤侵袭和转移。
92.因此,在某些实施方案中,施用包含usp7抑制剂的组合物通过调节emt(例如,通过减少emt)治疗癌症。
93.除了在细胞外基质建模中的作用外,成纤维细胞还有能力改变细胞外微环境,因此调节血管形成过程。成纤维细胞衍生的蛋白质,包括生长因子和基质蛋白,已被证明诱导、支持和调节内皮细胞出芽和毛细血管样网络(小管)的扩张。小管的形成有助于肿瘤的血管形成,并为肿瘤细胞的转移提供了进一步的手段。因此,通过usp7抑制减少成纤维细胞介导的上皮小管形成为治疗肿瘤和减少转移提供了进一步的途径。
94.因此,在某些实施方案中,施用usp7抑制剂通过抑制成纤维细胞介导的上皮小管形成来治疗癌症。在某些优选的实施方案中,施用usp7抑制剂通过抑制新生成纤维细胞介导的上皮小管形成来治疗癌症。
95.成纤维细胞,尤其是caf,也通过产生生长因子(如vegf)促进肿瘤相关血管生成。本文首次证明,usp7抑制降低了成纤维细胞的vegf生成,从而降低了全身以及肿瘤微环境中的vegf水平。vegf是癌症治疗的充分验证的靶标,抗vegf贝伐单抗(avastin
tm
)用于治疗至少结肠癌、肺癌、胶质母细胞瘤和肾癌。因此,抑制usp7以减少vegf提供了治疗癌症的进一步手段。
96.因此,在某些优选的实施方案中,施用包含usp7抑制剂的组合物通过降低受试者血清中的vegf水平来治疗癌症。
97.在某些优选的实施方案中,施用包含usp7抑制剂的组合物通过降低肿瘤微环境中的vegf水平来治疗癌症。
98.在某些优选的实施方案中,施用包含usp7抑制剂的组合物通过抑制癌症相关成纤维细胞(caf)产生vegf来治疗癌症。在某些实施方案中,抑制usp7抑制caf对vegf的分泌。在某些实施方案中,抑制usp7降低caf中的vegf mrna水平。
99.如本文所用,vegf是指由vegfa基因编码的vegfa。
100.通过抑制usp7抑制成纤维细胞(如caf)产生vegf是通过低氧诱导因子α(hifα),即vegf的转录因子的不稳定介导的。通过抑制usp7,usp7抑制剂减少成纤维细胞中的hifα的半衰期,从而减少vegf表达的驱动因素。
101.因此,在某些实施方案中,施用usp7抑制剂使低氧诱导转录因子(hif1α)不稳定,从而抑制癌症相关成纤维细胞产生vegf。在某些实施方案中,施用usp7抑制剂使癌症相关成纤维细胞中的低氧诱导转录因子(hif1α)不稳定,从而抑制肿瘤相关血管生成。
102.本文首次证明,抑制成纤维细胞中的usp7可抑制多种促肿瘤因子和效应,因此可减少肿瘤生长和/或侵袭。许多因素,例如成纤维细胞产生vegf和成纤维细胞介导的上皮小管形成可单独地或组合地促进血管生成。因此,抑制成纤维细胞中的usp7为抑制血管生成提供了新的手段,从而降低了肿瘤存活。
103.因此,在某些实施方案中,施用包含usp7抑制剂的组合物通过抑制血管生成来治疗癌症。在某些实施方案中,施用包含usp7抑制剂的组合物通过抑制新血管生成来治疗癌
症。在此类实施方案中,肿瘤微环境中的血管生成受到抑制。
104.在某些实施方案中,施用包含usp7抑制剂的组合物上调短(191aa)vegf-165 mrna的表达。在某些实施方案中,施用包含usp7抑制剂的组合物下调长(371aa)vegf-165mrna的表达。
105.在肿瘤微环境中具有肿瘤浸润淋巴细胞(til)对于肿瘤的有效免疫反应的重要性已得到充分证实。因此,值得注意的是,本文进一步证明,抑制usp7导致调节肿瘤免疫环境,以增加til浸润。
106.特别地,抑制usp7导致肿瘤浸润淋巴细胞(til)水平升高,特别是细胞毒性cd8 til。不希望受到理论约束,usp7抑制后细胞毒性cd8 til的增加可能是由于本文报道的usp7抑制对tme重塑的作用,例如通过癌症相关成纤维细胞,减少的caf介导的重塑允许更大的til浸润。
107.进一步值得注意的是,usp7抑制还降低了tme中treg细胞相对于cd8 til的比例。treg细胞对tme中局部免疫反应的阻尼效应是肿瘤细胞逃避免疫控制的已知机制。通过调节肿瘤免疫环境,特别是通过促进til浸润和减少tme中treg细胞的相对数量,usp7抑制提供了促进有效癌症治疗的进一步手段。
108.因此,在本发明的另一个方面,提供了一种通过抑制usp7活性治疗癌症的方法,该方法包括向有此需要的受试者施用包含usp7抑制剂的组合物,其中施用usp7抑制剂调节肿瘤免疫环境。肿瘤免疫环境的调节可以通过tme中存在的免疫细胞的数量和/或类型的变化来表征。例如,肿瘤免疫环境的调节可以tme中til的增加、tme中treg细胞的减少和/或tme中巨噬细胞的减少来表征。
109.在另一个方面,提供了一种通过抑制usp7活性来治疗癌症的方法,该方法包括向有此需要的受试者施用包含usp7抑制剂的组合物,其中施用包含uss7抑制剂的组合物增加了tme中til的数量,优选cd8 til的数量。
110.在另一个方面,提供了一种通过抑制usp7活性来治疗癌症的方法,该方法包括向有此需要的受试者施用包含usp7抑制剂的组合物,其中施用包含uss7抑制剂的组合物降低了tme中treg细胞相对于cd8 t细胞的比例。
111.在另一个方面,提供了一种通过抑制usp7活性来治疗癌症的方法,该方法包括向有此需要的受试者施用包含usp7抑制剂的组合物,其中施用包含uss7抑制剂的组合物减少tme中的巨噬细胞的数量。
112.在某些实施方案中,肿瘤免疫环境的调节(例如,til的增加、treg细胞和/或巨噬细胞的减少)通过抑制除肿瘤细胞外的细胞中的usp7来介导。在某些实施方案中,调节通过抑制成纤维细胞(例如caf)中的usp7来介导。
113.如随附实施例所证明的,usp7抑制剂与免疫检查点抑制剂(ici)的联合施用协同地组合以延长肿瘤模型中的存活。在不受理论约束的情况下,假设由usp7抑制剂介导的tme的调节导致til向肿瘤部位的更大募集和浸润,从而提供ici可以发挥作用的更大的潜在效应细胞群。
114.因此,在另一个方面,提供了一种通过向有此需要的受试者施用组合疗法治疗癌症的方法,该组合疗法包括包含usp7抑制剂的组合物和包含免疫检查点抑制剂的组合物。
115.在另一个方面,提供了一种用于治疗癌症的方法中的usp7抑制剂,该方法包括向
有此需要的受试者施用组合疗法,该组合疗法包括包含usp7抑制剂的组合物和包含免疫检查点抑制剂的组合物。
116.在另一个方面,提供了一种用于治疗癌症的方法中的免疫检查点抑制剂,该方法包括向有此需要的受试者施用组合疗法,该组合疗法包括包含usp7抑制剂的组合物和包含免疫检查点抑制剂的组合物。
117.在另一个方面,提供了一种用于治疗癌症的方法中的组合疗法,该方法包括向有此需要的受试者施用组合疗法,该组合疗法包括包含usp7抑制剂的组合物和包含免疫检查点抑制剂的组合物。
118.在优选的实施方案中,免疫检查点抑制剂选自pd1、pd-l1、ctla4、tigit、41bb、ox40、gitr的抑制剂。在某些实施方案中,检查点抑制剂选自抗pd1抗体、抗pd-l1抗体、抗ctla4抗体、抗41bb抗体、抗ox40抗体、抗gitr抗体和抗icos抗体。在某些实施方案中,检查点抑制剂选自抗pd1抗体、抗pd-l1抗体和抗ctla4抗体。在某些实施方案中,检查点抑制剂选自抗pd1抗体和抗pd-l1抗体。在某些实施方案中,检查点抑制剂是抗ctla4抗体。在某些实施方案中,检查点抑制剂选自:派姆单抗(keytruda
tm
)、纳武单抗(opdivo
tm
)、西米普利单抗(libtayo)
tm
)、阿特珠单抗(tecentriq
tm
)、阿维单抗(bavencio
tm
)、德瓦鲁单抗(imfinzi
tm
)、和伊匹单抗(yervoy
tm
)。
119.在本发明所有方面的优选实施方案中,以达到抑制肿瘤生长的剂量施用该组合物。
120.优选地,以达到抑制肿瘤侵袭的剂量施用该组合物。
121.优选地,以达到抑制肿瘤转移的剂量施用该组合物。
122.优选地,以实现肿瘤免疫环境调节的剂量施用该组合物。
123.优选地,以达到抑制肿瘤微环境中血管生成的剂量施用该组合物。
124.usp7抑制剂对成纤维细胞的新确定的作用的一个特别优点是,该作用独立于usp7抑制剂直接对癌细胞的任何作用(或缺乏作用)。即使当癌症对癌细胞中usp7的直接抑制没有反应,也可以通过抑制成纤维细胞(例如与癌症相关的成纤维细胞)中的usp7来实现抗肿瘤效果。
125.因此,在某些实施方案中,通过该方法治疗的癌症由癌细胞形成,并且癌细胞对usp7抑制剂具有抗性。
126.当癌细胞的存活和增殖不受癌细胞直接暴露于usp7抑制剂的影响时,癌细胞对usp7抑制剂具有抗性。癌细胞是否对usp7抑制剂具有抗性可以通过将癌细胞暴露于体外usp7抑制剂并监测其生长和存活来确定。
127.usp7抑制剂已被用于通过调节癌蛋白mdm2的泛素化来靶向癌症增殖。已假设mdm2在hifα表达中起作用;然而,本文证明,成纤维细胞中usp7抑制的作用不是通过mdm2介导的。因此,根据本发明,当治疗对mdm2途径抑制剂具有抗性的癌症时,通过抑制成纤维细胞中的usp7来治疗癌症是有效的。
128.因此,在某些实施方案中,癌症由癌细胞形成,并且癌细胞对mdm2途径的抑制剂具有抗性。在某些实施方案中,癌症由癌细胞形成并且癌细胞对mdm2抑制剂(例如mdm2调节p53的抑制剂)具有抗性。
129.在某些实施方案中,癌症由癌细胞形成,并且癌细胞对mdm2抑制剂具有抗性,该
mdm2抑制剂选自:rg7112、rg7388(依达奴林)、sar405838、mk-8242、amg232、cgm097、hdm201、cgm097、和alrn-6924。
130.如前所述,抑制成纤维细胞中的usp7可有效降低成纤维细胞的促肿瘤效应(例如,血管生成、ecm重塑、vegf生成)。由于本文提供的方法通过作用于成纤维细胞,特别是癌症相关成纤维细胞来治疗癌症,并且不需要对癌细胞本身产生任何影响,因此该方法适用于治疗广泛的癌症。
131.因此,在某些实施方案中,根据本发明治疗的癌症选自:肾癌(例如,肾细胞癌)、乳腺癌、脑肿瘤、淋巴瘤(例如,霍奇金淋巴瘤和非霍奇金淋巴癌、淋巴细胞性淋巴瘤、原发性cns淋巴瘤、b细胞淋巴瘤(例如,cll)、t细胞淋巴瘤(例如,塞扎里综合征))、鼻咽癌、黑素瘤(例如,转移性恶性黑素瘤)、前列腺癌、结肠癌、肺癌、骨癌、胰腺癌、皮肤癌、头颈癌(例如,头颈部鳞状细胞癌(hnscc))、皮肤上皮癌、皮肤或眼内恶性黑素瘤、子宫癌、卵巢癌、直肠癌、肛门区癌症、胃癌、睾丸癌、输卵管癌、子宫内膜癌、宫颈癌、阴道癌、外阴癌、食道癌、小肠癌、内分泌系统癌、甲状腺癌、甲状旁腺癌、肾上腺癌、软组织肉瘤、尿道癌、阴茎癌、膀胱癌、中枢神经系统肿瘤、脊柱轴肿瘤、脑干胶质瘤、垂体腺瘤、卡波西肉瘤、表皮样癌、鳞状细胞癌、间皮瘤。
132.在优选的实施方案中,癌症的特征在于肿瘤微环境中存在癌症相关成纤维细胞。
133.在某些实施方案中,癌症是癌或乳腺癌。在某些实施方案中,癌症是腺癌。在某些实施方案中,癌症是结直肠癌。在某些实施方案中,癌症是前列腺上皮癌。在某些实施方案中,癌症是结肠癌。在某些实施方案中,癌症是肺癌(例如,非小细胞肺癌)。在某些实施方案中,癌症是胶质母细胞瘤。在某些实施方案中,癌症是肾癌。
134.在某些优选的实施方案中,癌症是实体瘤。
135.如前所述,本发明的方法由于能够减少ecm的重塑而在治疗转移性癌症方面特别有效,从而减少癌细胞通过基底膜的迁移,这是癌症转移的已知因素。
136.因此,在某些优选的实施方案中,该方法是治疗转移性癌症的方法。在该上下文中治疗转移性癌症包括预防或减少转移、减缓转移进展和/或降低转移风险。在某些实施方案中,该方法包括治疗已经转移的癌症。在某些这样的实施方案中,所治疗的癌症是继发性肿瘤。
137.在某些实施方案中,所治疗的癌症是原发性肿瘤。
138.通过靶向成纤维细胞,例如caf,本发明的方法将有效治疗对旨在靶向癌细胞自身的治疗没有反应的癌症。因此,本发明的方法有利于治疗对替代癌症治疗(例如一线癌症治疗)无反应的患者。这些方法在治疗复发患者方面也是有效的,例如在一线治疗成功后。
139.因此,在某些实施方案中,受试者先前已被施用初始治疗剂,且未表现出反应。在某些实施方案中,受试者先前已被施用初始治疗剂并复发。
140.在某些此类实施方案中,初始治疗剂不是usp7抑制剂。在某些替代实施方案中,初始治疗剂是p53/mdm2途径的抑制剂。在某些实施方案中,初始治疗剂是p53/mdm2相互作用的抑制剂。
141.由于其tme重编程活性,usp7抑制剂有可能与其他已知调节tme的药剂(如免疫检查点抑制剂(如pd-1、pd-l1、ctla4抑制剂)、抗血管生成剂(如vegf抑制剂、vegfr抑制剂)或细胞外基质重编程剂)产生联合疗效。本发明的方法与检查点抑制剂组合将特别有效,因为
在本文中已证明,根据本发明在成纤维细胞中抑制usp7将导致cd8 t细胞更大程度地浸润到肿瘤部位,并且在存活率和肿瘤体积方面比加性改善更大。
142.因此,在某些实施方案中,该方法还包括施用附加治疗剂。
143.在某些实施方案中,附加治疗剂选自检查点抑制剂(也称为免疫检查点抑制剂)和抗血管生成剂。
144.在某些实施方案中,检查点抑制剂选自pd1、pd-l1、ctla4、tigit、41bb、ox40、gitr的抑制剂。在某些实施方案中,检查点抑制剂选自抗pd1抗体、抗pd-l1抗体、抗ctla4抗体、抗41bb抗体、抗ox40抗体、抗gitr抗体和抗icos抗体。在某些实施方案中,检查点抑制剂选自抗pd1抗体、抗pd-l1抗体和抗ctla4抗体。在某些实施方案中,检查点抑制剂选自抗pd1抗体和抗pd-l1抗体。在某些实施方案中,检查点抑制剂是抗ctla4抗体。在某些实施方案中,检查点抑制剂选自:派姆单抗(keytruda
tm
)、纳武单抗(opdivo
tm
)、西米普利单抗(libtayo)
tm
)、阿特珠单抗(tecentriq
tm
)、阿维单抗(bavencio
tm
)、德瓦鲁单抗(imfinzi
tm
)、和伊匹单抗(yervoy
tm
)。
145.在某些实施方案中,抗血管生成剂是vegf抑制剂或vegfr抑制剂。在某些实施方案中,抗血管生成剂选自:阿西替尼)、贝伐单抗卡博替尼依维莫司来那度胺乐伐替尼甲磺酸盐帕唑帕尼雷莫芦单抗瑞格非尼索拉非尼舒尼替尼沙利度胺(昔诺韦、)、凡德他尼和阿柏西普
146.在某些实施方案中,附加治疗剂与包含usp7抑制剂的组合物组合施用,例如作为组合疗法的一部分。在某些实施方案中,同时施用usp7抑制剂和附加治疗剂。在某些替代实施方案中,不同时施用usp7抑制剂和附加治疗剂。
147.在某些实施方案中,共同配制usp7抑制剂和附加治疗剂。在某些实施方案中,单独配制usp7抑制剂和附加治疗剂。
148.在某些实施方案中,包含usp7抑制剂的组合物进一步包含药学上可接受的赋形剂。
149.药物组合物可根据其特定用途和目的通过混合例如,赋形剂、粘合剂、润滑剂、崩解剂、包衣材料、乳化剂、悬浮剂、溶剂、稳定剂、吸收促进剂和/或软膏基质来配制。该组合物可适于口服、可注射、直肠或局部施用。
150.本领域技术人员已知合适的药学上可接受的赋形剂,例如:脂肪、水、生理盐水、醇(例如,乙醇)、甘油、多元醇、葡萄糖水溶液、增量剂、崩解剂、粘合剂、润滑剂、润湿剂、稳定剂、乳化剂、分散剂、防腐剂、甜味剂、着色剂、调味剂或芳香剂、浓缩剂、稀释剂、缓冲物质、溶剂或增溶剂、达到储存效果的化学品、改变渗透压的盐、包衣剂或抗氧化剂、糖,如乳糖或葡萄糖;玉米、小麦或水稻的淀粉;脂肪酸,如硬脂酸;无机盐,如偏硅酸铝酸镁或无水磷酸钙;合成聚合物,如聚乙烯吡咯烷酮或聚亚烷基二醇;醇,如硬脂醇或苯甲醇;合成纤维素衍生物,如甲基纤维素、羧甲基纤维素、乙基纤维素或羟丙基甲基纤维素;以及其他常规使用的添加剂,例如明胶、滑石、植物油和阿拉伯树胶。
151.例如,药物组合物可以口服施用,例如片剂、包衣片剂、硬或软明胶胶囊、溶液、乳
液或悬浮液的形式。也可以通过直肠进行施用,例如使用栓剂,局部或经皮施用,例如使用软膏、乳膏、凝胶或溶液,或肠胃外施用,例如使用可注射溶液。
152.对于片剂、包衣片剂或硬明胶胶囊的制备,本发明的化合物可与药学惰性、无机或有机赋形剂混合。合适的赋形剂的实例包括乳糖、玉米淀粉或其衍生物、滑石或硬脂酸或其盐。与软明胶胶囊一起使用的合适赋形剂包括,例如,植物油、蜡、脂肪和半固体或液体多元醇。
153.对于溶液和糖浆的制备,赋形剂包括例如,水、多元醇、蔗糖、转化糖和葡萄糖。
154.对于可注射溶液,赋形剂包括例如,水、醇、多元醇、甘油和植物油。
155.对于栓剂以及局部和经皮应用,赋形剂包括例如,天然或硬化油、蜡、脂肪和半固体或液体多元醇。
156.药物组合物还可包含防腐剂、增溶剂、稳定剂、润湿剂、乳化剂、甜味剂、着色剂、加臭剂、缓冲剂、包衣剂和/或抗氧化剂。
157.对于联合疗法,第二药物可以与usp7抑制剂以药物组合物的形式提供,或者可以单独提供。
158.在某些优选的实施方案中,口服施用包含usp7抑制剂的组合物。在某些优选实施方案中,通过注射(例如皮下或肌肉内)施用包含usp7抑制剂的组合物。
159.在本发明所有方面的优选实施方案中,待治疗的受试者是人类受试者。
160.在根据本发明提供的另一个方面中的是用于本文提供的治疗癌症的方法的usp7抑制剂。
161.usp7抑制剂
162.usp7抑制剂是本领域已知的,可用于本发明的方法。优选地,usp7抑制剂是小分子抑制剂。优选地,usp7抑制剂是分子量为900道尔顿或更小的有机化合物。
163.合适的usp7抑制剂的实例描述于wo2018/073602、us 2008/0103149 a1、wo 2010/114881 a1、wo 2010/081783 a1、wo 2011/086178 a1、wo 2013/030218 a1、ep 2565186 a1、ep 1749822 a1、wo 2016/109515 a1、wo 2016/109480 a1、wo 2016/126929 a1、wo 2016/126926 a1、wo 2016/126935 a1、wo 2016/150800 a1、wo2017/158381、wo2017/158388、wo2017/212010、wo2017/212012和us20190142834,其各自均通过引用并入本文。
164.合适的usp7抑制剂包括wo2018/073602中提供的那些,其全部内容通过引用并入,特别是关于权利要求保护的化合物、组合物和药物盐、优选实施方案以及其中例示的化合物。
165.在某些实施方案中,usp7抑制剂选自式(i)化合物:
[0166][0167]
包括药学上可接受的盐、互变异构体、立体异构体或其n-氧化物衍生物,其中:
[0168]
r1为h、oh或任选取代的烷基,优选地r1为h;
[0169]
r2为任选取代的c1-c6烷基、任选取代的c2-c6烯基、任选取代的c2-c6炔基、任选取代的c3-c6环烷基、任选取代的c4-c6烷基环烷基、任选取代的c4-c6芳基、任选取代的c3-c6杂芳基、任选取代的c4-c8芳氧基、任选取代的c7-c10芳基烷基或任选取代的c5-c10杂芳基烷基;以及
[0170]
q是任选取代的含氮杂环基。
[0171]
在某些实施方案中,q选自:
[0172][0173]
其中:
[0174]
w为n或c
[0175]
x为s、o、n、或ch
[0176]
y为cr
6a
、cr
9a
、n、或nr
6a
,
[0177]
z为cr
6b
、n、nr
6b
、nr
9b
、或o
[0178]
m不存在或cr
8a
[0179]
其中如果x为s、z为n且m不存在;并且其中如果m为cr
8a y不是n;
[0180]r5a
为h、卤基、任选取代的c1-c6烷基、或任选取代的氨基;
[0181]r5b
为h、卤基、任选取代的c1-c6烷基、任选取代的c2-c6炔基、苄基、任选单取代的c3-c6杂芳基、任选取代的c3-c6杂环烷基、任选取代的c1-c6烷氧基、nr’r”、或ranr’r”,
[0182]
其中ra为c1-c6烷基或c2-c6烯基;并且
[0183]
其中r’和r”各自独立地选自h、氧基取代的c1-c6烷基、羟基取代的c1-c6烷基、任选取代的c1-c6烷氧基、任选取代的c3-c6环烷基、任选取代的c1-c7烷基胺、任选取代的c2-c7烯基胺、任选取代的c3-c10杂环烷基、任选取代的c4-c10芳基、任选取代的c3-c10杂芳基、任选取代的c5-c10烷基芳基、任选取代的c4-c10烷基杂环烷基、和c4-c6烷基杂芳基、或其中r’和r”一起形成包括它们所连接的n的任选取代的c3-c8杂环烷基;
[0184]r6a
为h、任选取代的c1-c6烷基、任选取代的氨基、任选取代的c4-c6芳基、任选取代的c1-c6硫化物、任选取代的c1-c6磺酰基、或任选取代的氨基;
[0185]r6b
为h、氰基、卤基、任选取代的c1-c6烷基、任选取代的c2-c6烯基、任选取代的c2-c6炔基、任选取代的c3-c6环烷基、任选取代的c4-c6环烯基、任选取代的c2-c6炔醇、任选取代的c4-c6芳基、任选取代的c3-c6杂芳基、任选取代的氨基;
[0186]r7a
为h;
[0187]r7b
为h或任选取代的c4-c6芳基
[0188]
或其中r
7a
和r
7b
连同它们所连接的碳一起形成任选取代的c1-c6芳基;
[0189]r8a
为h或任选取代的c4-c6芳基;
[0190]r9a
为cl、f、br、i、或氰基;
[0191]r9b
为h、任选取代的c1-c6烷基、任选取代的c4-c6芳基、任选取代的c3-c8杂芳基、c1-c6烷氧基。
[0192]
在优选的实施方案中,usp7抑制剂是根据式(i)的化合物或其药学上可接受的盐,其中q选自:
[0193][0194]
其中:
[0195]
w为n或c
[0196]
x为s、o、n、或ch
[0197]
y为cr
6a
、cr
9a
、n、或nr
6a
,
[0198]
z为cr
6b
、n、nr
6b
、nr
9b
、或o
[0199]
m不存在或为cr
8a
[0200]
其中如果x为s,则z为n且m不存在;并且其中如果m为cr
8a
,则y不是n;
[0201]r5a
为h、卤基、任选取代的c1-c6烷基、或任选取代的氨基;
[0202]r5b
为h、卤基、任选取代的c1-c6烷基、任选取代的c2-c6炔基、苄基、任选单取代的c3-c6杂芳基、任选取代的c3-c6杂环烷基、任选取代的c1-c6烷氧基、nr’r”、或ranr’r”,
[0203]
其中ra为c1-c6烷基或c2-c6烯基;并且
[0204]
其中r’和r”各自独立地选自h、氧基取代的c1-c6烷基、羟基取代的c1-c6烷基、任选取代的c1-c6烷氧基、任选取代的c3-c6环烷基、任选取代的c1-c7烷基胺、任选取代的c2-c7烯基胺、任选取代的c3-c10杂环烷基、任选取代的c4-c10芳基、任选取代的c3-c10杂芳基、任选取代的c5-c10烷基芳基、任选取代的c4-c10烷基杂环烷基和c4-c6烷基杂芳基,或其中r’和r”一起形成包括它们所连接的n的任选取代的c3-c8杂环烷基;
[0205]r6a
为h、任选取代的c1-c6烷基、任选取代的氨基、任选取代的c4-c6芳基、任选取代的c1-c6硫化物、任选取代的c1-c6磺酰基、或任选取代的氨基;
[0206]r6b
为h、氰基、卤基、任选取代的c1-c6烷基、任选取代的c2-c6烯基、任选取代的c2-c6炔基、任选取代的c3-c6环烷基、任选取代的c4-c6环烯基、任选取代的c2-c6炔醇、任选取代的c4-c6芳基、任选取代的c3-c6杂芳基、任选取代的氨基;
[0207]r8a
为h或任选取代的c4-c6芳基;
[0208]r9a
为cl、f、br、i、或氰基;
[0209]r9b
为h、任选取代的c1-c6烷基、任选取代的c4-c6芳基、任选取代的c3-c8杂芳基、c1-c6烷氧基。
[0210]
在优选的实施方案中,usp7抑制剂是根据式(i)的化合物或其药学上可接受的盐,其中q选自:
[0211][0212]
其中r
5a
、r
5b
、r
6a
、r
6b
、r
9a
和r
9b
如上文所定义。
[0213]
在某些实施方案中,对于一个或多个取代基为任选的任何官能团,任选取代基独立地选自:oh、f、cl、br、i、cn、c1-c6烷基、cf3、chf2、ch2f、ch2oh、cooh、c(o)ch3、ch2nhc(o)och2ch3、c2-c6烯基、c2-c6炔基、c3-c6环烷基、c1-c6烷氧基、氨基、c1-c6烷基胺、c5-c6芳基、c3-c6杂芳基、苄基、氧代和酰胺、或两个相邻的取代基可以一起构成环。
[0214]
在优选的实施方案中,usp7抑制剂是根据式(i)的化合物或其药学上可接受的盐,其中q为:
[0215][0216]
且:
[0217]r5a
为h,
[0218]r5b
选自任选的甲基或乙胺取代的吡唑和nr’r”,其中r’和r”各自独立地选自h、甲基、环己胺、任选甲基-、氟-或氟苯基取代的c2-c7乙胺,任选取代的苯基,或其中r’和r”一起形成包括它们所连接的n的任选取代的c3-c8杂环烷基。
[0219]
在优选的实施方案中,r’为h,且r”是任选被甲基、氟或氟苯基取代的乙基吡咯烷。
[0220]
在优选的实施方案中,usp7抑制剂是根据式(i)的化合物或其药学上可接受的盐,其中q为:
[0221]
以及
[0222]r9a
为cl、f、br、i或氰基;
[0223]r9b
为h、任选取代的c1-c6烷基、或任选取代的c4-c6芳基;
[0224]
其中任选的取代基选自f、cl、br、甲氧基、oh、ch2oh、c1-c6烷基胺、环丙烷、四氢呋喃、二氧戊环、呋喃、任选被氟取代的甲基吡唑、和吗啉。
[0225]
在优选的实施方案中,r
9a
为cl、br、i、或氰基,且r
9b
为任选被f、cl、br、甲氧基、oh取代的苯基;c1-c6烷基胺、环丙烷、四氢呋喃、二氧戊环、呋喃、甲基吡唑。
[0226]
在某些优选的实施方案中,r
9a
为cl。
[0227]
在某些优选的此类实施方案中,r
9b
选自:
[0228][0229]
在优选的实施方案中,r
9b
为:
[0230][0231]
在优选的实施方案中,usp7抑制剂是根据式(i)的化合物或其药学上可接受的盐,其中q为:
[0232]
以及
[0233]r6a
为h或c1-c6烷基;
[0234]r6b
为h、卤基、任选取代的c2-c6烯基、任选取代的c2-c6炔基、任选取代的c3-c6环烷基、任选取代的c4-c6芳基、任选取代的c3-c6杂芳基;
[0235]
其中任选的取代基独立地选自f、cn、oh、ch2oh、酰胺、nh2、c1-c6烷基胺、c3-c6环烷基胺、cf3、cooh、甲基吗啉、ch(cf3)nh2、ch(chf2)nh2、ch2nhc(o)och2ch3。
[0236]
在优选的实施方案中,r
6a
为h、甲基或乙基;r
6b
为h、br、任选取代的丙烯基、乙炔基、任选取代的丙炔基、任选取代的戊炔基、任选取代的环己烷、任选取代的苯基、吡唑、吡啶;
[0237]
其中任选的取代基独立地选自f、cn、oh、ch2oh、酰胺、nh2、c1-c6烷基胺、c3-c6环烷基胺、cf3、ch(cf3)nh2、ch(chf2)nh2。
[0238]
在优选的实施方案中,r
6a
为甲基且r
6b
为任选被f、cn、oh、ch2oh、nh2、ch2nh2、ch2ch2nh2、ch(ch3)nh2、酰胺、环丙基胺、和环丁基胺中的一个或多个取代的苯基。
[0239]
在优选的实施方案中,r2为任选取代的噁唑或任选取代的c3-c6环烷基。在某些优选的实施方案中,r2为任选取代的噁唑或任选取代的环丙基。
[0240]
在某些此类实施方案中,每一个或多个任选的取代基独立地选自:c1-c6烷基和c3-c6环烷基。优选地,任选的取代基是甲基或环丙基。
[0241]
在某些优选的实施方案中,r2为被环丙烷取代的噁唑。在某些优选的实施方案中,r2为被甲基取代的环丙基。
[0242]
在某些优选的实施方案中,r2选自:
[0243][0244]
在usp7抑制剂为表现出立体异构性的化合物的优选实施方案中,该化合物为r-对映体。在usp7抑制剂为表现出立体异构性的化合物的优选实施方案中,该化合物为s-对映
体。
[0245]
在某些实施方案中,usp7抑制剂是根据式(i)的化合物或其药学上可接受的盐,选自wo2018/073602中例示的化合物(通过引用并入本文)。
[0246]
在某些实施方案中,usp7抑制剂是一种化合物或其药学上可接受的盐,选自以下:其中参考wo2018/073602给出实施例编号:
[0247]
实施例1:(r)-6-氯-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0248]
实施例2:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(吡啶-4-基)嘧啶-4(3h)-酮
[0249]
实施例3:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(1h-吡唑-5-基)嘧啶-4(3h)-酮
[0250]
实施例4:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(苯基氨基)嘧啶-4(3h)-酮
[0251]
实施例5:(r)-6-氨基-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0252]
实施例6:(r)-6-((2-(二甲基氨基)乙基)氨基)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0253]
实施例7:(r)-6-((2-(二甲基氨基)乙基)(甲基)氨基)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0254]
实施例8:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(4-甲基哌嗪-1-基)嘧啶-4(3h)-酮
[0255]
实施例9:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-甲氧基嘧啶-4(3h)-酮
[0256]
实施例10:(r)-6-(2-(二甲基氨基)乙氧基)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0257]
实施例11:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-((2-(吡咯烷-1-基)乙基)氨基)嘧啶-4(3h)-酮
[0258]
实施例12:6-((s)-3-氨基吡咯烷-1-基)-3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0259]
实施例13:(r)-6-(3-氨基氮杂环丁烷-1-基)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0260]
实施例14:(r)-5-氨基-6-氯-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0261]
实施例20:(r)-6-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0262]
实施例21:(r)-3-溴-6-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮(中间体b)
[0263]
实施例22:(r)-3-乙炔基-6-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
四氢-1,4-桥亚胺萘-6-基)氨基)嘧啶-4(3h)-酮盐酸盐
[0284]
实施例43:(r)-6-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-3-(苯基氨基)-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0285]
实施例44:(r)-6-氯-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-5-甲基嘧啶-4(3h)-酮
[0286]
实施例45:(r)-5-溴-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-((2-(吡咯烷-1-基)乙基)氨基)嘧啶-4(3h)-酮
[0287]
实施例46:(r)-3-(4-(氨基甲基)苯基)-6-((4-羟基-1-(4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0288]
实施例47:6-((s)-3-氨基哌啶-1-基)-3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮盐酸盐
[0289]
实施例48:(r)-6-(4-氨基哌啶-1-基)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮盐酸盐
[0290]
实施例49:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(2,8-二氮杂螺[4.5]癸-8-基)嘧啶-4(3h)-酮盐酸盐
[0291]
实施例50:6-((s)-3-(二甲基氨基)哌啶-1-基)-3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0292]
实施例51:(r,e)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(3-(吡咯烷-1-基)丙-1-烯-1-基)嘧啶-4(3h)-酮
[0293]
实施例52:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-((2-(哌啶-1-基)乙基)氨基)嘧啶-4(3h)-酮
[0294]
实施例53:(r,s)-3-(4-(氨基甲基)苯基)-6-((4-羟基-1-(4-甲氧基-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0295]
实施例54:(r)-6-(1-(2-(二甲基氨基)乙基)-1h-吡唑-4-基)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮,甲酸
[0296]
实施例55:6-((e)-3-((r)-3-氨基吡咯烷-1-基)丙-1-烯-1-基)-3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮,甲酸
[0297]
实施例56:6-((e)-3-((s)-3-氨基吡咯烷-1-基)丙-1-烯-1-基)-3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮,甲酸
[0298]
实施例57:3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)-6-(((s)-1-苯基-3-(吡咯烷-1-基)丙-2-基)氨基)嘧啶-4(3h)-酮,甲酸
[0299]
实施例58:3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)-6-(((r)-2-(吡咯烷-1-基)丙基)氨基)嘧啶-4(3h)-酮
[0300]
实施例59:3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)-6-(((s)-2-(吡咯烷-1-基)丙基)氨基)嘧啶-4(3h)-酮,甲酸
[0301]
实施例60:3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)-6-(((r)-1-(吡咯烷-1-基)丙-2-基)氨基)嘧啶-4(3h)-酮,甲酸
[0302]
实施例61:3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)-6-(((s)-1-(吡咯烷-1-基)丙-2-基)氨基)嘧啶-4(3h)-酮,甲酸
[0303]
实施例62:6-((s)-3-(二甲基氨基)吡咯烷-1-基)-3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0304]
实施例63:外消旋-6-(((
±‑
反式-1,2)-2-氨基环己基)氨基)-3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮,甲酸
[0305]
实施例64:6-(((
±‑
顺式-1,2)-2-氨基环己基)氨基)-3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮,甲酸
[0306]
实施例65:3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)-6-((((r)-1-甲基吡咯烷-2-基)甲基)氨基)嘧啶-4(3h)-酮,甲酸
[0307]
实施例66:3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)-6-((((s)-1-甲基吡咯烷-2-基)甲基)氨基)嘧啶-4(3h)-酮,甲酸
[0308]
实施例67:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0309]
实施例69:3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)-6-((2-((s)-2-甲基吡咯烷-1-基)乙基)氨基)嘧啶-4(3h)-酮,甲酸
[0310]
实施例70:3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)-6-((2-((r)-2-(甲氧基甲基)吡咯烷-1-基)乙基)氨基)嘧啶-4(3h)-酮
[0311]
实施例71:3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)-6-((2-((s)-3-甲基吡咯烷-1-基)乙基)氨基)嘧啶-4(3h)-酮,甲酸
[0312]
实施例73:6-((2-((r)-3-氟吡咯烷-1-基)乙基)氨基)-3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮,甲酸
[0313]
实施例74:6-((2-((s)-3-氟吡咯烷-1-基)乙基)氨基)-3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮,甲酸
[0314]
实施例75:3-(4-((r)-1-氨基乙基)苯基)-6-((4-羟基-1-((r)-4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0315]
实施例76:3-(4-((s)-1-氨基乙基)苯基)-6-((4-羟基-1-((r)-4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0316]
实施例79:(r)-3-(4-((二甲基氨基)甲基)苯基)-6-((4-羟基-1-(4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0317]
实施例80:(r)-3-(4-(氨基甲基)苯基)-6-((1-(4,4-二氟-3-苯基丁酰基)-4-羟基哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0318]
实施例81:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-异丙基嘧啶-4(3h)-酮
[0319]
实施例82:(r)-3-(4-(氨基甲基)-3-氟苯基)-6-((4-羟基-1-(4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0320]
实施例83:(r)-6-((4-羟基-1-(4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-3-(4-((甲基氨基)甲基)苯基)-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0321]
实施例84:(r)-3-(4-(氨基甲基)苯基)-6-((1-(3-(3,5-二氟苯基)-4,4,4-三氟丁酰基)-4-羟基哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0322]
实施例85:(r)-3-(4-(氨基甲基)苯基)-6-((4-羟基-1-(4,4,4-三氟-3-(4-氟苯基)丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0323]
实施例86:(r)-6-((2-(4-氟异吲哚啉-2-基)乙基)氨基)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮,甲酸
[0324]
实施例87:(r)-3-(4-(2-氨基乙基)苯基)-6-((4-羟基-1-(4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0325]
实施例88:(r)-3-(4-(1-氨基环丁基)苯基)-6-((4-羟基-1-(4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0326]
实施例89:(r)-3-((1-(4,4-二氟-3-苯基丁酰基)-4-羟基哌啶-4-基)甲基)-6-((2-(吡咯烷-1-基)乙基)氨基)嘧啶-4(3h)-酮,甲酸
[0327]
实施例95:6-((4-羟基-1-(3-苯基丙酰基)哌啶-4-基)甲基)-2-甲基-3-(三氟甲基)-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0328]
实施例97:3-(2-氟苯基)-6-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0329]
实施例98:(r)-3-(6-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-7-氧代-6,7-二氢-2h-吡唑并[4,3-d]嘧啶-3-基)苯甲腈
[0330]
实施例99:3-(2-氨基苯基)-6-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0331]
实施例100:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-吗啉代嘧啶-4(3h)-酮
[0332]
实施例102:(r)-1-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-[4,5
′‑
联嘧啶]-6(1h)-酮
[0333]
实施例103:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-((2-羟基乙基)氨基)嘧啶-4(3h)-酮
[0334]
实施例104:(r)-6-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-3-(3-羟基苯基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0335]
实施例106:(r)-4-(6-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-7-氧代-6,7-二氢-2h-吡唑并[4,3-d]嘧啶-3-基)苯甲酰胺
[0336]
实施例107:(r)-6-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-3-(4-(羟基甲基)苯基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0337]
实施例108:(r)-6-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-3-(3-(吗啉代甲基)苯基)-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0338]
实施例109:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-((2-甲氧基乙基)氨基)嘧啶-4(3h)-酮
[0339]
实施例110:3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)-6-(((r)-吡咯烷-3-基)氨基)嘧啶-4(3h)-酮
[0340]
实施例111:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-5-甲基-6-((2-(吡咯烷-1-基)乙基)氨基)嘧啶-4(3h)-酮
[0341]
实施例112:4-((3-(4-(氨基甲基)苯基)-2-甲基-7-氧代-2,7-二氢-6h-吡唑并[4,3-d]嘧啶-6-基)甲基)-4-羟基哌啶-1-羧酸苄基酯
[0342]
实施例113:(r)-n-(1-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-氧代-1,
6-二氢嘧啶-4-基)-n-(2-(吡咯烷-1-基)乙基)乙酰胺,甲酸
[0343]
实施例114:3-((4-羟基-1-((r)-3-苯基丁酰基)哌啶-4-基)甲基)-6-((2-((r)-2-甲基吡咯烷-1-基)乙基)氨基)嘧啶-4(3h)-酮,甲酸
[0344]
实施例116:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(苯基乙炔基)嘧啶-4(3h)-酮
[0345]
实施例117:(r)-6-苄基-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0346]
实施例118:3-(4-((r)-1-氨基-2,2,2-三氟乙基)苯基)-6-((4-羟基-1-((r)-4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0347]
实施例119:3-(4-((s)-1-氨基-2,2,2-三氟乙基)苯基)-6-((4-羟基-1-((r)-4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0348]
实施例120:(r)-3-(4-(氨基甲基)苯基)-2-乙基-6-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0349]
实施例121:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(苯基(2-(吡咯烷-1-基)乙基)氨基)嘧啶-4(3h)-酮,甲酸
[0350]
实施例122:3-(4-((s)-1-氨基-2,2-二氟乙基)苯基)-6-((4-羟基-1-((r)-4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0351]
实施例123:(r)-3-(4-(氨基甲基)-3-(三氟甲基)苯基)-6-((4-羟基-1-(4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0352]
实施例124:(r)-3-(4-(1-氨基环丙基)苯基)-6-((4-羟基-1-(4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0353]
实施例125:(s)-3-(4-(氨基甲基)苯基)-6-((4-羟基-1-(4,4,4-三氟-3-(5-甲基噻吩-2-基)丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0354]
实施例127:3-(4-(氨基甲基)苯基)-6-((1-(3,3-二环丙基丙酰基)-4-羟基哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0355]
实施例130:(r)-6-((4-羟基-1-(4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-3-(1-甲基-1,2,3,6-四氢吡啶-4-基)-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0356]
实施例131:6-((1-乙酰基-4-羟基哌啶-4-基)甲基)-2-甲基-3-苯基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0357]
实施例132:3-((1-乙酰基-4-羟基哌啶-4-基)甲基)-6-((2-(吡咯烷-1-基)乙基)氨基)嘧啶-4(3h)-酮
[0358]
实施例133:3-((1-(3,3-二环丙基丙酰基)-4-羟基哌啶-4-基)甲基)-6-((2-(吡咯烷-1-基)乙基)氨基)嘧啶-4(3h)-酮
[0359]
实施例134:(r)-3-(环己-1-烯-1-基)-6-((4-羟基-1-(4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0360]
实施例135:(r)-3-(3-(二甲基氨基)丙-1-炔-1-基)-6-((4-羟基-1-(4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
[0361]
实施例136:(r)-3-环己基-6-((4-羟基-1-(4,4,4-三氟-3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-2h-吡唑并[4,3-d]嘧啶-7(6h)-酮
基)甲基)嘧啶-4(3h)-酮
[0382]
实施例158:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(4-(吡啶-3-基甲基)哌嗪-1-基)嘧啶-4(3h)-酮
[0383]
实施例159:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(4-(吡啶-2-基甲基)哌嗪-1-基)嘧啶-4(3h)-酮
[0384]
实施例160:(r)-6-(4,4-二氟哌啶-1-基)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0385]
实施例161:(r)-2-((1-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-氧代-1,6-二氢嘧啶-4-基)氨基)-n,n-二甲基乙酰胺
[0386]
实施例162:(r)-6-(((2,3-二氢苯并[b][1,4]二恶英-6-基)甲基)氨基)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0387]
实施例163:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(((四氢-2h-吡喃-4-基)甲基)氨基)嘧啶-4(3h)-酮
[0388]
实施例164:(r)-n-(环丙基甲基)-1-(1-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-氧代-1,6-二氢嘧啶-4-基)氮杂环丁烷-3-甲酰胺
[0389]
实施例165:(r)-6-(3-氟氮杂环丁烷-1-基)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0390]
实施例166:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-((2-羟基乙基)(甲基)氨基)嘧啶-4(3h)-酮
[0391]
实施例167:(r)-6-(环戊基氨基)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0392]
实施例168:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(4-甲基-3-氧代哌嗪-1-基)嘧啶-4(3h)-酮
[0393]
实施例169:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(5-氧杂-2-氮杂螺[3.4]辛-2-基)嘧啶-4(3h)-酮
[0394]
实施例170:(r)-6-((1,3-二甲基-1h-吡唑-4-基)氨基)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0395]
实施例171:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(8-甲基-5-氧杂-2,8-二氮杂螺[3.5]壬-2-基)嘧啶-4(3h)-酮
[0396]
实施例172:(r)-6-(6-乙酰基-2,6-二氮杂螺[3.3]庚-2-基)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0397]
实施例173:(r)-6-(5,5-二氟-2-氮杂螺[3.3]庚-2-基)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)嘧啶-4(3h)-酮
[0398]
实施例174:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(7-氧杂-2-氮杂螺[3.5]壬-2-基)嘧啶-4(3h)-酮
[0399]
实施例175:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(2-氧杂-7-氮杂螺[3.5]壬-7-基)嘧啶-4(3h)-酮
[0400]
实施例176:(r)-3-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-6-(6-氧杂-2-氮杂螺[3.4]辛-2-基)嘧啶-4(3h)-酮
(4-氟苯基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0421]
实施例206:6-氯-3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-7-(4-氟-3-甲氧基苯基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0422]
实施例207:6-氯-7-(4-氟-3-甲氧基苯基)-3-((4-羟基-1-(1-甲基环丙烷羰基)哌啶-4-基)甲基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0423]
实施例208:6-氯-3-((4-羟基-1-(1-甲基环丙烷羰基)哌啶-4-基)甲基)-7-(3-(1-甲基-1h-吡唑-5-基)苯基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0424]
实施例209:6-氯-3-((4-羟基-1-(1-甲基环丙烷羰基)哌啶-4-基)甲基)-7-甲基-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0425]
实施例210:6-氯-3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-7-(3-(1-甲基-1h-吡唑-5-基)苯基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0426]
实施例211:6-氯-3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-7-(3-(1-甲基-1h-吡唑-4-基)苯基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0427]
实施例212:6-氯-3-((4-羟基-1-(1-甲基环丙烷羰基)哌啶-4-基)甲基)-7-(3-(1-甲基-1h-吡唑-4-基)苯基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0428]
实施例214:7-(3-溴苯基)-6-氯-3-((4-羟基-1-(1-甲基环丙烷羰基)哌啶-4-基)甲基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0429]
实施例215:6-氯-3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-7-苯基-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0430]
实施例216:6-氯-3-((4-羟基-1-(1-甲基-1h-吡唑-4-羰基)哌啶-4-基)甲基)-7-苯基-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0431]
实施例217:6-氯-3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-7-(3-环丙基苯基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0432]
实施例218:6-氯-7-(3-环丙基苯基)-3-((4-羟基-1-(1-甲基环丙烷羰基)哌啶-4-基)甲基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0433]
实施例219:6-氯-7-(3-环丙基苯基)-3-((4-羟基-1-(1-甲基-1h-吡唑-4-羰基)哌啶-4-基)甲基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0434]
实施例220:6-氯-7-(4-环丙基苯基)-3-((4-羟基-1-(1-甲基环丙烷羰基)哌啶-4-基)甲基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0435]
实施例221:6-氯-3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-7-(4-环丙基苯基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0436]
实施例223:6-溴-3-((4-羟基-1-(1-甲基环丙烷羰基)哌啶-4-基)甲基)-7-苯基-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0437]
实施例224:3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-6-碘代-7-苯基-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0438]
实施例225:6-氯-3-((4-羟基-1-(1-甲基环丙烷羰基)哌啶-4-基)甲基)-7-(3-(4-(三氟甲基)-1h-吡唑-1-基)苯基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0439]
实施例226:6-氯-3-((4-羟基-1-(1-甲基环丙烷羰基)哌啶-4-基)甲基)-7-(3-吗啉代苯基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0440]
实施例227:6-氯-7-(3-(4-氟-1h-吡唑-1-基)苯基)-3-((4-羟基-1-(1-甲基环丙烷羰基)哌啶-4-基)甲基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0441]
实施例228:6-氯-7-(4-氯苯基)-3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0442]
实施例229:6-氯-7-(4-氯苯基)-3-((4-羟基-1-(1-甲基-1h-吡唑-4-羰基)哌啶-4-基)甲基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0443]
实施例230:6-氯-3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-7-(4-氟-3-(1-甲基-1h-吡唑-5-基)苯基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0444]
实施例231:6-氯-7-(4-氟-3-(1-甲基-1h-吡唑-5-基)苯基)-3-((4-羟基-1-(1-甲基环丙烷羰基)哌啶-4-基)甲基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0445]
实施例232:6-氯-3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-7-(4-氟-3-(1-甲基-1h-吡唑-4-基)苯基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0446]
实施例233:6-氯-7-(4-氟-3-(1-甲基-1h-吡唑-4-基)苯基)-3-((4-羟基-1-(1-甲基环丙烷羰基)哌啶-4-基)甲基)-3h-吡咯并[2,3-d]嘧啶-4(7h)-酮
[0447]
实施例234:3-((4-羟基-1-(1-甲基环丙烷羰基)哌啶-4-基)甲基)-4-氧代-7-苯基-4,7-二氢-3h-吡咯并[2,3-d]嘧啶-6-甲腈
[0448]
实施例235:7-(苯并[d][1,3]二氧杂环戊烯-5-基-2,2-d2)-6-氯-3-((4-羟基-1-(1-甲基环丙烷-1-羰基)哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮
[0449]
实施例236:7-(苯并[d][1,3]二氧杂环戊烯-5-基-2,2-d2)-6-氯-3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮
[0450]
实施例237:3-(4-(氨基甲基)苯基)-6-((4-羟基-1-(1-甲基环丙烷-1-羰基)哌啶-4-基)甲基)-2-甲基-2,6-二氢-7h-吡唑并[4,3-d]嘧啶-7-酮
[0451]
实施例238:3-(4-(氨基甲基)苯基)-6-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-2-甲基-2,6-二氢-7h-吡唑并[4,3-d]嘧啶-7-酮
[0452]
实施例239:6-氯-7-(3,4-二甲氧基苯基)-3-((4-羟基-1-(1-甲基环丙烷-1-羰基)哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮
[0453]
实施例240:6-氯-3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-7-(3,4-二甲氧基苯基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮
[0454]
实施例241:7-(4-(氨基甲基)苯基)-6-氯-3-((4-羟基-1-(1-甲基环丙烷-1-羰基)哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮
[0455]
实施例242:7-(4-(氨基甲基)苯基)-6-氯-3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮
[0456]
实施例243:(r)-4-(6-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-7-氧代-6,7-二氢-2h-吡唑并[4,3-d]嘧啶-3-基)苯甲酸
[0457]
实施例244:(r)-3-(6-((4-羟基-1-(3-苯基丁酰基)哌啶-4-基)甲基)-2-甲基-7-氧代-6,7-二氢-2h-吡唑并[4,3-d]嘧啶-3-基)苯甲酸
[0458]
实施例245:4-(6-((4-羟基-1-(1-甲基环丙烷-1-羰基)哌啶-4-基)甲基)-2-甲基-7-氧代-6,7-二氢-2h-吡唑并[4,3-d]嘧啶-3-基)苯甲酸。
[0459]
在某些优选的实施方案中,usp7抑制剂为:
[0460][0461]
或其立体异构体或药学上可接受的盐。
[0462]
在某些优选的实施方案中,usp7抑制剂为:
[0463]
或其立体异构体或药学上可接受的盐。
[0464]
在某些优选的实施方案中,usp7抑制剂为:
[0465]
或其立体异构体或药学上可接受的盐。
[0466]
在某些优选的实施方案中,usp7抑制剂为:
[0467]
或其立体异构体或药学上可接受的盐。
[0468]
在某些优选的实施方案中,usp7抑制剂为:
[0469]
或其立体异构体或药学上可接受的盐。
[0470]
在某些优选的实施方案中,usp7抑制剂为:
[0471]
或其立体异构体或药学上可接受的盐。
[0472]
除了根据本发明的方法使用的合适的usp7抑制剂外,本文提供的所有usp7抑制剂化合物也作为化合物本身公开,包括其药学上可接受的盐、立体异构体、互变异构体及其n-氧化物衍生物。
[0473]
在某些替代实施方案中,usp7抑制剂选自wo2018/073602、us 2008/0103149 a1、wo 2010/114881 a1、wo 2010/081783 a1、wo 2011/086178 a1、wo 2013/030218 a1、ep 2565186 a1、ep 1749822 a1、wo 2016/109515 a1、wo 2016/109480 a1、wo 2016/126929 a1、wo 2016/126926 a1、wo 2016/126935 a1、wo 2016/150800 a1、wo2017/158381、wo2017/158388、wo2017/212010、wo2017/212012和us20190142834中的一种或多种中提供的usp7抑制剂,其各自均通过引用并入本文。
[0474]
关于本发明与根据本发明的化合物的治疗用途有关的方面,可向需要治疗的受试者施用
″
有效量”的化合物。术语“有效量”是指一种化合物的量或剂量,该化合物在向受试者单剂量或多剂量给药后,可在疾病的治疗中提供治疗效果。根据本发明的化合物的治疗有效量可以包括每单个剂量约0.1mg/kg至约20mg/kg范围内的量。任何个体患者的治疗有效量可由医疗专业人员通过本领域技术人员理解的方法确定。可以改变在任何给定时间点施用的化合物的量,以便在治疗过程中施用最佳量的化合物,无论是单独使用还是与任何其他治疗剂组合使用。
[0475]
当介绍本发明的要素或其优选实施方案(一个或多个)时,冠词“一
″
、“一个”、“该
″
和“所述
″
旨在意指存在一个或多个要素。术语“包括”、“包含”和“具有
″
旨在是包括性的,并且意指除所列要素外,可能还有其他要素。
[0476]
除非本文另有定义,否则与本发明相关的科学和技术术语应具有本领域普通技术人员通常理解的含义。然而,术语的含义和范围应明确,如果存在任何潜在的歧义,则本文提供的定义或如果本文未定义则在wo2018/073602中的定义优先于任何词典或外部定义。
[0477]
上述详细描述是通过解释和说明的方式提供的,并不旨在限制所附权利要求的范围。本文示出的当前优选实施方案中的许多变化对于本领域普通技术人员来说是显而易见的,并且仍在所附权利要求及其等效物的范围内。
[0478]
实施例
[0479]
1.1 usp7抑制降低原代激活的成纤维细胞中的vegf水平
[0480]
研究了usp7抑制对除肿瘤细胞本身外的肿瘤微环境中细胞类型的作用。ad-04(对应于wo2018/073602中的实施例30)是一种有效且特异的usp7抑制剂。ad-04是在基于细胞的biomap分析中筛选的,其是一种临床相关的生物标志物谱小组,其包括各种原代人类细胞衍生的共培养物。在多个细胞疾病系统中进行筛选:在与t细胞受体(tcr)配体刺激的pbmc共培养的原代人真皮成纤维细胞(hdf)以及与hdf和刺激的pbmcs共培养的癌细胞(ht29或h1299)中,ad-04显示出对细胞因子调节的显著影响。这些系统重现了肿瘤细胞、受
刺激的免疫细胞和宿主基质网络之间的相互作用。在不同浓度的ad-04下处理细胞48小时,然后通过elisa在共培养物上清液中进行细胞因子测量。与溶媒对照处理系统相关的生物标志物变化以对数转化比表示。ad-04的表型活性曲线显示各种生物标志物在统计学上的著减少,包括最引人注目的是,在300nm抑制剂浓度下,分泌型vegf(svegf)减少了3-log倍(图1和图2)。在与激活的pbmc和多个原代人成纤维细胞(包括原代肺成纤维细胞wi-38和imr-90或原代真皮成纤维细胞hdf)共同培养的同一癌细胞系ht29中,进一步证实了biomap固定样本数据调查结果。ent-ad-04对分泌型vegf水平没有显著影响(图3)。
[0481]
为了证实哪种细胞类型导致在共同培养物系统中观察到的响应于usp7抑制的vegf减少,我们独立测试了ad-04对每种细胞类型中vegf分泌的影响。首先,测试了四种癌细胞系:lncap和mcf-7对ad-04敏感;ht29和h1299对ad-04不敏感(数据未显示)。与溶媒处理的细胞相比,在来自所有测试的癌细胞系(敏感或不敏感)的上清液中,svegf水平未受到显著影响。(图4)接下来,研究了ad-04对激活的原代成纤维细胞中svegf水平的影响。在伤口愈合过程期间或在急性或慢性炎症信号刺激后,成纤维细胞获得激活表型(称为肌成纤维细胞)。一旦激活,肌成纤维细胞增殖和迁移,表现出收缩活性,施加物理力来改变组织结构,并变得转录活跃,导致细胞因子、趋化因子和细胞外基质(ecm)组分的分泌。在肿瘤微环境中,大多数残留的成纤维细胞被激活,称为癌症相关成纤维细胞(caf)。为了产生肌成纤维细胞表型,wi38、imr90和hdf与抗cd3/抗cd28刺激的pbmc共培养,或由fgf-2或tgfβ刺激。激活后,用ad-04或其对映体处理细胞,并通过elisa测定svegf水平。在usp7抑制后,激活的原代肺和真皮成纤维细胞显示svegf蛋白水平呈剂量依赖性降低(图5)。ad-04处理后,在原发性结直肠腺癌相关成纤维细胞中观察到svegf的类似下降(图6)。usp7抑制剂介导的vegf分泌减少仅在原代成纤维细胞中观察到;永生化肺成纤维细胞(wi38-va13)对usp7抑制剂处理不敏感,可能是由于sv-40永生化成纤维细胞中不稳定的p53(图7)。
[0482]
类似地,usp7抑制降低了激活的成纤维细胞和caf中的细胞内vegf蛋白水平,然而在癌细胞和sv-40转化的肺成纤维细胞中,这种变化没有统计学意义。(图8)综上所述,这些结果证明,usp7抑制不仅调节分泌型vegf蛋白,而且还调节激活的成纤维细胞(tme内的主要基质细胞群)中的细胞内vegf水平。
[0483]
1.2 usp7抑制依赖性vegf减少通过调节hif-1α信号传导介导
[0484]
响应于创伤和在肿瘤生长期间,激活的成纤维细胞分泌ecm的合成物和修饰物,但也分泌可溶性血管生成生长因子,如vegf。尽管vegf基因表达可由多种转录因子控制,但其主要调节因子是低氧诱导型因子1(hif-1)。hif-1介导实体瘤和各种恶性细胞系中响应于低氧的vegf转录激活。为了通过低氧水平触发vegf表达,在低氧室内生长原代成纤维细胞,并用溶媒dmso、ad-04或ent-ad-04处理48小时。正如预期的那样,低氧显著诱导原代人成纤维细胞中的vegf分泌;通过用ad-04处理,vegf分泌以剂量依赖性方式减少。与常氧一样,在低氧条件下,ent-4对分泌型vegf蛋白没有显著影响(图9)。
[0485]
为了阐明usp7抑制对激活的成纤维细胞中hif-1α稳定性的作用,检查了ad-04对hif-1α半衰期的影响。hdf用放线菌酮预处理,然后用ad-04或作为对照的dmso处理,并置于低氧室中。随后在不同时间点收集样品,用于探测hif-1α水平的免疫印迹分析。在这些实验条件下并按照光密度测定分析,确定未处理细胞中的hif-1α半衰期为6分36秒。用ad-04处理可将该值显著降低40%至4分钟(图10),证实了usp7抑制对hif-1α稳定性的直接调节。
[0486]
鉴于已知hif-1α稳定性在蛋白质水平上通过泛素化调节,我们研究了usp7抑制是否调节hif-1α多泛素化。简言之,hdf在低氧中孵育3小时,然后用dmso、ad-04或mg132在指定浓度下处理另外1小时。收获细胞,裂解并使用两种类型的四泛素结合实体(tubes)下拉多泛素化蛋白质;对k48/k63具有相等的亲和力或对k63的亲和力提高10倍。使用hif-1α抗体通过免疫印迹分析样品。如图11所示,mg132有效阻止蛋白酶体降解,导致hif-1α的多泛素化增加。用usp7抑制剂ad-04处理增强k63/k48多泛素化hif-1α的积累(图11,上图)。相反,在dmso对照和用ad-04治疗之间,hif-1α的k63特异性多泛素化没有差异(图11,下图)。这些发现表明usp7抑制导致hif-1α的k48多泛素化,因此导致其蛋白酶体介导的降解。
[0487]
1.3 usp7抑制原代激活的成纤维细胞中的vegf分泌的作用机制
[0488]
为了了解usp7抑制剂ad-04的作用机制并评估usp7抑制低氧激活的成纤维细胞的途径,进行了rna-seq。hdf用溶媒dmso和ad-04处理,然后在常氧或低氧条件下孵育6或24小时。然后提取rna并通过rna-seq分析。vegf mrna水平的比较表明,其表达在低氧条件下诱导,并在用ad-04处理后下调(图13)。这些发现表明usp7抑制剂在蛋白质和mrna水平上介导vegf调节。为了评估低氧激活的成纤维细胞中诱导的信号传导途径,对来自非激活(常氧)和低氧激活的原代成纤维细胞的样品进行了比较途径分析。结果表明,在低氧激活的成纤维细胞样品中,hif1a mrna表达网络是使用nci自然规范途径数据库最显著富集的途径(图12)。此外,相同样本的ad-04处理导致hif1a表达网络中已知基因的调节,包括vegf基因(图14)。有趣的是,低氧激活和usp7抑制剂处理的原代人成纤维细胞显示vegfa选择性剪接的选择性模式。在24小时时,抗血管生成短(191aa)亚型vegf-165的mrna表达上调。vegf-165的促血管生成长(371aa)亚型在24小时时强烈下调(图15)。这些数据表明usp7抑制后对vegf表达的补偿反馈环,并进一步强调usp7介导的vegf表达调节的重要性。
[0489]
1.4原代人成纤维细胞中vegf分泌的调节是usp7特异性的
[0490]
众所周知,抑制usp7导致癌蛋白e3连接酶mdm2降解和p53水平升高。为了证实激活的成纤维细胞中vegf蛋白的调节不是通过mdm2-p53轴介导的,对临床相关的mdm2拮抗剂进行了基准测试实验。癌细胞、永生化成纤维细胞和fgf-2激活的原代成纤维细胞在常氧或低氧条件下生长,然后用溶媒、usp7抑制剂ad-04、其非活性对映体(ent-4)、nutlin-3a、rg7112或sar405838在指定浓度下处理48小时。收集细胞培养上清液,通过elisa测定svegf。同时,如上所述在常氧和低氧条件下处理细胞,然后在72小时后进行细胞活力测量。与ad-04相比,测试的mdm2拮抗剂不能调节vegf的分泌(图16和17)。这些结果证实,在usp7抑制后,原代人成纤维细胞中vegf分泌的调节不是通过mdm2介导的。此外,与mdm2拮抗剂相比,ad-04及其非活性对映体在高浓度下不会影响细胞活力(图18)。为了确保usp7抑制剂的作用通过直接usp7调节来介导,我们使用基于活性的探针竞争分析来监测usp7细胞靶接合。用浓度增加的抑制剂处理细胞,裂解,并加入泛素-丙炔胺(ub-pa)探针。ad-04以浓度依赖性的方式与ub-pa探针有效竞争(ec
50
=11-24nm,
±
5s.d),证明了在原代成纤维细胞、永生化成纤维细胞和癌细胞中的高效靶接合(图19和20)。我们的选择性usp7抑制剂ad-04对原代人成纤维细胞中vegf分泌的特异性通过usp7 crispr敲除得到证实。crispr/cas9诱导的原代hdf中usp7的敲低通过使用抗usp7抗体的免疫印迹分析进行验证,并导致低氧培养48小时后细胞培养物上清液中检测到的svegf蛋白几乎完全消失(图21)。
[0491]
1.5 ad-04抑制成纤维细胞侵袭,并下调侵袭成纤维细胞中的mmp-7分泌
[0492]
为了研究usp7抑制对原代成纤维细胞、永生化成纤维细胞,内皮细胞和癌细胞增殖的影响,使用活细胞延时成像。结果显示ad-04对癌细胞、成纤维细胞或内皮细胞增殖没有影响(图22)。接下来,使用划痕损伤细胞迁移试验研究了ad-04对细胞迁移的影响。用ad-04处理没有调节任何测试细胞中的迁移(划痕伤口愈合过程),表明usp7抑制对癌细胞、成纤维细胞或内皮细胞中的细胞迁移没有影响(图23)。最后,在划痕损伤细胞侵袭试验中检测了ad-04在细胞侵袭能力中的功能。在ad-04处理后,侵袭癌细胞和永生化成纤维细胞的数量没有改变。然而,与溶媒处理的细胞相比,usp7抑制后原代成纤维细胞通过基底膜的侵袭大大减少(图24)。这些发现证明了ad-04抑制原代激活的成纤维细胞基底膜重塑的能力。
[0493]
已经证明,除vegf外,基质金属蛋白酶还通过降解ecm组分参与肿瘤侵袭和转移。为了进一步探讨usp7抑制是否调节侵袭成纤维细胞中的mmp水平,收集hdf侵袭分析的细胞培养上清液,并使用多重分析检测各种mmp的存在。结果显示,与溶媒处理样品相比,用usp7抑制剂处理的样品中mmp-7水平显著降低。ent ad-04和avastin对mmp-7水平没有显著影响(图25)。
[0494]
1.6 usp7抑制减少体外小管形成并降低内皮成纤维细胞共培养物中的mmp-2水平
[0495]
除了在细胞外基质的合成和维持中起主要作用外,成纤维细胞还具有改变机械细胞外微环境的能力,从而调节血管生成过程。成纤维细胞衍生的蛋白质,包括生长因子和基质蛋白,已被证明诱导、支持和调节内皮细胞出芽和体外毛细血管样网络(小管)的扩张。在与原代人内皮细胞(huvec)的共培养物系统中评估了usp7抑制对hdf支持毛细血管样结构形成能力的影响。为了研究ad-04对从头小管形成的影响,接种后立即处理共培养物。通过在接种后几天处理共培养物(当小管已经形成时),研究ad-04对预先形成的小管的影响。在孵育14天后,hdf和huvec共培养导致血管小管形成。然而,usp7抑制以剂量依赖的方式导致小管长度显著减少(图26,上图)。有趣的是,当预先形成的毛细血管样网络用ad-04处理时,对小管形成的抑制明显不那么明显(图26,下图)。测定了毛细管样结构的平均长度(图26,图表)。综上所述,这些结果表明,usp7抑制可防止hdf和huvec共培养物的新生小管形成。除了成纤维细胞和内皮细胞共培养物外,当huvec在上生长时,小管也可以在6小时内形成。因此,研究了usp7抑制对huvec小管形成的直接影响。ad-04处理未破坏上的huvec小管形成(图27),支持先前的发现,其中usp7抑制仅影响激活的成纤维细胞,并且观察到的对小管形成的影响原代成纤维细胞中由usp7抑制剂介导。
[0496]
接下来,研究了小管形成抑制的潜在机制。据报道,mmp-2的诱导在体外增加内皮细胞的小管形成,而mmp-2缺陷小鼠显示出对肿瘤诱导的血管生成的抑制。为了确定mmp-2的分泌是否受usp7抑制调节,14天后收集huvec和hdf共培养物的上清液。如图28所示,观察到usp7抑制后mmp-2水平的剂量依赖性下降。ent-ad-04和avastin对mmp-2水平没有显著影响。
[0497]
1.7 us7抑制抑制肿瘤异种移植物的体内生长,降低血清vegf水平,并促进免疫细胞募集到tme中
[0498]
为了检测usp7抑制的体内抗肿瘤活性及其对肿瘤基质的影响,建立了balb/c小鼠中ct-26细胞的同基因小鼠异种移植模型。作为阳性对照,使用了索拉非尼,一种具有抗肿
瘤和抗血管生成特性的多激酶抑制剂。ct-26小鼠癌细胞系在体外对ad-04抑制不敏感,这使其成为研究usp7抑制对tme影响的良好模型(图29)。如图30所示,ad-04以剂量依赖性方式抑制ct26异种移植物的生长,以肿瘤体积减小为证据。最高ad-04剂量具有与阳性对照索拉非尼(58%)处理相似的肿瘤生长抑制作用(53%)。ad-04和索拉非尼治疗均显示出低毒性,因为任一种处理都未导致荷瘤小鼠的显著体重下降(图29,左图)。正如在体外原代成纤维细胞中观察到的,在用ad-04处理的动物中,小鼠血清中循环vegf的测量显示出显著的剂量依赖性降低(图31)。
[0499]
tme已被证明在肿瘤进展和预后中起关键作用。抗癌疗法的疗效取决于tme概况。例如,肿瘤免疫细胞浸润被认为是决定免疫检查点抑制成功的重要因素,并与患者生存率的提高相关。为了了解ad-04是否调节免疫细胞向肿瘤中的募集,使用流式细胞术分析了几个免疫群体。我们首先评估了肿瘤浸润淋巴细胞(til)中各种t细胞亚群的频率。结果显示,ad-04处理后细胞毒性t淋巴细胞显著增加,而观察到cd4淋巴细胞中无明显变化。相反,在usp7抑制后观察到treg/cd8比例下降的趋势。骨髓细胞浸润分析显示,在用usp7抑制剂治疗后,巨噬细胞减少(图32)。
[0500]
最后,通过rna序列分析ad-04在体内的作用模式。对同基因ct-26肿瘤样品的分析显示肿瘤vegfa mrna的不同剪接;在用usp7抑制剂ad-04处理后,观察到vegfa短亚型减少和长vegfa亚型增加。途径分析证明,在用100mg/kg的usp7抑制剂处理10天的肿瘤样品中,hif1a mrna表达网络位于前10个最显著富集的途径(nci自然定义)中。此外,hif1a途径中的单个基因在usp7抑制后受到差异调节。这些结果支持了先前的发现,并证实了usp7通过hif-1α途径在体内和体外都调节tme的概念。
[0501]
1.7对于多种usp7抑制剂,观察到us7抑制的成纤维细胞介导的抗癌效果
[0502]
为了证明usp7抑制剂的作用不限于ad-04,评估了替代usp7抑制剂。adc-159是usp7抑制剂,其效力和选择性与ad-04相当。adc-159具有结构:
[0503][0504]
为了在功能分析中评估其效力,hdf用溶媒或不同浓度的usp7抑制剂adc-159处理,并在低氧室中孵育48小时。收集细胞培养上清液,并通过elisa测量分泌型vegf。如图33所示,adc-159以剂量依赖性方式降低vegf水平,显示出与ad-04相当的效力。在其他usp7抑制剂adc-160、adc-198、adc-199和adc-556中观察到相当的结果(图34)。此外,在hdf和huvec小管形成分析中,adc-159对新形成的小管显示出剂量依赖性方式的抑制作用,而对已建立的小管没有显著影响(图35)。adc-159对小管形成的抑制作用与ad-04相当。
[0505]
为了检测adc-159抑制usp7的体内抗肿瘤活性及其对肿瘤基质的影响,在balb/c小鼠的ct-26细胞的同基因小鼠异种移植模型中测试了adc-159。与ad-04类似,ct-26小鼠癌细胞系在体外对adc-159不敏感,这使其成为研究usp7抑制对tme影响的良好模型。
[0506]
对ct-26同基因小鼠模型的研究表明,adc-159可在体内防止肿瘤血管形成。用溶媒处理的对照动物仅在肿瘤部位显示肿瘤血管形成,其特征是成熟血管的细分支,通过内
皮标志物cd31和周细胞标志物ng2的阳性染色检测细分支(图36a)。相反,adc-159治疗阻止了肿瘤部位成熟血管的细分支(图36b)。
[0507]
尽管ct-26细胞对adc-159的直接抑制usp7有抵抗力,但与用单独的溶媒处理的对照动物相比,adc-59处理导致ct-26体内模型中的肿瘤面积显著减少(图37a)。与溶媒对照相比,adc-159治疗还导致肿瘤部位的坏死面积的显著增加(图37b),进一步证明了usp7抑制对tme的影响,导致减少的肿瘤生长。
[0508]
1.8 usp抑制剂与免疫检查点抑制剂(ici)协同结合
[0509]
由于其tme重编程活性,usp7抑制剂有可能与其他已知调节tme的药物(如免疫检查点抑制剂)发挥联合功效。在同基因ct-26小鼠模型中,与溶媒对照相比,usp7抑制(adc-159)单独导致肿瘤体积减小(图38a)。用usp7抑制剂(adc-159)和免疫检查点抑制剂(抗pd-l1或抗ctla4抗体)的组合处理的小鼠导致肿瘤体积显著进一步减少。特别地,adc-159产生23%的肿瘤生长抑制(tgi),与抗-pd-l1(22%)达到的水平非常相似。adc-159和抗pd-l1的组合显示出协同效应,导致肿瘤生长进一步减少(tgi=43%)。用抗-ctla-4处理导致43%的tgi,这与adc-159和抗-ctra-4组合产生的tgi相似(图38a)。未观察到体重的显著变化,表明处理耐受性良好(图38b)。
[0510]
usp7抑制剂和ici之间的协同作用显著提高了体内肿瘤模型的存活率。图39显示了本文描述的同基因ct-26小鼠模型的存活图,其中小鼠用单独的usp7抑制剂adc-159、单独的抗pd-l1或抗ctla4或usp7抑制剂adc-159和抗pd-l1或抗ctla4的组合处理。溶媒处理仅用作对照。
[0511]
图39显示,与单独使用usp7抑制剂和ici处理的小鼠相比,使用usp7抑制剂和ici的组合处理的小鼠存活率显著提高。该效果大于两种疗法的相加效果,如用单独的抗pd-l1处理与溶媒对照相比,存活率无改善所证实。adc-159处理可使存活率适度但显著增加。相比之下,adc-159与抗pd-l1的组合导致生存率提高更多。adc-159和抗ctla4也观察到同样的协同组合效应,其中50天的存活率高达80%(图39a)。每种处理对肿瘤生长的影响如图39b所示。
[0512]
与usp7抑制剂的组合导致用免疫检查点抑制剂处理的小鼠表现出细胞毒性t淋巴细胞向肿瘤的浸润增加(图40)。在不受理论约束的情况下,假设组合的协同效应来自于由于usp7抑制而增加的til募集,从而在肿瘤部位提供了能够被icis激活的更大的效应细胞群体。
[0513]
1.9结论
[0514]
usp7抑制剂先前已显示可调节癌蛋白mdm2的泛素化并抑制癌细胞增殖。据我们所知,本文报道的数据代表了usp7在tme重编程中的首次公开作用,靶向主要基质细胞群成纤维细胞。
[0515]
我们在与疾病相关的共培养物系统中进行了usp7抑制剂的表型筛选,并确定vegf是受usp7抑制调节的主要生物标志物。我们发现这种效应仅对激活的成纤维细胞特异,而不是癌细胞或永生化成纤维细胞。为了激活并获得肌成纤维细胞表型,可以用不同的分子刺激正常成纤维细胞,包括fgf-2、tgfβ或免疫细胞。在患者癌症相关成纤维细胞中观察到usp7抑制后分泌型vegf的减少,证实对肌成纤维细胞的强大作用,无论其激活方式如何(图1-7)。此外,vegf减少不仅限于分泌蛋白,而且在细胞内水平也观察到(图8)。
[0516]
通过crispr/cas9 usp7敲除证实usp7抑制对vegf调节的特异性,其中来自激活的成纤维细胞的分泌型vegf水平的降低与小分子抑制剂观察到的降低相当(图21)。此外,在原代成纤维细胞的细胞靶接合分析中,对usp7的高效力(ec
50
=11-24nm,图20)证实,usp7抑制剂的作用是通过直接usp7调节介导的。
[0517]
vegf基因表达的主要激活因子是hif-1α转录因子。多种刺激,如多个生长因子,可在常氧细胞中以hif-1依赖性方式诱导vegf表达。然而,由于泛素-蛋白酶体系统的快速降解,在大多数细胞类型中无法检测到hif-1α蛋白。相反,低氧诱导hif-1α蛋白的积累,从而诱导vegf蛋白的积累。事实上,低氧水平诱导成纤维细胞中的vegf分泌,如图9所示。我们确认激活的成纤维细胞中usp7抑制的作用机制是通过调节hif-1α蛋白。我们证实usp7抑制导致hif-1α半衰期减少和k48特异性多泛素化,从而导致其蛋白酶体介导的降解(图11和13)。低氧激活成纤维细胞的rna-seq显示,在usp7抑制后,除了vegf蛋白水平外,vegf mrna也降低(图12)。这可能表明hif1途径的反馈调节,支持usp7在tme中调节基本应激反应途径的关键功能。此外,途径分析的结果表明,hif-1α信号传导途径在响应usp7抑制时排名很高,参与hif-2α信号传导的已知基因受到调控(图14和15)。这些发现支持usp7在tme中直接调节hif-1α蛋白和hif1途径中的作用。
[0518]
我们针对对已建立的mdm2拮抗剂对usp7抑制剂(ad-04)观察效果进行了基准测试,结果表明,与ad-04相比,所有三种测试的mdm2拮抗剂均不调节激活的成纤维细胞中分泌型vegf蛋白水平(图17和18)。这些结果证明与mdm2化合物有很强的区别。我们还证明了与抗血管生成化合物贝伐单抗、抗vegf单克隆抗体的差异(图26)。
[0519]
成纤维细胞、免疫细胞、内皮细胞和癌细胞之间的串扰导致肿瘤细胞增殖和转移所需的生长因子、细胞因子、细胞外基质蛋白和基质降解酶金属蛋白酶的诱导。我们试图探索usp7抑制是否对tme内的细胞功能产生影响,如增殖、迁移和侵袭。ad-04对tme内任何细胞类型的增殖和迁移没有任何显著影响(图22和23),但显示出对成纤维细胞通过基底膜侵袭的强烈抑制(图24)。鉴于成纤维细胞具有降解ecm和促进癌细胞侵袭的能力,这些发现非常重要。已经报道了一种肿瘤侵袭机制,其中癌细胞通过整合素/纤维连接蛋白相互作用结合成纤维细胞来侵袭胶原基质。usp7抑制剂处理后抑制成纤维细胞侵袭的可能机制是通过观察到的在侵袭成纤维细胞中mmp7水平的降低(图25),因为mmp-7降解管状基底膜的主要组分,如弹性蛋白、iv型胶原和层粘连蛋白,以诱导上皮间充质细胞转化(emt)。成纤维细胞在tme重编程中的另一个作用是调节血管化过程。我们表明,usp7抑制防止体外成纤维细胞和内皮细胞共培养中的新生小管形成(图26)。然而,usp7抑制并不直接影响内皮细胞小管形成,因为在没有成纤维细胞的情况下,ad-04无法调节新生内皮细胞形成(图27),这表明usp7抑制剂ad-04仅靶向tme内的成纤维细胞群。已知mmp-2在体外小管形成中很重要。从成纤维细胞和内皮细胞共培养物中观察到的usp7抑制后mmp-2水平的降低指向小管形成抑制的可能作用机制。在结肠腺瘤和结肠癌组织样本中,mmp-2和vegf的mrna表达水平与hif-1α呈正相关,支持我们与usp7抑制相关的新发现。综上所述,这些结果清楚地表明,在usp7抑制后,成纤维细胞经历ecm重塑,从而重新编程tme。
[0520]
虽然usp7抑制不影响体外ct-26细胞系增殖,也不对分泌型vegf水平有显著影响(图29),但体内实验显示usp-7抑制剂介导ct-26同基因异种移植物中的肿瘤生长抑制(图30和37-39)。usp7抑制对体内肿瘤生长抑制的效果与索拉非尼阳性对照处理组相似(图
30)。在同基因肿瘤模型中,usp7抑制与免疫检查点抑制剂处理协同组合,可显著提高生存率(图39)。
[0521]
我们从小鼠血清样品中观察到循环vegf蛋白水平显著降低(图31),表明usp7抑制对vegf生长因子的整体全身水平具有强烈的总体效应。在用usp7抑制剂处理后,肿瘤内细胞毒性淋巴细胞数量的增加和巨噬细胞的减少进一步证实了tme通过浸润免疫细胞重编程(图32和40)。此外,对肿瘤样本的rna-seq分析证实,usp7抑制剂调节已知强烈影响体内tme的途径,包括hif-1α途径。
[0522]
总之,本文证明了usp7通过直接影响成纤维细胞中的vegf生长因子的水平和调节肿瘤免疫微环境来调节和重编程tme的全新作用模式。usp7在tme重编程中的作用与之前描述的usp7调节癌蛋白mdm2的作用无关。本文描述的是usp7在原代或癌相关成纤维细胞中的独特作用,而不是在tme中存在的癌细胞或其他原代细胞中观察到的作用机制。本文提供的数据支持了usp7抑制剂通过抑制成纤维细胞,尤其是caf中的usp7功能的新治疗策略。
[0523]
材料和方法
[0524]
细胞和培养条件
[0525]
所有原代细胞和细胞系均来自美国典型培养物保藏中心(atcc),经str分析(promega)验证,并使用mycoalert支原体检测(lonza;lt07-318)显示为无支原体。为了生长,细胞在含5%co2的潮湿大气中保持在37℃。ht29(结直肠)细胞在补充有10%(v/v)的fbs、1%(v/v)的青霉素-链霉素、1%(v/v)的l-谷氨酰胺的mccoy培养基5a中培养。lncap(前列腺)细胞在补充有10%(v/v)的fbs、1%(v/v)的青霉素-链霉素、1%(v/v)的l-谷氨酰胺的rpmi中培养。h1299(肺)和ct26(小鼠结肠癌)细胞在补充有10%(v/v)的fbs和1%(v/w)的青霉素-链霉素的rpmi中培养。mcf7(乳腺)细胞在补充有10%(v/v)的fbs、0.01mg/ml的人重组胰岛素和1%(v/w)的青霉素-链霉素的伊格尔氏极限必需培养基中培养。hdf在补充有成纤维细胞生长试剂盒-低血清的成纤维细胞基础培养基中培养(每种组分的最终浓度如下:l-谷氨酰胺7.5mm;rh fgf基础5ng/ml;rh胰岛素5μg/ml;氢化可的松1μg/ml;抗坏血酸50μg/ml;胎牛血清2%)。wi38、wi38-va13和imr-90在补充有10%(v/v)的fbs的伊格尔氏极限必需培养基中培养。huvec在涂有0.2%明胶的烧瓶上在补充有内皮细胞生长试剂盒-bbe的血管细胞基础培养基中培养(每种组分的最终浓度如下:牛脑提取物(bbe)0.2%;rh egf 5ng/ml;l-谷氨酰胺10mm;硫酸肝素0.75单位/ml;氢化可的松1μg/ml;抗坏血酸50μg/ml和胎牛血清:2%)。培养基和补充剂从life technologies和atcc购买,除非另有说明。
[0526]
靶接合分析
[0527]
ht-29、ct-26、imr-90和hdf细胞用溶媒(dmso)或usp7抑制剂处理2小时。将hdf置于低氧室中,而其他细胞保持在常氧状态。孵育后,细胞用1
×
pbs广泛洗涤三次,并在含有50mm的tris-hcl(ph7.4)、150mm的nacl、5mm的mgcl2、0.5mm的edta、0.5%的np40、10%的甘油、2mm的dtt的te裂解缓冲液中收获,在含50mm的tris-hcl(ph7.6)、5mm的mgcl2、250mm的蔗糖、0.5mm的edta和2mm的dtt的分析缓冲液中与ub-pa(终浓度为8μg/ml)孵育澄清的细胞裂解物(30μg)30分钟。通过加入lds样品缓冲液(life technologies)并加热至70℃终止反应。然后使用细胞信号传导抗-usp7 ab(#4833;1:1,000稀释度)通过蛋白质印迹分析样品。ec
50
值通过光密度测定分析确定。使用imagej软件对谱带强度进行量化,其中上谱带(usp7-ub)和下谱带(usp7)计算为相应dmso对照的百分比(-/ ub-pa),然后将值归一化为每个浓
度的上谱带和上谱带之和。
[0528]
细胞增殖分析
[0529]
细胞以96孔板格式接种(通常为4000个细胞/孔,并在24小时后在常氧或低氧条件下用浓度增加的化合物(100μm至1nm)处理,如图所示。在72小时后,使用synergy 4酶标仪(biotek)通过celltiter-glo评估细胞活力。使用graphpad prism(graphpad软件,inc,la jolla,ca;四参数logistic函数)推导分析和ec
50
值。数据以平均值
±
s.d.表示。(n≥3)。
[0530]
体外共培养小管形成分析和免疫染色
[0531]
将hdf接种在96孔板(2500个细胞/孔)中。一旦hdf形成单层,移除培养基并将huvec(2500个细胞/孔)接种在hdf顶部。为了测定对新血管形成的影响,培养物在接种后24小时用ad-04(10nm-1μm)、其非活性对映体、dmso和avastin处理。通过在处理前形成小管来分析对现有血管的影响。在孵育14天期间,每3天处理细胞,然后用pbs洗涤并在室温(rt)下用4%甲醛固定15分钟。用含有0.1%triton x-100的1
×
pbs在rt下渗透细胞5分钟,并在1%bsa/pbs中封闭30分钟。随后,将细胞与cd31抗体(thermo fisher,ma5-13188)1/50在1%bsa/0.1%吐温-20/pbs中在4℃ 1/50下孵育过夜,然后与二级alexa fluor 488f山羊抗小鼠(thermo fisher,a28175)在rt下以1/2000稀释度孵育3小时。免疫标记的样品用hoechst 33342核染料(thermo fisher,62249)以1/1000稀释度复染15分钟。用细胞内分析仪2000使用2x物镜观察小管。使用angiotool软件测量和量化每个成像场的小管长度(可在公共域中获取,网址为:https://ccrod.cancer.gov/confluence/display/rob2/downloads。
[0532]
小管形成分析
[0533]
预冷却的96孔板的每个孔涂有50μl的未聚合生长因子还原的(9.2mg/ml),并在37℃下在5%co2中中孵育2小时。用胰蛋白酶收获huvec,将1.5
×
104个细胞重新悬浮在100μl的完整内皮细胞生长培养基中。在将细胞铺板到包衣板上之前,用不同浓度的溶媒(dmso)、ad-04、ent-ad-04和avastin处理细胞。在37℃下和5%co2中孵育约6小时后,以4倍放大倍数拍摄每个板中心图像。
[0534]
泛素化分析
[0535]
细胞在低氧条件下孵育3小时,然后用10μm的蛋白酶体抑制剂mg132(sigma),1μm的ad-04或dmso处理1小时。细胞在含有50mm的tris-hcl、ph 7.5、150mm的nacl、1mm的edta、1%的np-40、10%的甘油、50mm的naf、5mm的焦磷酸钠、10mm的磷酸甘油、1mm的原钒酸钠、蛋白酶和磷酸酶抑制剂片剂和50μm的pr-619的缓冲液中裂解。通过在4℃下以16000
×
g离心15分钟预清除裂解物,并使用bca法测定蛋白质浓度。取20ul的样品作为输入。将含有0.5mg的总蛋白的预清除上清液添加到20μl的平衡的琼脂糖试管1或试管2珠(life sensors)中,并在摇杆平台上在4℃下孵育2小时。通过低速离心(1000-5000
×
g,4℃)5分钟收集珠粒,并取上清液作为ft。用1ml的tbs-t(20mm tris-hcl,ph 8.0,0.15m nacl,0.1%吐温-20)洗涤珠粒两次,并通过低速离心收集。最后,将珠粒重新悬浮在30μl的sds还原样品缓冲液中,并在95℃下煮沸10分钟。使用抗hif-1a抗体(d2u3t,兔mab#14179细胞信号传导)对样品进行蛋白质免疫印迹分析。
[0536]
环己酰亚胺追逐分析
[0537]
为了测定细胞中hif1α的半衰期,hdf细胞在低氧条件下孵育3小时,然后在环己酰亚胺(100μg/ml)存在下用dmso或1μm的ad-04处理以阻断新生蛋白质合成。收获细胞并在不同时间点裂解,并使用抗hif-1a抗体(d2u3t,兔mab#14179细胞信号传导)和作为加载对照的β-肌动蛋白(santa cruz:a5316;1:5000)进行蛋白质免疫印迹分析。
[0538]
mmp检测
[0539]
根据制造商的方案,使用luminex人细胞因子/趋化因子复合试剂盒(millipore,st.charles,mo,usa)一式三份地测定细胞培养上清液中mmp2和mmp7的浓度。
[0540]
使用酶联免疫吸附试验(elisa)检测vegf
[0541]
对于共培养实验,将5
×
104个癌细胞接种在0.5ml完全生长培养基中的transwell中并放置在1ml低血清生长培养基中在12孔中铺板的0.1
×
105成纤维细胞的顶部过夜。第二天,用pbs洗涤癌细胞,并用减少的(1%)血清生长培养基替换培养基。将0.5
×
106的pbmc铺板在成纤维细胞顶部上,孵育45分钟,并用cd3(1μg/ml终浓度)和cd28(5μg/ml终浓度)激活。随后,用溶媒(dmso)、ad-04和ent-ad-04在37℃下和5%co2中处理共培养物48小时。单一培养实验,将0.1
×
105个细胞接种在1ml完全生长培养基(癌细胞)或低血清生长培养基中(成纤维细胞)中的12孔中,并孵育过夜。第二天,用pbs洗涤癌细胞,并用减少的(1%)血清生长培养基替换培养基,然后在37℃下的5%co2中中在常氧或低氧条件下用溶媒(dmso)、ad-04、ent-ad-04、mdm-2拮抗剂nutlin-3a(tocris;#3984)、sar405838(medchemexpress;mi-773、#hy-17493)和rg7112(medchemexpress;35hy-10959)处理48小时。然后,收集细胞培养基,通过离心去除细胞碎片。根据制造商的说明,使用人/小鼠vegf免疫测定quantikine elisa试剂盒(r&d systems,minneapolis,mn,usa)测量细胞培养上清液中的vegf浓度。对于细胞内vegf检测,通过bca(pierce chemical,usa)测定总蛋白浓度,并使用上述相同试剂盒测定vegf水平。
[0542]
biomap表型筛选
[0543]
用原代人类细胞使用biomap系统。这些研究遵循美国卫生和公众服务部人体受试者条例(45cfr第46部分)下的受试者研究指南。根据供应商(lonza,inc.,allendale,nj)的建议,收集并培养来自3名供体的人新生儿包皮成纤维细胞(hdfn)。外周血单核细胞(pbmc)根据标准方法从正常人供体的血沉棕黄层制备。自身免疫hdfsag系统由原代人真皮成纤维细胞(hdf)与t细胞受体(sag)刺激的pbmc共培养组成,以模拟慢性t细胞激活和炎症。基质肿瘤学结直肠癌和非小细胞肺癌(nsclc)面板由癌细胞(ht29或h1299)、hdf和用sag刺激的pbmc组成。该模型捕捉肿瘤细胞、受刺激的免疫细胞和宿主基质网络之间的相互作用。用sag(20ng/ml)激活共培养物,并用溶媒和浓度为10、3.3、1.1和0.37μm的ad-04处理48小时。然后,使用elisa测量来自共培养物的上清液中的生物标志物,如下所示:mcp-1、vcam-1、胶原i、ip-10、mmp-1、sil-10、sil-17a、sil-17f、sil-2、sil-6、srb、stgfb、stnfa、svegf、il-8、mig、mcsf、upar、col-iii、ip-10、egfr、hgf、pal-1、pbmc细胞毒性、tpa、upa、s颗粒酶、spge2、sifg、sil-13、smdc、胶原iii、mmp-9、timp-2、ceacam5、角蛋白20。生物标志物水平以对数转化比表示。
[0544]
crispr/cas9 rnp敲除
[0545]
usp7特异性crrna和非特异性tracrrna在微量离心管中以等摩尔浓度混合,形成
tracrrna:crrna双链(引导rna)。样品在95℃下加热5分钟,并使其在室温下冷却。为了形成与引导rna偶联的重组cas9的核糖核蛋白(rnp)复合物,将cas9酶(21um终浓度)添加到tracrna:crrna双链中。将rnp复合物在室温下孵育20分钟。在电穿孔之前,通过胰蛋白酶化收获hdf并用pbs洗涤。将含有5
×
105个细胞的颗粒与94μl的核转染剂溶液(amaxa人真皮成纤维细胞核转染剂,lonza)混合,并将5μl的正确rnp或2μg的总pmaxgfp和1μl的alt-r cas9电穿孔促进剂(终浓度1μm)加入每个试管中,混合并转移到电穿孔液槽。随后,使用来自核转染装置(转染剂iib装置)的u-020程序对细胞进行核感染,并立即向细胞中添加500μl的预热培养基。细胞生长9天,使表型发育。
[0546]
迁移和侵袭分析
[0547]
96孔imagelock板包覆有0.1mg/ml的生长因子减少的并在37℃下孵育1小时。将细胞以10,000
–
40,000个细胞/孔的密度接种在100μl/孔中,并孵育过夜。第二天,使用伤口划痕器在所有孔中同时制造伤口。在创伤后,从每个孔中抽吸培养基,并用pbs洗涤细胞两次。对于侵袭,细胞以3mg/ml覆盖50μl的顶层,并在37℃下孵育30分钟。随后,向每个孔中加入100μl的含有溶媒(dmso)、ad-04、ent-ad-04和avastin的培养基。将细胞板放入incucyte活细胞分析系统中,每2小时使用10倍物镜对每个孔成像,共5天。使用incucyte刮伤方案分析图像,结果以伤口汇合百分比表示。
[0548]
肿瘤模型
[0549]
对于皮下肿瘤植入,在8-10周龄balb/c雌性小鼠的右侧皮下注射ct26细胞(200μl的rpmi中的1
×
106个细胞)。小鼠接受皮下输注溶媒、使用泵以30mg/kg/天和100mg/kg/天的剂量皮下输注ad-04,或以50mg/kg/天的剂量口服施用索拉非尼,每天施用一次,共十天。用数字卡尺每周测量两次肿瘤体积(以mm3计),并通过以下公式计算:体积=(宽度)2×
长度/2。每周测量两次体重。在植入后第10天,处死小鼠,收获肿瘤进行下游实验。
[0550]
对于免疫检查点抑制剂体内实验,在balb/c小鼠的右后侧皮下注射0.1ml的pbs中的5
×
105个活ct-26肿瘤细胞。每天给药adc-159(75mg/kg和100mg/kg),而每3天给药抗-pd-l1或抗-ctla4(10mg/kg)。使用卡钳每周测量三次肿瘤体积(mm3),使用以下等式:肿瘤体积(tv)=0.5((w)2×
l),其中w为最短肿瘤直径(宽度),且l为最长垂直直径(长度),以毫米计。在给药阶段期间的同一天测量体重。使用以下公式计算第13天的肿瘤生长抑制(tgi):tgi(%)=1-(tvt/tvv)*100%,其中tv
t
为治疗组第13天的肿瘤体积,且tvv为溶媒对照组第13天的肿瘤体积。使用单因素方差分析,然后dunnett多重比较检验对肿瘤体积进行绘图和统计分析。除第13天指定进行样品收集的五只动物外,其余动物在其单个肿瘤体积超过1800mm3时终止。tv不超过1800mm3的动物在第57天研究结束后停止样品收集,并在生存分析中被视为存活。使用经批准的人道方法对小鼠实施安乐死。所有涉及动物护理和使用的程序均由当地iacuc小组批准,并由训练有素的人员根据aaalac法规和良好兽医实践进行。
[0551]
流式细胞术实验。
[0552]
收获的肿瘤收集在hbss培养基中,切碎并在37℃的非酶细胞解离缓冲液中孵育30分钟,然后通过70μm过滤器进行机械解离。然后使用梯度富集活细胞。对所有细胞
悬浮液进行计数,并将100万活细胞接种在100μl的染色缓冲液中的96孔板中进行采集。使用小鼠fcr阻断试剂进行非特异性结合。viobility 405/452可固定染料(miltenyi biotec)用于评估细胞活力。加入针对cd45、cd335(nkp46)、foxp3 cd8a、cd4、cd3e、cd19、f4/80、cd11c、cd11b(miltenyi biotec)、ly6g和ly6c(biolegend)的抗体。用fortessa x20血细胞计数器(bd biosciences)分析染色细胞。
[0553]
组织学和免疫荧光
[0554]
将新收集的肿瘤组织(5只动物/组)置于10%nbf中,并在室温下固定24小时,然后修剪至不超过3-5mm的厚度。在用流动水冲洗后,将试样转移到真空组织处理器(histocore pearl,leica)进行脱水,然后使用组织嵌入中心(eg1150,leica)嵌入ffpe块中。ffpe块用手动旋转切片机(rm2235,leica)切片,4μm厚度/切片。将切片处理以进行苏木精和伊红染色(h&e)或进行免疫荧光(if)分析。对于if切片,用cd31(abcam)、ng2(sigma)特异性一级抗体染色,细胞核用dapi复染。所有染色的切片均使用pannoramic数字载玻片扫描仪进行40倍放大扫描(3dhistech,pannoram ic scan)。所有图像均经halotm平台分析肿瘤面积,并量化大的坏死面积。排除周围的非肿瘤组织。
[0555]
化合物合成
[0556]
缩写和首字母缩略词
[0557]
aq:水溶液;boc:叔丁氧基羰基;br:宽;dcm:二氯甲烷;d:双峰(光谱);dipea:二异丙基乙胺;dmf:n,n-二甲基甲酰胺;dmso:二甲基亚砜;etoac:乙酸乙酯;esi:电喷雾电离;h:小时;hatu:n-[(二甲基氨基)-1h-1,2,3-三唑并-[4,5-b]吡啶-1-基亚甲基]-n-甲基甲铵六氟磷酸盐n-氧化物;hplc:高压液相色谱法;lc:液相色谱法;lcms:液相色谱-质谱法;m:摩尔;m/z:质荷比;meoh:甲醇;min:分钟;ms:质谱;m:多重峰(光谱);nmr:核磁共振;ph:苯基;ppm:百万分之一:q:四重峰(光谱);r
t
:保留时间;rt:室温;s:单峰;tfa:三氟乙酸;t:三重峰;uv:紫外;v/v:每单位体积的体积。
[0558]
一般实验条件
[0559]
溶剂和试剂
[0560]
反应中使用的常见有机溶剂(如dmf、dcm和meoh)以无水形式从购买,装在sure/seal
tm
瓶中,并在氮气下进行适当处理。使用elga purelab option-q对水进行去离子。所有其他使用的溶剂(即,用于后处理程序和纯化)通常为hplc级,并从各种商业来源供应。除非另有说明,所有使用的起始材料均从商业供应商处购买,并按供货时状态使用。
[0561]
微波合成
[0562]
使用biotage initiator
tm eight instrument进行微波实验。该系统在60-250℃的温度范围和高达最大20巴的压力下提供良好的再现性和控制性。
[0563]
快速色谱法
[0564]
使用biotage-isola-four系统通过快速色谱法实现化合物纯化。除非另有说明,biotage kp-sil snap盒式柱(10-340g)或grace graceresolv盒式柱(4-330g)与所述溶剂体系和适当的溶剂梯度一起使用,取决于化合物极性。对于更强极性和碱性的化合物,使用biotage kp-nh snap盒式柱(11g)。
[0565]
nmr光谱学
[0566]
使用bruker ascend(500mhz)光谱仪在环境温度下记录1hnmr光谱。所有化学位移(δ)均以ppm表示。使用残留溶剂信号作为内标,并将特征溶剂峰校正为j.org.chem.,1997,62,p7512-7515中概述的参考数据;在其他情况下,nmr溶剂含有四甲基硅烷,其用作内标。
[0567]
液相色谱-质谱(lcms)
[0568]
使用以下方法进行液相色谱-质谱(lcms)实验以确定保留时间(r
t
)和相关质量离子:
[0569]
方法a:该系统由agilent technologies 6130四极杆质谱仪组成,与带有紫外二极管阵列检测器和自动取样器的agilent technologies 1290infinity lc系统相连。光谱仪由正离子和负离子模式运行的电喷雾电离源组成。使用以下条件对提交的每个样品进行lcms实验:lc柱:保持在40℃下的agilent eclipse plus c18 rrhd,1.8μm,50x 2.1mm。流动相:a)0.1%(v/v)甲酸的水溶液;b)0.1%(v/v)甲酸的乙腈溶液。
[0570][0571]
方法b:该系统由agilent technologies 6140单一四极杆质谱仪组成,与带有紫外二极管阵列检测器和自动取样器的agilent technologies 1290 infinity lc系统相连。光谱仪由以正离子和负离子模式工作的多模电离源(电喷雾和大气压化学电离)组成。使用以下条件对提交的每个样品进行lcms实验:lc柱:保持在40℃下的zorbax eclipse plus c18 rrhd,1.8μm,50
×
2.1mm。流动相:a)0.1%(v/v)甲酸的水溶液;b)0.1%(v/v)甲酸的乙腈溶液。
[0572]
[0573]
实施例1(adc-159):6-氯-7-(2,3-二氢苯并呋喃-5-基)-3-((4-羟基-1-(1-甲基环丙烷-1-羰基)哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮
[0574][0575]
步骤1:4,6-二氯-7-(2,3-二氢苯并呋喃-5-基)-7h-吡咯并[2,3-d]嘧啶:在空气下将4,6-二氯-7h-吡咯并[2,3-d]嘧啶(500mg,2.66mmol)[市售]、2,3-二氢苯并呋喃-5-硼酸(1.31g,7.98mmol)、1,10-菲咯啉(958mg,5.32mmol)和醋酸铜(ii)(966mg,5.32mmol)在dmf(30ml)的悬浮液在室温下搅拌过夜。所得混合物用etoac(100ml)稀释,并用1:1饱和盐水/水溶液(3
×
100ml)洗涤。水相用etoac(50ml)萃取。将合并的有机相干燥(相分离器)并真空浓缩。残余物通过快速色谱法纯化(0-40%etoac的环己烷),得到白色固体形式的标题化合物(392mg,46%)。lcms(方法a):r
t
=1.43min,m/z=306[m h]
。
[0576]
步骤2:6-氯-7-(2,3-二氢苯并呋喃-5-基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮:将4,6-二氯-7-(2,3-二氢苯并呋喃-5-基)-7h-吡咯并[2,3-d]嘧啶(392mg,1.24mmol)在2m hcl(水)溶液(2.5ml,4.96mmol)和1,4-二噁烷(5ml)中的悬浮液在120℃下在微波辐射下加热2小时,然后混合物真空浓缩,然后在真空炉中干燥过夜,得到粗标题化合物(358mg,84%),为粉色/棕色固体,无需进一步纯化即可使用。lcms(方法a):r
t
=0.94min,m/z=288[m h]
。
[0577]
步骤3:4-((6-氯-7-(2,3-二氢苯并呋喃-5-基)-4-氧代-4,7-二氢-3h-吡咯并[2,3-d]嘧啶-3-基)甲基)-4-羟基哌啶-1-羧酸叔丁酯:在80℃下,将6-氯-7-(2,3-二氢苯并呋喃-5-基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮(357mg,1.04mmol)、1-氧杂-6-氮杂螺[2.5]辛烷-6-羧酸叔丁酯(442mg,2.08mmol)[市售]和碳酸铯(372mg,1.14mmol)在dmf(8ml)的悬浮液加热4小时。冷却后,用etoac稀释反应混合物,并用盐水溶液洗涤两次。水相用etoac萃取两次,合并的有机物干燥(相分离器)并真空浓缩。通过快速色谱法(0-80%etoac的环己烷溶液)纯化残余物,得到标题化合物(169mg,33%)。lcms(方法a):r
t
=1.45min,m/z=501[m h]
。
[0578]
步骤4:6-氯-7-(2,3-二氢苯并呋喃-5-基)-3-((4-羟基哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮:4-((6-氯-7-(2,3-二氢苯并呋喃-5-基)-4-氧代-4,7-二氢-3h-吡咯并[2,3-d]嘧啶-3-基)甲基)-4-羟基哌啶-1-羧酸叔丁基酯(169mg,0.337mmol)在dcm(3ml)和tfa (1.5ml,19.5mmol)的溶液在室温下搅拌1小时。反应混合物加入到预处理的(使用1:4meoh/dcm)5g scx-2柱中。用20ml的1:4meoh/dcm洗涤结合产物,然后用20ml的1:4 7n nh3的meoh/dcm溶液洗脱。在减压下蒸发含有产物的馏分。残余物用乙腈/水冷冻干燥,得到淡白色固体形式的标题化合物(118mg,86%)。lcms(方法a):r
t
=0.76min,m/z=401[m h]
。
[0579]
步骤5:6-氯-7-(2,3-二氢苯并呋喃-5-基)-3-((4-羟基-1-(1-甲基环丙烷-1-羰基)哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮:向6-氯-7-(2,3-二氢苯并
呋喃-5-基)-3-((4-羟基哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮(59.6mg,0.149mmol)、1-甲基环丙烷-1-羧酸(14.9mg,0.149mmol)和hatu(67.8mg,0.178mmol)的无水dcm(1ml)的搅拌溶液加入dipea(78ul,0.446mmol)并将溶液搅拌30分钟。混合物用饱和碳酸氢钠(水)溶液(4ml)洗涤。分离水层并用dcm(2x 2ml)萃取。将合并的有机相干燥(分相器)并真空浓缩。将残余物通过快速色谱法(50-100%etoac的环己烷溶液;然后0-15%meoh的etoac溶液)纯化,并冷冻干燥,得到标题化合物(53.9mg,74%)。lcms(方法b):r
t
=1.08min,m/z=483[m h]
。1hnmr(500mhz,dmso-d6):δ8.07(s,1h),7.28(s,1h),7.12(d,1h),6.91(d,1h),6.76(s,1h),4.90(s,1h),4.64(t,2h),4.01(s,2h),3.98-3.90(m,2h),3.26-3.02(m,4h),1.58-1.45(m,2h),1.45-1.35(m,2h),1.21(s,3h),0.80-0.73(m,2h),0.55-0.49(m,2h).
[0580]
adc-159是特别有利的,因为它表现出与ad-04相当的有效和选择性usp7抑制作用(参见图33和35),但也表现出比ad-04更好的口服可用性。
[0581]
表1进一步表征了adc-159的特性:
[0582]
[0583][0584]
表1
[0585]
实施例2(adc-160):6-氯-3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-7-(2,3-二氢苯并呋喃-5-基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮
[0586][0587]
向6-氯-7-(2,3-二氢苯并呋喃-5-基)-3-((4-羟基哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮(26.0mg,0.065mmol)、2-环丙基噁唑-5-羧酸(10.9mg,0.071mmol)和hatu(29.6mg,0.078mmol)的无水dcm(1ml)的搅拌溶液中加入dipea (34ul,0.195mmol)并将溶液搅拌30分钟。混合物用饱和碳酸氢钠(水)溶液(1ml)洗涤。分离水层并用dcm(2x 1ml)萃取。将合并的有机相干燥(分相器)并真空浓缩。将残余物通过快速色谱法(0-100%etoac的环己烷溶液;然后0-5%meoh的etoac溶液)纯化,并冷冻干燥,得到标题化合物(27.0mg,76%)。lcms(方法b):r
t
=1.09min,m/z=536[m h]
。1hnmr(500mhz,dmso-d6):δ8.07(s,1h),7.49(s,1h),7.28(d,1h),7.12(dd,1h),6.91(d,1h),6.75(s,1h),4.98(s,1h),4.64(t,2h),4.17-3.88(m,4h),3.54-3.30(m,1h),3.28-3.04(m,3h),2.20-2.13(m,1h),1.66-1.54(m,2h),1.50-1.41(m,2h),1.12-1.06(m,2h),1.01-0.96(m,2h).
[0588]
实施例3(adc-198):6-氯-7-(2,3-二氢苯并呋喃-6-基)-3-[[4-羟基-1-(1-甲基
环丙烷羰基)-4-哌啶基]甲基]吡咯并[2,3-d]嘧啶-4-酮
[0589][0590]
步骤1:4,6-二氯-7-(2,3-二氢苯并呋喃-6-基)-7h-吡咯并[2,3-d]嘧啶:在空气下将4,6-二氯-7h-吡咯并[2,3-d]嘧啶(100mg,0.532mmol)、2,3-二氢苯并呋喃-6-硼酸(262mg,1.60mmol)、1,10-菲咯啉(192mg,1.06mmol)和醋酸铜(ii)(193mg,1.06mmol)在dmf(10ml)的悬浮液在室温下搅拌64小时。将所得混合物用水(30ml)稀释并用etoac(3x 50ml)萃取。将合并的有机相干燥(无水mgso4)并真空浓缩。残余物通过快速色谱法纯化(2-50%etoac的环己烷溶液),得到粘性透明油形式的标题化合物(160mg,88%)。lcms(方法a):r
t
=1.44min,m/z=306[m h]
。
[0591]
步骤2:6-氯-7-(2,3-二氢苯并呋喃-6-基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮:将4,6-二氯-7-(2,3-二氢苯并呋喃-6-基)-7h-吡咯并[2,3-d]嘧啶(160mg,0.471mmol)在2m hcl(水)溶液(1.3ml,2.59mmol)和1,4-二噁烷(3ml)中的悬浮液在120℃下在微波辐射下加热2小时,然后混合物真空浓缩,然后在真空炉中干燥过夜,得到深黄色固体形式的粗标题化合物(150mg,92%),无需进一步纯化即可使用。lcms(方法a):r
t
=0.96min,m/z=288[m h]
。
[0592]
步骤3:4-((6-氯-7-(2,3-二氢苯并呋喃-6-基)-4-氧代-4,7-二氢-3h-吡咯并[2,3-d]嘧啶-3-基)甲基)-4-羟基哌啶-1-羧酸叔丁酯:在80℃下,将6-氯-7-(2,3-二氢苯并呋喃-6-基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮(150mg,0.434mmol)、1-氧杂-6-氮杂螺[2.5]辛烷-6-羧酸叔丁酯(201mg,0.943mmol)和碳酸铯(169mg,0.519mmol)在dmf(4ml)的悬浮液加热4小时。冷却后,用etoac稀释反应混合物,并用盐水溶液洗涤两次。水相用etoac萃取两次,合并的有机相干燥(相分离器)并真空浓缩。通过快速色谱法(0-100%etoac的环己烷溶液)纯化残余物,得到标题化合物(80.4mg,37%)。lcms(方法a):r
t
=1.45min,m/z=501[m h]
。
[0593]
步骤4:6-氯-7-(2,3-二氢苯并呋喃-6-基)-3-((4-羟基哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮:4-((6-氯-7-(2,3-二氢苯并呋喃-6-基)-4-氧代-4,7-二氢-3h-吡咯并[2,3-d]嘧啶-3-基)甲基)-4-羟基哌啶-1-羧酸叔丁基酯(80mg,0.160mmol)在dcm(1.5ml)和tfa(0.75ml,9.73mmol)的溶液在室温下搅拌1小时。反应混合物加入到预处理的(使用1:4meoh/dcm)5g scx-2柱中。用20ml的1:4meoh/dcm洗涤结合产物,然后用20ml的1:4 7n nh3的meoh/dcm溶液洗脱。在减压下蒸发含有产物的馏分,得到淡白色固体形式的标题化合物(66.1mg,定量)。lcms(方法a):r
t
=0.63min,m/z=401[m h]
。
[0594]
步骤5:6-氯-7-(2,3-二氢苯并呋喃-6-基)-3-((4-羟基-1-(1-甲基环丙烷-1-羰基)哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮:向6-氯-7-(2,3-二氢苯并呋喃-6-基)-3-((4-羟基哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮(66.1mg,0.165mmol)、1-甲基环丙烷-1-羧酸(17.5mg,0.175mmol)和hatu(66.4mg,
0.175mmol)的无水dcm(2ml)的搅拌悬浮液加入dipea(122ul,0.699mmol)并将溶液搅拌1小时。反应混合物用dcm稀释,并用饱和碳酸氢钠(水)溶液洗涤两次。分离水相并用dcm萃取。将合并的有机相干燥(分相器)并真空浓缩。将残余物通过快速色谱法(0-100%etoac的环己烷溶液;然后0-5%meoh的etoac溶液)纯化,并冷冻干燥,在真空烘箱中干燥,得到标题化合物(62.4mg,78%)。lcms(方法a):r
t
=1.10min,m/z=483[m h]
。1hnmr(500mhz,dmso-d6):δ8.07(s,1h),7.42-7.37(m,1h),6.88-6.82(m,2h),6.77(s,1h),4.90(s,1h),4.68-4.61(m,2h),4.01(s,2h),3.98-3.90(m,2h),3.29-3.25(m,2h),3.24-3.05(br s,2h),1.56-1.46(m,2h),1.43-1.36(m,2h),1.21(s,3h),0.79-0.75(m,2h),0.54-0.49(m,2h).
[0595]
实施例4(adc-199):6-氯-3-((1-(2-环丙基噁唑-5-羰基)-4-羟基哌啶-4-基)甲基)-7-(2,3-二氢苯并呋喃-6-基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮
[0596][0597]
将2-环丙基噁唑-5-羧酸(15.2mg,0.100mmol)、hatu(45.5mg,0.120mmol)和dipea(52μl,0.299mmol)的无水dcm(2.5ml)的溶液搅拌5分钟,然后加入6-氯-7-(2,3-二氢苯并呋喃-6-基)-3-((4-羟基哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮(40.0mg,0.100mmol),溶液在室温下搅拌3小时。混合物用饱和碳酸氢钠(水)溶液稀释,并用dcm萃取三次。将合并的有机相干燥(分相器)并真空浓缩。将残余物通过快速色谱法(0-100%etoac的环己烷溶液;然后0-10%meoh的etoac溶液,kp-nh)纯化,并冷冻干燥,得到标题化合物(28.5mg,53%)。lcms(方法b):r
t
=1.10min,m/z=536[m h]
。1hnmr(500mhz,dmso-d6):δ8.08(s,1h),7.50(s,1h),7.42-7.39(m,1h),6.88-6.83(m,2h),6.77(s,1h),5.00(s,1h),4.65(t,2h),4.03(br s,4h),3.41(br s,1h),3.28(d,2h),3.18(br s,1h),2.17(tt,1h),1.60(t,2h),1.46(d,2h),1.12-1.07(m,2h),1.01-0.97(m,2h).
[0598]
实施例5(adx-556):7-(苯并[d][1,3]二氧杂环戊烯-5-基)-6-氯-3-((4-羟基-1-(1-甲基环丙烷-1-羰基)哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮
[0599][0600]
该化合物通过wo 2018/073602中描述的方法制备(实施例202)。
[0601]
实施例6:7-(1-苯并呋喃-5-基)-6-氯-3-((4-羟基-1-(1-甲基环丙烷-1-羰基)哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮
[0602][0603]
步骤1:7-(1-苯并呋喃-5-基)-4,6-二氯-7h-吡咯并[2,3-d]嘧啶:将4,6-二氯-7h-吡咯并[2,3-d]嘧啶(385mg,2.05mmol)、1-苯并呋喃-5-基硼酸(1g,6.17mmol)、硼酸(506mg,8.19mmol)、醋酸铜(ii)(744mg,4.10mmol)和1,10-菲咯啉(738mg,4.10mmol)在dmf(20.5ml)中的溶液在50℃下搅拌5天。在冷却至室温后,用10%氢氧化铵水溶液(40ml)稀释反应混合物,并使用biotage相分离器用dcm(3x 40ml)萃取。在减压下浓缩合并的有机相,并通过快速色谱法(0%、2%、4%然后6%的etoac的环己烷溶液(等度))纯化所得残余物,得到白色固体形式的标题化合物(31.3mg,5%)。lcms(方法b):r
t
=1.45min,m/z=304,306[m h]
。
[0604]
步骤2:7-(1-苯并呋喃-5-基)-6-氯-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮盐酸盐:7-(1-苯并呋喃-5-基)-4,6-二氯-7h-吡咯并[2,3-d]嘧啶(31.3mg,0.103mmol)在2m hcl(水溶液)(0.21ml,0.420mmol)和1.4-二噁烷(1.1ml)中的悬浮液在微波辐射120℃下加热2小时。反应混合物在减压下浓缩,残余物在50℃的真空烘箱中干燥,得到红色/棕色固体形式的标题化合物(34mg,102%)。将该材料无需进一步纯化而使用。lcms(方法b):r
t
=0.99min,m/z=286,288[m h]
。
[0605]
步骤3:7-(1-苯并呋喃-5-基)-6-氯-3-((4-羟基-1-(1-甲基环丙烷-1-羰基)哌啶-4-基)甲基)-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮:将7-(1-苯并呋喃-5-基)-6-氯-3,7-二氢-4h-吡咯并[2,3-d]嘧啶-4-酮盐酸盐(34mg,0.106mmol)、(1-甲基环丙基)(1-氧杂-6-氮杂螺[2.5]辛-6-基)甲酮(wo 2018/073602 a1,通过引用并入本文)(30.9mg,0.1583mmol)和碳酸铯(75.7mg,0.232mmol)在dmf(1.1ml)的悬浮液在80℃下搅拌18小时。冷却至室温后,用饱和氯化铵(15ml)水溶液稀释反应混合物,所得悬浮液使用biotage相分离器用dcm(3x 10ml)萃取。合并的有机相在减压下浓缩,残余物通过快速色谱法(0-100%etoac的环己烷溶液,然后0-10%meoh的etoac溶液)和制备型hplc纯化,冻干后得到白色固体形式的标题化合物(1.9mg,3.7%)。lcms(方法b):r
t
=1.13min,m/z=481,483[m h]
。1hnmr(500mhz,dmso-d6):8.16(d,j=2.2hz,1h),8.07(s,1h),7.80(d,j=8.8hz,1h),7.77(d,j=2.1hz,1h),7.36(dd,j=8.7,2.1hz,1h),7.08(dd,j=2.2,0.9hz,1h),6.82(s,1h),4.92(s,1h),4.02(s,2h),3.98-3.92(m,2h),3.17(br s,2h),1.58-1.46(m,2h),1.44-1.36(m,2h),1.21(s,3h),0.79-0.75(m,2h),0.54-0.50(m,2h).
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
