一种高折射率树枝状(甲基)丙烯酸酯类单体及其制备方法和应用
技术领域
1.本发明涉及光功能材料领域。更具体地,涉及一种高折射率树枝状(甲基)丙烯酸酯类单体及其制备方法和应用。
背景技术:
2.全息技术是一种可以记录光波的振幅和相位等全部信息的技术,其在高密度数据存储、全息光学元件(hoes)、全息防伪、全息传感、全息光刻等领域拥有独特的优势。全息图是三维干涉图案的图像,这些图案可以由两个光束在光敏介质中的交叉产生。体积全息记录相对于表面基存储格式的差别在于可以使用多路技术,以重叠方式在相同体积光敏介质中储存大量全息图,从而实现海量的信息存储。hoes作为一种衍射光学元件(doe)可以取代传统重而复杂的光学元件,尤其在头戴式显示器(hmd)中具有很好的应用潜力。但是,实现全息记录作为可行技术的阻碍之一是研发合适的全息记录介质材料。目前,典型的全息记录介质材料主要有卤化银乳胶、重铬酸盐明胶、光降解高分子材料、光导热塑料材料、光折变材料、光致聚合物材料、光致异构化材料和超表面材料这几种。其中,光致聚合物材料凭借其感光灵敏度高、分辨率高、制备简单、成本低、无需化学/热后处理等诸多优势被认为是未来全息记录介质材料的首选对象。
3.光致聚合物材料通常包含低折射率的基底树脂和高折射率的书写单体。在实际应用中,单体聚合会产生难以避免的体积收缩,对于厚膜型体全息记录介质而言,这将导致全息光栅形变,影响图像再现的清晰度等难题。因此,为了进一步改善全息记录介质的灵敏度、体积收缩率等全息性能,提供一种性能更优异的树枝状可聚合单体是十分必要的。
技术实现要素:
4.本发明的第一个目的在于提供一种树枝状(甲基)丙烯酸酯类单体。该树枝状(甲基)丙烯酸酯类单体具有低的体积收缩和较高的折射率。
5.本发明的第二个目的在于提供上述树枝状(甲基)丙烯酸酯类单体的制备方法。
6.本发明的第三个目的在于提供一种含有所述树枝状(甲基)丙烯酸酯类单体的光致聚合物型全息记录介质。
7.本发明的第四个目的在于提供一种含有上述光致聚合物型全息记录介质的全息光学元件。
8.本发明的第五个目的在于提供一种含有上述光致聚合物型全息记录介质的光致聚合物型全息存储光盘。
9.为达到上述目的,本发明采用下述技术方案:
10.第一方面,本发明提供一种树枝状(甲基)丙烯酸酯类单体,其结构式如g1-g9任一所示;
[0011][0012]
其中,在g1-g9的每一个中:r1为甲基或氢;r2为氢、溴、苯基、c1~c
10
烷基、和中的任一种;
[0013]
在中,a1为氢、苯基、c1~c
10
烷基、c1~c
10
烷硫基和c1~c
10
烷氧基中的任一种。
[0014]
优选地,r1为甲基或氢;r2为氢、br、苯基、c1~c4烷基、中的任一种,在中,a1各自代表苯基、c1~c4烷基。
[0015]
更优选地,在g4中,r2为br、苯基或c1~c4烷基。
[0016]
第二方面,本发明提供一种上述树枝状(甲基)丙烯酸酯类单体的制备方法。
[0017]
具体地,所述树枝状(甲基)丙烯酸酯类单体的结构式为g1时,包括以下步骤:
[0018]
s1、将式a所示化合物、[3,5-二(巯基)苯基]甲醇和naoh在溶剂中混合,加热至70~120℃,反应5~10h,提纯、分离得到式b所示化合物;
[0019]
s2、冰浴下,向含有式b所示化合物和三乙胺的溶液中滴加含丙烯酰氯或甲基丙烯
酰氯的溶剂,反应0.5~1h,提纯、分离即得;
[0020][0021]
优选地,步骤s1中,原料在溶剂中混合后,加热至80~100℃反应6-7h。
[0022]
当所述树枝状(甲基)丙烯酸酯类单体的结构式为g2时,包括以下步骤:
[0023]
s1、冰浴下,向含有式b所示化合物的溶液中滴加含pbr3的溶剂,反应0.5-6h,提纯、分离得式c所示化合物;
[0024]
s2、将式c所示化合物,[3,5-二(巯基)苯基]甲醇和naoh依次加入到溶剂中,加热至70~120℃,反应5~10h,提纯、分离得到式d所示化合物;
[0025]
s3、冰浴下,向含有式d所示化合物和三乙胺的混合液中滴加含丙烯酰氯或甲基丙烯酰氯的溶剂,反应0.5~1h,提纯、分离即得;
[0026][0027]
优选地,步骤s2中,原料在溶剂中混合后,加热至80~100℃反应6-7h。
[0028]
当所述树枝状(甲基)丙烯酸酯类单体的结构式为g3时,包括以下步骤:
[0029]
s1、冰浴下,向含有式d所示化合物的溶剂中滴加含pbr3的溶剂,反应0.5-6h,提纯、分离得式e所示化合物;
[0030]
s2、将式e所示化合物,[3,5-二(巯基)苯基]甲醇和naoh依次加入到溶剂中,加热至70~120℃,反应5~10h,提纯、分离得到式f所示化合物;
[0031]
s3、冰浴下,向含有式f所示化合物和三乙胺的混合液中滴加含丙烯酰氯或甲基丙烯酰氯的溶剂,反应0.5~1h,提纯、分离即得;
[0032][0033]
优选地,步骤s2中,原料在溶剂中混合后,加热至80~100℃反应6-7h。
[0034]
当所述树枝状(甲基)丙烯酸酯类单体的结构式为g4时,包括以下步骤:
[0035]
s1、将式g所述化合物和1,8-二氮杂环[5,4,0]十一烯-7加入到溶剂中,室温搅拌10~15min,然后滴加环氧氯丙烷,反应15~20h,提纯、分离得式h所述化合物;
[0036]
s2、冰浴下,向含有式h所述化合物和三乙胺的混合液中滴加含丙烯酰氯或甲基丙烯酰氯的溶剂,反应0.5~1h,提纯、分离即得;
[0037][0038]
当所述树枝状(甲基)丙烯酸酯类单体的结构式为g5时,包括如下步骤:
[0039]
s1、冰浴下,向含有式h所示化合物的溶液中滴加含pbr3的溶剂,反应0.5-6h,提纯、分离得式i所示化合物;
[0040]
s2、将式i所示化合物,[3,5-二(巯基)苯基]甲醇和naoh依次加入到溶剂中,加热至70~120℃,反应5~10h,提纯、分离得到式j所示化合物;
[0041]
s3、冰浴下,向含有式j所示化合物和三乙胺的混合液中滴加含丙烯酰氯或甲基丙烯酰氯的溶剂,反应0.5~1h,提纯、分离即得;
[0042][0043]
优选地,步骤s2中,原料在溶剂中混合后,加热至80~100℃反应6-7h。
[0044]
当所述树枝状(甲基)丙烯酸酯类单体的结构式为g6时,包括如下步骤:
[0045]
s1、冰浴下,向含有式j所示化合物的溶液中滴加含pbr3的溶剂,反应0.5-6h,提纯、分离得式k所示化合物;
[0046]
s2、将式k所示化合物,[3,5-二(巯基)苯基]甲醇和naoh依次加入到溶剂中,加热至70~120℃,反应5~10h,提纯、分离得到式l所示化合物;
[0047]
s3、冰浴下,向含有式l所示化合物和三乙胺的混合液中滴加含丙烯酰氯或甲基丙烯酰氯的溶剂,反应0.5~1h,提纯、即得;
[0048][0049]
优选地,步骤s2中,原料在溶剂中混合后,加热至80~100℃反应6-7h。
[0050]
当所述树枝状(甲基)丙烯酸酯类单体的结构式为g7时,包括如下步骤:
[0051]
s1、将式a所示化合物,2,3-二巯基丙醇和naoh依次加入到溶剂中,加热至70~120
℃,反应5~10h,提纯、分离得到式m所示化合物;
[0052]
s2、冰浴下,向含有式m所示化合物和三乙胺的溶液中滴加含丙烯酰氯或甲基丙烯酰氯的溶剂,反应0.5~1h,提纯、分离即得;
[0053][0054]
优选地,步骤s1中,原料在溶剂中混合后,加热至80~100℃反应6-7h。
[0055]
当所述树枝状(甲基)丙烯酸酯类单体的结构式为式g8时,包括如下步骤:
[0056]
s1、冰浴下,向含有式m所示化合物的溶液中滴加含pbr3的溶剂,反应0.5-6h,提纯、分离得式n所示化合物;
[0057]
s2、将式n所示化合物,[3,5-二(巯基)苯基]甲醇和naoh依次加入到溶剂中,加热至70~120℃,反应5~10h,提纯、分离得式p所示化合物;
[0058]
s3、冰浴下,向含有式p所示化合物和三乙胺的混合液中滴加含丙烯酰氯或甲基丙烯酰氯的溶剂,反应0.5~1h,提纯、分离即得;
[0059][0060]
优选地,步骤s2中,原料在溶剂中混合后,加热至80~100℃反应6-7h。
[0061]
当所述树枝状(甲基)丙烯酸酯类单体的结构式为g9时,包括如下步骤:
[0062]
s1、冰浴下,向含有式p所示化合物的溶液中滴加含pbr3的溶剂,反应0.5-6h,提纯、分离得式q所示化合物;
[0063]
s2、将式q所示化合物,[3,5-二(巯基)苯基]甲醇和naoh依次加入到溶剂中,加热至70~120℃,反应5~10h,提纯、分离得到式r所示化合物;
[0064]
s3、冰浴下,向含有式r所示化合物和三乙胺的混合液中滴加含丙烯酰氯或甲基丙烯酰氯的溶剂,反应0.5~1h,提纯、分离即得;
[0065][0066]
优选地,步骤s2中,原料在溶剂中混合后,加热至80~100℃反应6-7h。
[0067]
优选地,在g1-g9的制备方法中,所述溶剂包括但不限于乙醇,石油醚,二氯甲烷,三氯甲烷,乙酸乙酯,四氢呋喃,甲苯,乙腈,n,n-二甲基甲酰胺或二甲基亚砜中的一种或多种。
[0068]
可以理解,在g1-g9的制备方法中,选择添加丙烯酰氯或甲基丙烯酰,对应的结果是g1-g9结构式中r1为氢或甲基。
[0069]
优选地,在上述方法中,所述[3,5-二(巯基)苯基]甲醇、式a所示化合物和naoh的摩尔比为1:2~4:2~4;
[0070]
所述[3,5-二(巯基)苯基]甲醇、式c所示化合物和naoh的摩尔比为1:2~4:2~4;
[0071]
所述[3,5-二(巯基)苯基]甲醇、式e所示化合物和naoh的摩尔比为1:2~4:2~4;
[0072]
所述式g所示化合物、环氧氯丙烷和1,8-二氮杂环[5,4,0]十一烯的摩尔比为1:0.4~0.5:1~1.2;
[0073]
所述[3,5-二(巯基)苯基]甲醇、式i所示化合物和naoh的摩尔比为1:2~4:2~4;
[0074]
所述[3,5-二(巯基)苯基]甲醇、式k所示化合物和naoh的摩尔比为1:2~4:2~4;
[0075]
所述2,3-二巯基丙醇、式a所示化合物和naoh的摩尔比为1:2~4:2~4;
[0076]
所述[3,5-二(巯基)苯基]甲醇、式n所示化合物和naoh的摩尔比为1:2~4:2~4;
[0077]
所述[3,5-二(巯基)苯基]甲醇、式q所示化合物和naoh的摩尔比为1:2~4:2~4;所述式b所示化合物、丙烯酰氯或甲基丙烯酰氯、三乙胺的摩尔比为1:1~2:1~5;
[0078]
所述式d所示化合物、丙烯酰氯或甲基丙烯酰氯、三乙胺的摩尔比为1:1~2:1~5;
[0079]
所述式f所示化合物、丙烯酰氯或甲基丙烯酰氯、三乙胺的摩尔比为1:1~2:1~5;
[0080]
所述式h所示化合物、丙烯酰氯或甲基丙烯酰氯、三乙胺的摩尔比为1:1~2:1~5;
[0081]
所述式j所示化合物、丙烯酰氯或甲基丙烯酰氯、三乙胺的摩尔比为1:1~2:1~5;
[0082]
所述式l所示化合物、丙烯酰氯或甲基丙烯酰氯、三乙胺的摩尔比为1:1~2:1~5;
[0083]
所述式m所示化合物、丙烯酰氯或甲基丙烯酰氯、三乙胺的摩尔比为1:1~2:1~5;
[0084]
所述式p所示化合物、丙烯酰氯或甲基丙烯酰氯、三乙胺的摩尔比为1:1~2:1~5;
[0085]
所述式r所示化合物、丙烯酰氯或甲基丙烯酰氯、三乙胺的摩尔比为1:1~2:1~5。
[0086]
所述式b所示化合物和pbr3的摩尔比为1:1~4;
[0087]
所述式d所示化合物和pbr3的摩尔比为1:1~4;
[0088]
所述式h所示化合物和pbr3的摩尔比为1:1~4;
[0089]
所述式j所示化合物和pbr3的摩尔比为1:1~4;
[0090]
所述式m所示化合物和pbr3的摩尔比为1:1~4;
[0091]
所述式p所示化合物和pbr3的摩尔比为1:1~4。
[0092]
第三方面,本发明提供一种光致聚合物型全息记录介质,其原料包含下述组分a)-组分h);
[0093]
组分a)具有多个异氰酸酯反应性官能团的化合物;
[0094]
组分b)多异氰酸酯基化合物;
[0095]
组分c)树枝状(甲基)丙烯酸酯类单体;
[0096]
组分d)其他可聚合单体;
[0097]
组分e)光引发剂;
[0098]
组分f)链转移剂;
[0099]
组分g)任选地催化剂;
[0100]
组分h)任选地添加剂;
[0101]
其中,所述树枝状(甲基)丙烯酸酯类单体为权利要求1所述的g1-g9中的任意一种或几种的组合。
[0102]
优选地,以光致聚合物型全息记录介质的总质量计,所述组分c)的含量为0.1~45%;更优选为5~35%。
[0103]
优选地,所述组分a)、组分b)、组分c)、组分d)、组分e)、组分f)、组分g)和组分h)的质量比为20~70:0.1~50:0.1~45:0.1~60:0.1~4:0.1~3:0.1~3:0.1~7。
[0104]
优选地,所述光致聚合物型全息记录介质还包括:组分i)光敏剂;优选地,所述组分i)与组分f)的质量比为0.001~1:0.1~3。
[0105]
优选地,所述光敏剂为光照下具有较高电子转移效率的染料,其中,通过调控光敏剂种类可实现不同宽波段响应。但是当本发明光致聚合物型全息记录介质原料中选用了适配波长的光引发剂时,光敏剂可以不添加。
[0106]
优选地,所述光敏剂包括但不限于菁类染料、荧光素类染料、香豆素酮类染料、含氮芳杂环化合物、芳香族胺类化合物、苄叉环烷烃酮类化合物或这些化合物的任意比例混合物。例如包括新亚甲基蓝、硫堇、碱性黄、氯化频那氰醇、罗丹明6g、棓花青、乙基紫、维多利亚蓝r、天青石蓝、亚甲基蓝、astrazon orange g、darrow红、吡咯红y、碱性红29、喹哪啶红、结晶紫、乙基紫、亮绿、pyri11lium i、天青a、结晶紫白腈、孔雀绿白腈等中的一种或多种。
[0107]
优选地,所述添加剂包括消泡剂、流平剂和增塑剂中的一种或多种。
[0108]
优选地,所述具有多个异氰酸酯反应性官能团的化合物中,异氰酸酯反应性官能团为羟基;更优选为低折射率并带有两个及以上羟基管能团化合物;更优选为:四甘醇、三羟甲基乙烷、甘油、三乙醇胺、分子量为200~2000的聚酯多元醇、聚碳酸酯多元醇、聚醚多元醇。
[0109]
优选地,所述多异氰酸酯基化合物为低折射率并带有两个及以上异氰酸酯基团的化合物;更优选为六亚甲基二异氰酸酯、三甲基六亚甲基二异氰酸酯、(2,4,6-三氧代三嗪-1,3,5(2h,4h,6h)-三基)三(六亚甲基)异氰酸酯、丁烷-1,4-二异氰酸酯、异佛尔酮二异氰酸酯、二环己基甲烷二异氰酸酯。
[0110]
优选地,所述其他可聚合单体选自烯基萘类化合物、烯基蒽类化合物、烯基苯类化合物、丙烯酸类化合物、甲基丙烯酸类化合物、丙烯酸酯类化合物、甲基丙烯酸酯类化合物、n-乙烯基吡咯、n-乙烯基咔唑、n-乙烯基咪唑、n-乙烯基吲哚、n-乙烯基吡咯烷酮、反式n-3-炔丁烯基咔唑中的至少一种。
[0111]
更优选地,所述烯基苯类化合物可以选自苯乙烯、2-氯代苯乙烯、2-溴代苯乙烯、3-氯代苯乙烯、3-溴代苯乙烯、4-氯代苯乙烯、4-溴代苯乙烯、对-(氯代甲基)苯乙烯、对-(溴代甲基)苯乙烯等。
[0112]
所述甲基丙烯酸类化合物可以为甲基丙烯酸及其衍生物;示例性地,丙烯酸酯类化合物可以选自丙烯酸五溴苯基酯、丙烯酸五氯苯基酯、丙烯酸苯氧乙基酯、丙烯酸五溴苄基酯、丙烯酸2-萘酯、丙烯酸1,4-二(2-硫代萘基)2-丁基酯、丙烯酸苯氧基乙氧基乙酯、双酚a二丙烯酸酯、四溴双酚a二丙烯酸酯、2-苯氧基乙基丙烯酸酯、苄基丙烯酸酯、丙烯酸对
氯苯基酯、丙烯酸2,4,6-三氯苯基酯、丙烯酸对溴苯基酯、丙烯酸2,4,6-三溴苯基酯、丙烷-2,2-二基双[(2,6-二溴-4,1-亚苯基)氧(2-{[3,3,3-三(4-氯苯基)丙酰基]氧}丙烷-3,1-二基)氧乙烷-2,1-二基]二丙烯酸酯等。
[0113]
所述甲基丙烯酸酯类化合物可以选自2-苯氧基乙基甲基丙烯酸酯、苄基甲基丙烯酸酯、甲基丙烯酸对溴苯基酯、甲基丙烯酸对氯苯基酯、甲基丙烯酸2,4,6-三氯苯基酯、甲基丙烯酸五溴苯基酯、甲基丙烯酸五氯苯基酯、甲基丙烯酸苯氧乙基酯、甲基丙烯酸苯氧基乙氧基乙酯、甲基丙烯酸1,4-二(2-硫代萘基)2-丁基酯、甲基丙烯酸五溴苄基酯、甲基丙烯酸2-萘酯、双酚a二甲基丙烯酸酯、四溴双酚a二甲基丙烯酸酯等。
[0114]
所述乙烯基蒽类化合物可以选自2-乙烯基蒽、9-乙烯基蒽等。
[0115]
所述乙烯基萘类化合物可以选自1-乙烯基萘、2-乙烯基萘等。
[0116]
优选地,所述光引发剂是能够被光化辐射激活并引发相应的可聚合基团发生聚合反应的引发剂。包括但不限于芳香酮类化合物、安息香及其衍生物、苯偶酰缩酮、酰基膦氧化物、芳基硼酸铵、铬盐、芳基重氮盐、鎓盐、有机金属化合物或这些化合物的任意比例混合物。
[0117]
优选地,所述催化剂为叔胺类催化剂和有机金属类催化剂,包括但不限于三亚乙基二胺、双(二甲氨基乙基)醚、二甲基乙醇胺、2-(2-二甲氨基-乙氧基)乙醇、三甲基羟乙基丙二胺、n,n-双(二甲胺丙基)异丙醇胺、二月桂酸二丁基锡、辛酸亚锡、羧酸钾类催化剂和羧酸铋类催化剂。
[0118]
优选地,所述消泡剂为有机硅消泡剂,例如毕克公司生产的byk-011、byk-012、byk-014、byk-023、byk-051n、byk-085、byk-1610、byk-1707、byk-1740、byk-1760,道康宁公司生产的dc65、afe-7820或这些消泡剂的任意比例混合物。
[0119]
优选地,所述流平剂为有机硅表面助剂,例如毕克公司生产的byk-302、byk-306、byk-307、byk-327、byk-329、byk-333、byk-356、byk-358、byk-378、byk-3455、byk-3566或这些表面助剂的任意比例混合物。
[0120]
优选地,所述增塑剂为甲苯、二甲苯、二甲基甲酰胺、二甲基乙酰胺、甘油、邻苯二甲酸酯或这些化合物的任意比例混合物。
[0121]
优选地,所述除水剂包括但不限于对甲基苯磺酰异氰酸酯、原甲酸三乙酯、广州优润合成材料有限公司cuwr-wb20除水剂、安乡艾利特化工有限公司alt-201除水剂、上海鲁尔化工贸易有限公司pcci除水剂等。
[0122]
第四方面,本发明提供一种全息光学元件,该全息光学元件的原料包括上述光致聚合物型全息记录介质。
[0123]
第五方面,本发明提供一种光致聚合物型全息存储光盘,该光致聚合物型全息存储光盘的原料包括上述光致聚合物型全息记录介质。
[0124]
示例性地,所述光致聚合物型全息存储光盘的制备参照专利cn200910237040.7。
[0125]
另外,如无特殊说明,本发明中所用原料均可通过市售商购获得,本发明所记载的任何范围包括端值以及端值之间的任何数值以及端值或者端值之间的任意数值所构成的任意子范围。所述百分比如无特殊说明均为质量百分比,所述溶液若无特殊说明均为水溶液。
[0126]
本发明的有益效果如下:
[0127]
本发明提供的树枝状(甲基)丙烯酸酯类单体具有折射率高、体积收缩低的优势,将其引入光致聚合物型全息记录介质中,可以获得兼具高灵敏度、高衍射效率、高折射率调制度和低收缩的全息记录介质。
[0128]
本发明提供的光致聚合物型全息记录介质性能稳定,在避光条件下可长期保存,曝光后可实现信息的高质量存储与再现,在高密度光存储和全息光学元件等领域具有广泛应用前景。
附图说明
[0129]
下面结合附图对本发明的具体实施方式作进一步详细的说明。
[0130]
图1示出实施例7以及对比例1的0.005mm厚光致聚合物型全息记录介质的532nm激光曝光特性曲线对比图。
[0131]
图2示出实施例8以及对比例2的0.5mm厚光致聚合物型全息记录介质的405nm激光曝光特性曲线对比图。
[0132]
图3示出实施例9以及对比例3的0.05mm厚光致聚合物型全息记录介质的633nm激光曝光特性曲线对比图。
[0133]
图4示出实施例7的光致聚合物型全息记录介质在不同空间频率下的曝光特性曲线对比图。
[0134]
图5示出实施例10~13的光致聚合物型全息记录介质的532nm激光曝光特性曲线对比图。
[0135]
图6示出实施例8的光致聚合物型全息记录介质在空间频率2600lines/mm曝光下全息光栅bragg选择角曲线。
[0136]
图7示出实施例9的光致聚合物型全息记录介质在空间频率2600lines/mm曝光下全息光栅bragg选择角曲线。
[0137]
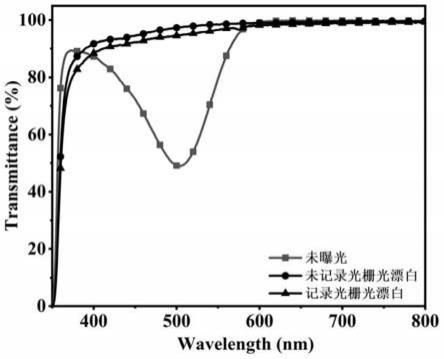
图8示出由实施例7的光致聚合物型全息记录介质制备的全息光栅、透明全息光栅的紫外可见透过率曲线对比图。
具体实施方式
[0138]
下面对本发明的实施方式进行详细说明。以下所例示出的物质、方法等为本发明的实施方式的一例(代表例),只要不脱离其要点,本发明并不限定于这些内容。
[0139]
实施例1
[0140]
树枝状(甲基)丙烯酸酯类单体g1、g2、g3的合成
[0141]
(1)将化合物1(40mmol)、化合物2(100mmol)和naoh(150mmol)溶于乙醇和水的混合溶液中,加热至90℃反应6h,反应完成后用二氯甲烷萃取,合并的有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为化合物3,产率约90%。
[0142][0143]
(2)冰浴下化合物3(20mmol)和三乙胺(40mmol)溶于50ml二氯甲烷中,搅拌10min
后将丙烯酰氯(30mmol)溶于30ml二氯甲烷,在0℃下滴加到化合物3和三乙胺的混合溶液中。反应完全后,滴加稀盐酸,除去过量的丙烯酰氯,依次使用饱和nacl溶液、饱和nahco3溶液和去离子水洗涤,有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为g1,产率约95%。表征数据如下:1h nmr(400mhz,cdcl3)δ7.23(d,4h),7.07
–
6.99(m,7h),6.41(d,1h),6.12(dd,1h),5.83(d,1h),5.12(s,2h),4.33(s,4h),2.38(s,6h).
13
c nmr(101mhz,cdcl3)δ166.5,141.6,140.9,136.8,133.5,130.0,129.8,128.7,127.9,121.1,118.6,65.1,40.0,14.8.
[0144][0145]
(3)冰浴下将化合物3(20mmol)溶于150ml二氯甲烷中,将pbr3(20mmol)溶于50ml二氯甲烷,在0℃下滴加到上述混合溶液中,反应3h后,滴加饱和nahco3溶液除去未反应的pbr3,依次用nahco3溶液和水洗涤,有机相经硫酸钠干燥后,旋蒸除去溶剂,柱层析分离得无色油状液体为化合物4,产率约93%。
[0146][0147]
(4)将化合物5(40mmol)、化合物4(100mmol)和naoh(150mmol)溶于乙醇和水的混合溶液中,加热至90℃反应6h,反应完成后用二氯甲烷萃取,合并的有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为化合物6,产率约90%。
[0148][0149]
(5)冰浴下化合物6(20mmol)和三乙胺(40mmol)溶于50ml二氯甲烷中,搅拌10min后将甲基丙烯酰氯(30mmol)溶于30ml二氯甲烷,在0℃下滴加到化合物6和三乙胺的混合溶液中。反应完全后,滴加稀盐酸,除去过量的甲基丙烯酰氯,依次使用饱和nacl溶液、饱和nahco3溶液和去离子水洗涤,有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为g2,产率约95%。表征数据如下:1h nmr(400mhz,cdcl3)δ7.23(d,8h),7.07
–
6.99(m,13h),6.87(s,4h),6.48(d,1h),6.40(d,1h),5.12(s,2h),4.33(s,12h),2.38(s,12h),2.01(s,3h).
13
c nmr(101mhz,cdcl3)δ167.2,141.6,140.9,140.0,137.8,136.8,136.6,133.5,128.7,127.9,123.7,121.1,120.6,119.2,118.6,65.4,40.0,39.2,17.9,14.8.
[0150][0151]
(6)冰浴下将化合物6(20mmol)溶于150ml二氯甲烷中,将pbr3(20mmol)溶于50ml二氯甲烷,在0℃下滴加到上述混合溶液中,反应3h后,滴加饱和nahco3溶液除去未反应的pbr3,依次用nahco3溶液和水洗涤,有机相经硫酸钠干燥后,旋蒸除去溶剂,柱层析分离得无色油状液体为化合物7,产率约93%。
[0152][0153]
(7)将化合物5(40mmol)、化合物7(100mmol)和naoh(150mmol)溶于乙醇和水的混合溶液中,加热至90℃反应6h,反应完成后用二氯甲烷萃取,合并的有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为化合物8,产率约90%。
[0154][0155]
(8)冰浴下化合物8(20mmol)和三乙胺(40mmol)溶于50ml二氯甲烷中,搅拌10min后将丙烯酰氯(30mmol)溶于30ml二氯甲烷,在0℃下滴加到化合物8和三乙胺的混合溶液中。反应完全后,滴加稀盐酸,除去过量的丙烯酰氯,依次使用饱和nacl溶液、饱和nahco3溶液和去离子水洗涤,有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为g3,产率约95%。表征数据如下:1h nmr(400mhz,cdcl3)δ7.23(d,16h),7.07
–
6.99(m,25h),6.87(s,12h),6.41(d,1h),6.12(dd,1h),5.83(d,1h),5.12(s,2h),4.33(s,28h),2.38(s,24h).
13
c nmr(101mhz,cdcl3)δ166.6,148.9,141.6,140.9,140.0,136.8,136.6,133.5,130.0,129.8,128.7,127.9,123.7,121.1,120.6,119.2,118.6,116.1,105.7,65.4,40.0,39.2,36.8,16.2,14.8.
[0156][0157]
实施例2
[0158]
树枝状(甲基)丙烯酸酯类单体g4、g5、g6的合成
[0159]
(1)将化合物9(20mmol)和1,8-二氮杂环[5,4,0]十一烯-7(20mmol)溶于100ml甲苯中,室温搅拌10~15min,然后滴加环氧氯丙烷(9mmol),室温下反应15~20h,反应结束后旋蒸除去溶剂,再将浓缩液用二氯甲烷溶解,并依次用1mol/l的稀盐酸、饱和nacl溶液、去离子水洗涤,合并的有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得化合物10,产率约90%。
[0160][0161]
(2)将化合物9(10mmol)和三乙胺(40mmol)溶于100ml二氯甲烷,冰浴条件下滴加含丙烯酰氯(15mmol)的二氯甲烷溶剂,反应0.5~1h,反应结束后滴加稀盐酸,依次用nacl溶液,nahco3溶液和水洗涤,有机相经硫酸钠干燥后,旋蒸除去溶剂,柱层析分离得g4。表征数据如下:1h nmr(400mhz,cdcl3)δ7.60(d,4h),7.43(d,4h),6.41(d,1h),6.12(dd,1h),5.83(d,1h),5.27(m,1h),3.01(d,4h).
13
c nmr(101mhz,cdcl3)δ165.2,135.4,131.8,131.3,129.0,128.2,119.5,78.9,40.9.
[0162][0163]
(3)冰浴下,将化合物9(10mmol)溶于150ml二氯甲烷中,将pbr3(10mmol)溶于50ml二氯甲烷,冰浴下滴加到上述混合溶液中,反应3h后,滴加饱和nahco3溶液除去未反应的pbr3,依次用nahco3溶液和水洗涤,有机相经硫酸钠干燥后,旋蒸除去溶剂,柱层析分离得化合物10,产率约93%。
[0164][0165]
(4)将化合物5(5mmol)、化合物10(12mmol)和naoh(20mmol)溶于乙醇和水的混合溶液中,加热至90℃反应6h,反应完成后用二氯甲烷萃取,合并的有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得化合物11,产率约90%。
[0166][0167]
(5)冰浴下,化合物11(20mmol)和三乙胺(40mmol)溶于50ml二氯甲烷中,搅拌10min后将甲基丙烯酰氯(30mmol)溶于30ml二氯甲烷,在0℃下滴加到化合物11和三乙胺的混合溶液中。反应完全后,滴加稀盐酸,除去过量的甲基丙烯酰氯,依次使用饱和nacl溶液、饱和nahco3溶液和去离子水洗涤,有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得g5,产率约95%。表征数据如下:1h nmr(400mhz,cdcl3)δ7.60(d,8h),7.43(d,8h),7.07(s,1h),6.99(s,2h),6.48(d,1h),6.40(d,1h),5.12(s,2h),3.49(m,2h),3.13(d,8h),2.01(s,3h).
13
c nmr(101mhz,cdcl3)δ167.2,141.6,137.8,136.8,135.4,131.8,129.0,123.7,121.1,119.5,118.6,65.4,47.3,40.4,17.9.
[0168][0169]
(6)冰浴下,将化合物11(10mmol)溶于150ml二氯甲烷中,将pbr3(10mmol)溶于50ml二氯甲烷,冰浴下滴加到上述混合溶液中,反应3h后,滴加饱和nahco3溶液除去未反应的pbr3,依次用nahco3溶液和水洗涤,有机相经硫酸钠干燥后,旋蒸除去溶剂,柱层析分离得12,产率约93%。
[0170][0171]
(7)将化合物5(5mmol)、化合物12(12mmol)和naoh(20mmol)溶于乙醇和水的混合溶液中,加热至90℃反应6h,反应完成后用二氯甲烷萃取,合并的有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得13,产率约90%。
[0172]
[0173]
(8)冰浴下,化合物13(20mmol)和三乙胺(40mmol)溶于50ml二氯甲烷中,搅拌10min后将丙烯酰氯(30mmol)溶于30ml二氯甲烷,在0℃下滴加到化合物13和三乙胺的混合溶液中。反应完全后,滴加稀盐酸,除去过量的丙烯酰氯,依次使用饱和nacl溶液、饱和nahco3溶液和去离子水洗涤,有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得g6,产率约95%。表征数据如下:1h nmr(400mhz,cdcl3)δ7.60(d,16h),7.43(d,16h),7.07(s,1h),7.00(s,2h),6.87(s,4h),6.41(d,1h),6.12(dd,1h),5.83(d,1h),5.12(s,2h),4.33(s,4h),3.49(m,4h),3.13(d,16h).
13
c nmr(101mhz,cdcl3)δ166.5,141.6,136.8,136.6,135.4,131.8,129.8,129.0,123.7,121.1,120.6,119.5,119.2,118.6,65.1,47.3,40.4,39.2.
[0174][0175]
实施例3
[0176]
树枝状(甲基)丙烯酸酯类单体g7、g8、g9的合成
[0177]
(1)将化合物14(40mmol)、化合物15(100mmol)和naoh(150mmol)溶于乙醇和水的混合溶液中,加热至90℃反应6h,反应完成后用二氯甲烷萃取,合并的有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为化合物16,产率约90%。
[0178][0179]
(2)冰浴下,化合物16(20mmol)和三乙胺(40mmol)溶于50ml二氯甲烷中,搅拌10min后将丙烯酰氯(30mmol)溶于30ml二氯甲烷,在0℃下滴加到化合物16和三乙胺的混合溶液中。反应完全后,滴加稀盐酸,除去过量的丙烯酰氯,依次使用饱和nacl溶液、饱和nahco3溶液和去离子水洗涤,有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为g7,产率约95%。表征数据如下:1h nmr(400mhz,cdcl3)δ7.40(d,4h),7.29
–
7.25(m,6h),6.41(d,1h),6.12(dd,1h),5.83(d,1h),4.58
–
4.35(m,2h),3.70(s,4h),3.40(m,1h),2.88
–
2.62(m,2h).
13
c nmr(101mhz,cdcl3)δ166.5,138.8,131.3,128.7,128.6,127.1,67.5,43.3,36.4,35.6,35.4.
[0180][0181]
(3)冰浴下,将化合物16(20mmol)溶于150ml二氯甲烷中,将pbr3(20mmol)溶于50ml二氯甲烷,在0℃下滴加到上述混合溶液中,反应3h后,滴加饱和nahco3溶液除去未反应的pbr3,依次用nahco3溶液和水洗涤,有机相经硫酸钠干燥后,旋蒸除去溶剂,柱层析分离
得无色油状液体为化合物17,产率约93%。
[0182][0183]
(4)将化合物5(40mmol)、化合物17(100mmol)和naoh(150mmol)溶于乙醇和水的混合溶液中,加热至90℃反应6h,反应完成后用二氯甲烷萃取,合并的有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为化合物18,产率约90%。
[0184][0185]
(5)冰浴下,化合物18(20mmol)和三乙胺(40mmol)溶于50ml二氯甲烷中,搅拌10min后将甲基丙烯酰氯(30mmol)溶于30ml二氯甲烷,在0℃下滴加到化合物18和三乙胺的混合溶液中。反应完全后,滴加稀盐酸,除去过量的甲基丙烯酰氯,依次使用饱和nacl溶液、饱和nahco3溶液和去离子水洗涤,有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为g8,产率约95%。表征数据如下:1h nmr(400mhz,cdcl3)δ7.40(d,8h),7.29
–
7.25(m,12h),7.07(s,1h),6.99(s,2h),6.48(d,1h),6.40(d,1h),5.12(s,2h),3.70(s,8h),3.23-2.98(m,6h),2.88-2.62(m,4h),2.01(s,3h).
13
c nmr(101mhz,cdcl3)δ167.2,141.6,138.8,137.8,136.8,128.7,128.6,127.1,123.7,121.1,118.6,65.4,45.2,40.8,38.3,36.4,35.0,17.9.
[0186][0187]
(6)冰浴下,将化合物18(20mmol)溶于150ml二氯甲烷中,将pbr3(20mmol)溶于50ml二氯甲烷,在0℃下滴加到上述混合溶液中,反应3h后,滴加饱和nahco3溶液除去未反应的pbr3,依次用nahco3溶液和水洗涤,有机相经硫酸钠干燥后,旋蒸除去溶剂,柱层析分离得无色油状液体为化合物19,产率约93%。
[0188][0189]
(7)将化合物5(40mmol)、化合物19(100mmol)和naoh(150mmol)溶于乙醇和水的混合溶液中,加热至90℃反应6h,反应完成后用二氯甲烷萃取,合并的有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为化合物20,产率约90%。
[0190][0191]
(8)冰浴下,化合物20(20mmol)和三乙胺(40mmol)溶于50ml二氯甲烷中,搅拌10min后将丙烯酰氯(30mmol)溶于30ml二氯甲烷,在0℃下滴加到化合物20和三乙胺的混合溶液中。反应完全后,滴加稀盐酸,除去过量的丙烯酰氯,依次使用饱和nacl溶液、饱和nahco3溶液和去离子水洗涤,有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为g9,产率约95%。表征数据如下:1h nmr(400mhz,cdcl3)δ7.40(d,16h),7.29
–
7.25(m,24h),7.07(s,1h),7.00
–
6.99(m,4h),6.87(d,4h),6.41(d,1h),6.12(dd,1h),5.83(d,1h),5.12(s,2h),4.33(s,4h),3.70(s,16h),3.23-2.98(m,12h),2.88-2.62(m,8h).
13
c nmr(101mhz,cdcl3)δ166.5,141.6,140.4,138.8,136.8,136.6,130.3,129.8,128.7,128.6,127.1,121.1,120.6,119.2,118.6,65.1,45.2,40.8,39.2,38.3,36.4,35.0.
[0192][0193]
实施例4
[0194]
树枝状(甲基)丙烯酸酯类单体g2-1、g3-1的合成
[0195]
(1)冰浴下将化合物21(20mmol)溶于150ml二氯甲烷中,将pbr3(20mmol)溶于50ml二氯甲烷,在0℃下滴加到上述混合溶液中,反应3h后,滴加饱和nahco3溶液除去未反应的pbr3,依次用nahco3溶液和水洗涤,有机相经硫酸钠干燥后,旋蒸除去溶剂,柱层析分离得无色油状液体为化合物22,产率约93%。
[0196][0197]
(2)将化合物5(40mmol)、化合物22(100mmol)和naoh(150mmol)溶于乙醇和水的混合溶液中,加热至90℃反应6h,反应完成后用二氯甲烷萃取,合并的有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为化合物23,产率约90%。
[0198][0199]
(3)冰浴下化合物23(20mmol)和三乙胺(40mmol)溶于50ml二氯甲烷中,搅拌10min后将甲基丙烯酰氯(30mmol)溶于30ml二氯甲烷,在0℃下滴加到化合物23和三乙胺的混合溶液中。反应完全后,滴加稀盐酸,除去过量的甲基丙烯酰氯,依次使用饱和nacl溶液、饱和nahco3溶液和去离子水洗涤,有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为g2-1,产率约95%。表征数据如下:1h nmr(400mhz,cdcl3)δ7.47-7.40(m,28h),7.07
–
6.99(m,13h),6.87(s,4h),6.48(d,1h),6.40(d,1h),5.12(s,2h),4.33(s,12h),2.01(s,3h).
13
c nmr(101mhz,cdcl3)δ167.2,141.6,140.0,137.8,136.8,136.6,135.7,135.6,134.1,131.2,129.3,128.3,127.2,121.1,120.6,119.2,118.6,123.7,65.4,40.0,39.2,17.9.
[0200][0201]
(4)冰浴下将化合物24(20mmol)溶于150ml二氯甲烷中,将pbr3(20mmol)溶于50ml二氯甲烷,在0℃下滴加到上述混合溶液中,反应3h后,滴加饱和nahco3溶液除去未反应的pbr3,依次用nahco3溶液和水洗涤,有机相经硫酸钠干燥后,旋蒸除去溶剂,柱层析分离得无色油状液体为化合物25,产率约93%。
[0202][0203]
(5)将化合物5(40mmol)、化合物25(100mmol)和naoh(150mmol)溶于乙醇和水的混合溶液中,加热至90℃反应6h,反应完成后用二氯甲烷萃取,合并的有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为化合物26,产率约90%。
[0204][0205]
(6)冰浴下化合物26(20mmol)和三乙胺(40mmol)溶于50ml二氯甲烷中,搅拌10min后将丙烯酰氯(30mmol)溶于30ml二氯甲烷,在0℃下滴加到化合物26和三乙胺的混合溶液中。反应完全后,滴加稀盐酸,除去过量的丙烯酰氯,依次使用饱和nacl溶液、饱和nahco3溶液和去离子水洗涤,有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为g3-1,产率约95%。表征数据如下:1h nmr(400mhz,cdcl3)δ7.25-7.27(d,24h),7.07
–
6.99(m,25h),6.87(s,12h),6.41(d,1h),6.12(dd,1h),5.83(d,1h),5.12(s,2h),4.33(s,28h).
13
c nmr(101mhz,cdcl3)δ166.5,141.6,140.0,137.1,136.8,136.6,130.0,129.8,128.7,127.7,127.1,121.1,120.6,119.2,118.6,65.1,40.0,39.2.
[0206][0207]
实施例5
[0208]
树枝状(甲基)丙烯酸酯类单体g4-1、g6-1的合成
[0209]
(1)将化合物27(20mmol)和1,8-二氮杂环[5,4,0]十一烯-7(20mmol)溶于100ml甲苯中,室温搅拌10~15min,然后滴加环氧氯丙烷(9mmol),室温下反应15~20h,反应结束后旋蒸除去溶剂,再将浓缩液用二氯甲烷溶解,并依次用1mol/l的稀盐酸、饱和nacl溶液、去离子水洗涤,合并的有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得化合物28,产率约90%。
[0210][0211]
(2)将化合物28(10mmol)和三乙胺(40mmol)溶于100ml二氯甲烷,冰浴条件下滴加含丙烯酰氯(15mmol)的二氯甲烷溶液,反应0.5~1h,反应结束后滴加稀盐酸,依次用nacl溶液,nahco3溶液和水洗涤,有机相经硫酸钠干燥后,旋蒸除去溶剂,柱层析分离得g4-1。表征数据如下:1h nmr(400mhz,cdcl3)δ7.26(s,8h),6.41(d,1h),6.12(dd,1h),5.83(d,1h),5.27(m,1h),3.01(d,4h),2.38(s,6h).
13
c nmr(101mhz,cdcl3)δ165.2,135.8,132.8,
131.3,128.1,119.5,78.9,40.9,14.8.
[0212][0213]
(3)冰浴下,将化合物29(10mmol)溶于150ml二氯甲烷中,将pbr3(10mmol)溶于50ml二氯甲烷,冰浴下滴加到上述混合溶液中,反应3h后,滴加饱和nahco3溶液除去未反应的pbr3,依次用nahco3溶液和水洗涤,有机相经硫酸钠干燥后,旋蒸除去溶剂,柱层析分离得30,产率约93%。
[0214][0215]
(4)将化合物5(5mmol)、化合物30(12mmol)和naoh(20mmol)溶于乙醇和水的混合溶液中,加热至90℃反应6h,反应完成后用二氯甲烷萃取,合并的有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得31,产率约90%。
[0216][0217]
(5)冰浴下,化合物31(20mmol)和三乙胺(40mmol)溶于50ml二氯甲烷中,搅拌10min后将丙烯酰氯(30mmol)溶于30ml二氯甲烷,在0℃下滴加到化合物34和三乙胺的混合溶液中。反应完全后,滴加稀盐酸,除去过量的丙烯酰氯,依次使用饱和nacl溶液、饱和nahco3溶液和去离子水洗涤,有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得g6-1,产率约95%。表征数据如下:1h nmr(400mhz,cdcl3)δ7.29(d,32h),7.20(d,8h),7.07(s,1h),7.00(s,4h),6.87(s,4h),6.41(d,1h),6.12(dd,1h),5.83(d,1h),5.12(s,2h),4.33(s,4h),3.49(m,4h),3.13(d,16h).
13
c nmr(101mhz,cdcl3)δ166.5,141.6,140.0,136.8,136.6,136.4,130.0,129.3,129.8,128.9,125.1,121.1,120.6,119.2,118.6,65.1,47.3,40.4,39.2.
[0218][0219]
实施例6
[0220]
树枝状(甲基)丙烯酸酯类单体g7-1的合成
[0221]
(1)将化合物32(40mmol)、化合物14(100mmol)和naoh(150mmol)溶于乙醇和水的混合溶液中,加热至90℃反应6h,反应完成后用二氯甲烷萃取,合并的有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为化合物33,产率约90%。
[0222][0223]
(2)冰浴下,化合物33(20mmol)和三乙胺(40mmol)溶于50ml二氯甲烷中,搅拌10min后将丙烯酰氯(30mmol)溶于30ml二氯甲烷,在0℃下滴加到化合物36和三乙胺的混合溶液中。反应完全后,滴加稀盐酸,除去过量的丙烯酰氯,依次使用饱和nacl溶液、饱和nahco3溶液和去离子水洗涤,有机相经无水硫酸钠干燥后,旋蒸除去多余溶剂,柱层析分离得到无色油状液体为g7-1,产率约95%。表征数据如下:1h nmr(400mhz,cdcl3)δ7.23(d,4h),7.06(d,4h),6.41(d,1h),6.12(dd,1h),5.83(d,1h),4.58
–
4.35(m,2h),3.70(s,4h),3.40(m,1h),2.88
–
2.62(m,2h),2.38(s,6h).
13
c nmr(101mhz,cdcl3)δ166.5,138.8,131.3,128.7,128.6,127.1,67.5,43.3,36.4,35.6,35.4,14.8.
[0224][0225]
实施例7
[0226]
提供一种光致聚合物型全息记录介质,其原料组分详见表1;其中g3-1为实施例4所得的树枝状(甲基)丙烯酸酯类单体。
[0227]
其制备方法,包括如下步骤:在红光条件下,往500ml有搅拌设备的容器中,依次加入表1各成分,室温下充分搅拌15min,通过孔径为0.45微米过滤器除去灰尘等杂质,将混合液分别注入80mm
×
35mm
×
0.5mm/0.05mm/0.005mm的玻璃容器内,室温固化,即得不同厚度的光致聚合物型全息记录介质。
[0228]
表1
[0229]
[0230][0231]
实施例8
[0232]
提供一种光致聚合物型全息记录介质,其原料组分详见表2;其中,g5为实施例2所得的树枝状(甲基)丙烯酸酯类单体。
[0233]
其制备方法同实施例7。
[0234]
表2
[0235][0236]
实施例9
[0237]
提供一种光致聚合物型全息记录介质,其原料组分详见表2;其中,g7-1为实施例6所得的树枝状(甲基)丙烯酸酯类单体。
[0238]
其制备方法同实施例7。
[0239]
表3
[0240]
[0241][0242]
实施例10~13
[0243]
提供一种光致聚合物型全息记录介质,其原料组分详见表4所示。其中,g1为实施例1所得的树枝状(甲基)丙烯酸酯类单体g1。实施例10~13中,g1所占比例分别为7.84%、15.68%、23.52%、31.36%。
[0244]
其制备方法同实施例7。
[0245]
表4
[0246][0247][0248]
对比例1
[0249]
提供一种光致聚合物型全息记录介质,其原料组分详见表5。
[0250]
其制备方法同实施例7。
[0251]
表5
[0252][0253]
对比例2
[0254]
提供一种光致聚合物型全息记录介质,其原料组分详见表6。
[0255]
其制备方法同实施例7。
[0256]
表6
[0257][0258]
对比例3
[0259]
提供一种光致聚合物型全息记录介质,其原料组分详见表7。
[0260]
其制备方法同实施例7。
[0261]
表7
[0262][0263]
试验例
[0264]
(1)测试实施例1~6合成的树枝状(甲基)丙烯酸酯类单体的折射率及单体聚合后收缩率,结果如表8所示。
[0265]
(2)测试并对比实施例7~9,对比例1~3的光致聚合物型全息记录介质的全息性能,结果如图1~3或表9所示。
[0266]
测试方法,包括以下步骤:
[0267]
用405nm、532nm、633nm波长的固体激光器作为光源,经扩束器、分束器和半波片后得到两束光强相同直径为8mm的光束。两光束相交于全息记录介质内进行曝光,记录介质的法线平分两光束,两束光的夹角为58~90
°
,对应的分辨率为1800~2600lp/mm,光强度为2.91mw/cm2。检测光源采用不与记录介质发生反应的785nm波长固体激光器,检测光从bragg角入射到曝光区,通过光电检测器对透射光和衍射光进行实时监测,并通过公式(1)~(3)计算得出不同厚的光致聚合物样片的单光栅衍射效率(η)和记录介质的感光灵敏度(s)。利用旋转台改变检测光的入射角,在bragg角附近读出衍射光,测得全息光栅bragg选择角。
[0268][0269][0270][0271]
式中,η为衍射效率,η
max
为最高衍射效率,id为衍射光,i
t
为透射光,s为感光灵敏度,e为曝光能量,δe为达到最高衍射效率时的曝光能量。
[0272]
(3)测评实施例7的光致聚合物型全息记录介质(厚度0.05mm)在不同空间频率下(分别为:1800lp/mm、2300lp/mm、2600lp/mm)的曝光率,结果如图4所示,图4显示:在不同空间频率下,实施例7的光致聚合物型全息记录介质均具有90%以上的衍射效率。
[0273]
(4)按测评(1)的方法测评实施例10~13的光致聚合物型全息记录介质的532nm激光曝光特性曲线,结果如图5所示,图5显示:实施例10~13的光致聚合物型全息记录介质均
具备较优的全息记录性能。
[0274]
(5)测评实施例8的光致聚合物型全息记录介质(厚度0.005mm)、实施例9的光致聚合物型全息记录介质(厚度0.5mm)在空间频率2600lines/mm曝光下全息光栅bragg选择角曲线,结果如图6、图7所示。如图所示,0.005mm厚的样片选择角高达15.2
°
,可以为使用者提供大视场角,完全满足光致聚合物型全息记录介质的应用要求;0.5mm厚的样片选择角仅为0.16
°
,在进行全息光存储时可以实现多角度复用,完全满足实际应用要求。
[0275]
(6)将实施例7的光致聚合物型全息记录介质曝光制得全息光栅,然后光漂白制得透明全息光栅。
[0276]
对比光致聚合物型全息记录介质曝光前、全息光栅(未光漂白)及透明全息光栅(光漂白后)的紫外可见光(100mw/cm2,2h)透过率,结果如图8所示。
[0277]
(7)对比实施例10~13的全息记录介质的全息性能参数及稳定性,结果见表10所示。
[0278]
表8实施例1~6合成单体的折射率及单体聚合后收缩率
[0279][0280][0281]
表9实施例7~9,对比例1~3的全息记录介质的全息性能参数
[0282] 实施例7实施例8实施例9对比例1对比例2对比例3曝光量(mj/cm2)14.810.18.722.99.522.0衍射效率(%)98.896.096.824.310.642.3感光灵敏度(10-3
cm2/mj)67.297.0113.121.534.329.6收缩率(%)0.060.050.081.021.230.95
[0283]
表10实施例10~13的全息记录介质的全息性能参数及稳定性
[0284][0285]
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。