1.本发明涉及氧氮氢化物、包含氧氮氢化物的金属担载物、以及氨合成用催化剂。
2.本技术基于2020年2月26日在日本提出申请的特愿2020-030730号主张优先权,将其内容援引于此。
背景技术:
3.作为代表性的氨合成法的哈伯-博施法(haber-bosch process)为下述方法:使用在fe3o4中包含数质量%的al2o3和k2o的双重促进铁(doubly promoted iron)催化剂,使氮与氢的混合气体在高温高压条件下于该催化剂上直接反应,从而制造氨的方法。该技术即使在当前也几乎仍以完成当时的制造工序应用在工业上。
4.另一方面,研究了在比哈伯-博施法的反应温度低的温度下合成氨的方法。研究了通过该使氮和氢接触而能够合成氨的催化剂,并研究了将过渡金属作为其催化剂活性成分。其中,作为效率高的方法,提出了将钌(ru)作为催化剂活性成分担载于各种载体而用作氨合成用催化剂的方法(例如专利文献1)。
5.此外,报道了将具有钙钛矿型晶体结构的氧氮氢化物bati(o
3-zhx
ny)、具有钙钛矿型晶体结构的金属氧化物baceo3作为载体,担载ru而形成的金属担载催化剂(例如专利文献2、非专利文献1)。此外,报道了将掺杂有zr的baceo3作为载体,担载ru而形成的金属担载催化剂(ru/bace
1-xyxo3-y
)(例如非专利文献2)。
6.另一方面,虽然报道了氮掺杂的baceo3(钙钛矿型氧氮化物、bace2(o,n)4)的合成方法等,但没有公开将其用于氨合成用催化剂(例如非专利文献3)。
7.现有技术文献
8.专利文献
9.专利文献1:日本特开2006-231229号公报
10.专利文献2:国际公开第2015/136953号
11.非专利文献
12.非专利文献1:yang,xiao-long et al.,“catalysis communications”11,p.867-870(2010)
13.非专利文献2:shimoda,naohiro et al.,“international journal of hydrogen energy”42,p.29745-29755(2017)
14.非专利文献3:liu,guo et al.“journal of solid state chemistry”89,p.366-371(1990)
技术实现要素:
15.发明所要解决的课题
16.基于主要使用双重促进铁催化剂的哈伯-博施法的氨合成虽已被实用化,但需要
高温高压条件,因此存在在装置方面、成本方面的负担大的问题。
17.专利文献1中记载的担载金属催化剂通常使用活性炭等碳质载体、无机氧化物载体。然而,这些担载金属催化剂的反应活性低,仅具有在实际应用中并不充分的性能。专利文献2、非专利文献1中记载那样的担载金属催化剂的催化剂活性不充分。
18.即,需要即使在与哈伯-博施法的反应条件相比为更低温、低压的条件下也具有充分的反应活性的氨合成用催化剂。
19.[用于解决课题的手段]
[0020]
本技术的发明人发现通过将过渡金属担载于包含氧氮氢化物的组合物能够同时实现催化剂性能的提高和稳定化的本发明的氨合成用催化剂,从而完成了本发明。
[0021]
即,本发明的要旨为:
[0022]
[1]下述通式(1)表示的氧氮氢化物。
[0023]anbmol-xnyhz
ꢀꢀ
(1)
[0024]
(上述通式(1)中,a为选自由ba及sr组成的组中的至少1种,b为选自由al、ga、in、si、ge及sn组成的组中的至少1种,n为1~3的整数,m为1或2,l为4或5,x表示由0.1≤x≤3.5表示的数;y表示由0.1≤y≤2.0表示的数;z表示由0.1≤z≤2.0表示的数。)
[0025]
[2]如[1]所述的氧氮氢化物,其中,上述通式(1)中,b为选自al、ga及in组成的组中的至少1种,n=1,m=2,且l=4。
[0026]
[3]如[1]所述的氧氮氢化物,其中,上述通式(1)中,b为选自si、ge及sn组成的组中的至少1种,n=3,m=1,且l=5。
[0027]
[4]金属担载物,其是将过渡金属(m)担载于载体的金属担载物,其特征在于,上述载体为包含[1]~[3]中任一项所述氧氮氢化物的组合物。
[0028]
[5]如[4]所述的金属担载物,其中,相对于上述载体100质量份,上述过渡金属(m)的担载量为0.01质量份以上、50质量份以下。
[0029]
[6]如[4]或[5]所述的金属担载物,其中,上述过渡金属(m)为选自由ru、co及fe组成的组中的至少1种。
[0030]
[7]担载金属催化剂,其包含[4]~[6]中任一项所述的金属担载物。
[0031]
[8]氨合成用催化剂,其包含[4]~[6]中任一项所述的金属担载物。[9]氨合成用催化剂,其特征在于,上述氨合成用催化剂为包含[1]~[3]中任一项所述的氧氮氢化物的组合物。
[0032]
[10]氨的制造方法,其特征在于,在[7]所述的担载金属催化剂的存在下,使氮和氢反应。
[0033]
[11]氧氮氢化物的制造方法,其为制造下述通式(1)表示的氧氮氢化物的制造方法,其特征在于,包括在氨气氛下对下述通式(2)表示的化合物与下述通式(3)表示的化合物进行加热的工序。
[0034]anbmol-xnyhz
ꢀꢀꢀ
(1)
[0035]
a(nh2)2ꢀꢀꢀ
(2)
[0036]bk
ojꢀꢀꢀ
(3)
[0037]
(上述通式(1)~(3)中,a为选自由ba及sr组成的组中的至少1种,b为选自由al、ga、in、si、ge及sn组成的组中的至少1种,n为1~3的整数,m为1或2,l为4或5,k为1或2,j为1
至3,x表示由0.1≤x≤3.5表示的数;y表示由0.1≤y≤2.0表示的数;z表示由0.1≤z≤2.0表示的数。)
[0038]
[12]如[11]所述的氧氮氢化物的制造方法,其中,上述通式(1)中,b为选自al、ga及in组成的组中的至少1种,n=1,m=2,且l=4。
[0039]
[13]如[11]所述的氧氮氢化物的制造方法,其中,所述通式(1)中,b为选自si、ge及sn组成的组中的至少1种,n=3,m=1,且l=5。
[0040]
发明效果
[0041]
在将本发明的氧氮氢化物用作氨合成用催化剂的情况下,即使在低反应温度且低反应压力下也具有高氨合成活性,并且即使重复进行合成反应也没有发现催化剂活性的降低,因此适合用作氨合成用催化剂。
[0042]
此外,与以往的氧氮化物或氧氢化物的合成方法相比较,包含本发明的氧氮氢化物的组合物能够在低温加热处理工序下进行合成,从生产性和成本方面出发也优异。
附图说明
[0043]
[图1]为实施例1中,利用各种各样的加热处理温度及处理时间作为粉末的处理条件合成的氧氮氢化物baal2o
4-xnyhz
粉末的xrd图案。
[0044]
[图2]为实施例1中得到的baal2o
4-xnyhz
的升温脱附谱。
[0045]
[图3]为示出实施例1及2、比较例1中的氨合成速率的反应温度依赖性的图。
[0046]
[图4]为实施例2、3~5中,利用各种各样的加热处理温度及处理时间作为粉末的处理条件合成的氧氮氢化物ba3sio
5-xnyhz
粉末的xrd图案。
具体实施方式
[0047]
(氧氮氢化物)
[0048]
本发明的氧氮氢化物为将氮与氢掺杂于下述通式(5)表示的复合氧化物的氧位点的氧氮氢化物。本发明的氧氮氢化物为下述通式(1)表示的化合物。具有与未掺杂氮、氢的下述通式(5)表示的复合氧化物相同类型的晶体结构是优选的。也即,优选地,本实施方式的氧氮氢化物是在将氮与氢掺杂于下述通式(5)表示的复合氧化物的氧位点的同时,维持该复合氧化物的晶体结构的物质。
[0049]anbmol
ꢀꢀ
(5)
[0050]
上述通式(5)中,a为选自由ba及sr组成的组中的至少1种,b为选自由al、ga、in、si、ge及sn组成的组中的至少1种。
[0051]anbmol-xnyhz
ꢀꢀ
(1)
[0052]
上述通式(1)中,a为选自由ba及sr组成的组中的至少1种,b为选自由al、ga、in、si、ge及sn组成的组中的至少1种,n为1~3的整数,m为1或2,l为4或5,x表示由0.1≤x≤3.5表示的数;y表示由0.1≤y≤2.0表示的数;z表示由0.1≤z≤2.0表示的数。
[0053]
掺杂于本发明的氧氮氢化物中的氮的量及氢的量没有特别限定。维持通式(5)表示的复合氧化物的晶体结构是优选的。
[0054]
就x、y、z的关系而言,例如,优选2x-(3y z)=0以使得氧氮氢化物为电荷中性。
[0055]
上述通式(1)中,x表示由0.5≤x≤3.5表示的数;y表示由0.25≤y≤1.8表示的数;
z表示由0.25≤z≤1.8表示的数是优选的。
[0056]
上述通式(1)中,x表示由1.0≤x≤3.4表示的数;y表示由1.0≤y≤1.7表示的数;z表示由1.0≤z≤1.8表示的数是更优选的。
[0057]
就上述掺杂的氮和氢而言,只要不损害本发明的效果,其一部分可以进一步被氮和氢以外的原子取代,具体而言,可以包含电子、碳、卤原子等。
[0058]
作为本发明的氧氮氢化物的具体例,可举出例如,a为ba、b为al的通式(1)表示的氧氮氢化物;a为ba、b为ga的通式(1)表示的氧氮氢化物;a为ba、b为in的通式(1)表示的氧氮氢化物;a为ba、b为si的通式(1)表示的氧氮氢化物;a为ba、b为ge的通式(1)表示的氧氮氢化物;a为ba、b为sn的通式(1)表示的氧氮氢化物;a为sr、b为选自由al、ga、si及ge组成的组中的至少1种的通式(1)表示的氧氮氢化物等。其中,a为ba、b为al的通式(1)表示的氧氮氢化物、a为ba、b为si的通式(1)表示的氧氮氢化物是优选的,a为ba、b为al的通式(1)表示的氧氮氢化物是更优选的。
[0059]
作为a为ba、b为al的通式(1)表示的氧氮氢化物,可举出baal2o
4-xnyhz
、ba3al2o
6-xnyhz
、ba3al
17o20-xnyhz
、ba4al2o
7-xnyhz
、ba
17
al3o
7-xnyhz
、baal4o
7-xnyhz
、baal
12o19-xnyhz
等。其中,baal2o
4-xnyhz
是更优选的。
[0060]
作为a为ba、b为ga的通式(1)表示的氧氮氢化物,可举出baga2o
4-xnyhz
、baga4o
7-xnyhz
、ba4ga2o
7-xnyhz
、ba3ga2o
6-xnyhz
等。
[0061]
作为a为ba、b为in的通式(1)表示的氧氮氢化物,可举出baino
2.5-xnyhz
、ba3in2o
6-xnyhz
、ba8in6o
17-xnyhz
、ba2in2o
5-xnyhz
、ba4in2o
7-xnyhz
、ba4in6o
13-xnyhz
等。
[0062]
作为a为ba、b为si的通式(1)表示的氧氮氢化物,可举出ba3sio
5-xnyhz
、ba2sio
4-xnyhz
、ba2si4o
10-xnyhz
、ba4si6o
16-xnyhz
、basi4o
9-xnyhz
、ba5si8o
21-xnyhz
、ba6si
10o26-xnyhz
、basio
3-xnyhz
、basi2o
5-xnyhz
等。其中,ba3sio
5-xnyhz
是更优选的。
[0063]
作为a为ba、b为ge的通式(1)表示的氧氮氢化物,可举出ba3geo
1-xnyhz
、bage4o
9-xnyhz
、bage2o
5-xnyhz
、ba
10
ge7o
3-xnyhz
、bageo
3-xnyhz
、ba3ge3o
9-xnyhz
等。
[0064]
作为a为ba、b为sn的通式(1)表示的氧氮氢化物,可举出ba2sno
4-xnyhz
、basno
3-xnyhz
、ba3sno
1-xnyhz
等。
[0065]
作为a为sr、b为选自由al、ga、si及ge组成的组中的至少1种的通式(1)表示的氧氮氢化物,可举出sral2o
4-xnyhz
、srga2o
4-xnyhz
、sr3sio
5-xnyhz
、sr3geo
5-xnyhz
等。
[0066]
(氧氮氢化物的制造方法)
[0067]
制造本发明的下述通式(1)表示的、氧氮氢化物的方法包括在氨气氛下对下述通式(2)表示的化合物与下述通式(3)表示的化合物进行加热的工序。
[0068]anbmol-xnyhz
ꢀꢀꢀ
(1)
[0069]
a(nh2)2ꢀꢀꢀ
(2)
[0070]bk
ojꢀꢀꢀ
(3)
[0071]
(上述通式(1)~(3)中,a为选自由ba及sr组成的组中的至少1种,b为选自由al、ga、in、si、ge及sn组成的组中的至少1种,n为1~3的整数,m为1或2,l为4或5,k为1或2,j为1至3,x表示由0.1≤x≤3.5表示的数;y表示由0.1≤y≤2.0表示的数;z表示由0.1≤z≤2.0表示的数。)
[0072]
作为本发明的制造方法的生成物的上述通式(1)表示的、氧氮氢化物的优选方式
以及具体例与上述的本发明的氧氮氢化物相同。
[0073]
作为本发明的制造方法的原料的上述通式(2)表示的化合物,可举出例如ba(nh2)2、sr(nh2)2。
[0074]
作为本发明的制造方法的原料的上述通式(3)表示的化合物,可举出例如al2o3、sio2、sno、in2o3、geo2等。
[0075]
本发明的一实施方式的上述通式(1)表示的氧氮氢化物的制造方法包括在氨气氛下对ba(nh2)2或sr(nh2)2的金属酰胺化合物与上述通式(1)中由b表示的元素的氧化物(例如,al2o3、sio2)进行加热的工序。详细的制造方法在后述的各实施方式中进行说明。例如,包括将上述金属酰胺化合物与上述氧化物混合的混合工序、将通过混合工序得到的混合物在氨气氛中进行加热处理的加热处理工序。此外,混合工序前,还包括进一步将上述金属酰胺化合物进行脱水处理的前处理工序是优选的。在这种情况下,优选地,上述混合工序为在ar手套箱中等的稀有气体气氛下对脱水处理后的上述氧化物进行混合的工序。
[0076]
作为上述脱水处理工序,可举出例如于300℃以上且低于900℃、优选地于400℃以上且低于800℃,更优选地于500℃以上且低于700℃进行真空加热处理的方法。
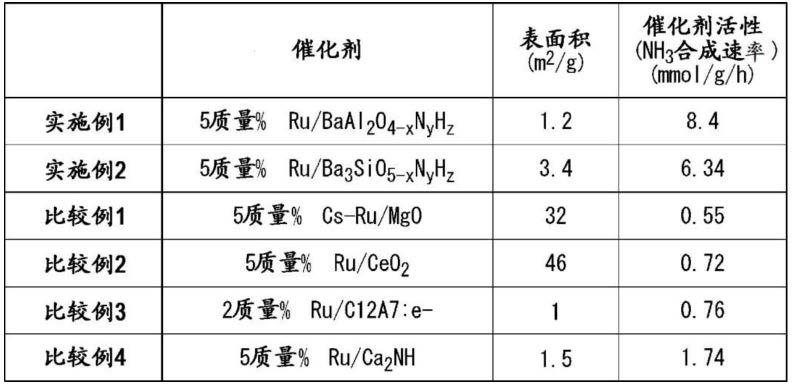
[0077]
作为上述加热处理,可举出例如在氨气流中于300℃以上且低于900℃加热处理1~50小时、优选地于300℃以上且低于900℃加热处理2~30小时、更优选地于400℃以上且低于900℃加热处理5~20小时的方法。
[0078]
在制造上述通式(1)表示的化合物的情况下,作为上述氧化物与金属酰胺化合物的原料加入比,使元素a与元素b的摩尔比(a:b)成为0.5
×
n:m~1.5
×
n:m(mol/mol)的方式进行混合是优选的,0.8
×
n:m~1.2
×
n:m是更优选的,0.9
×
n:m~1.1
×
n:m是进一步优选的。此处的a、b、n、m与上述通式(1)的a、b、n、m含义相同。
[0079]
所使用的上述金属酰胺的原料的形态及上述氧化物的原料的形态为粉末是优选的。例如,上述金属酰胺可举出例如市售的物质。上述金属酰胺可以通过以下的步骤进行合成。将纯度99.99%的金属a(a选自由ba及sr组成的组中的至少1种。)放入耐压容器中,在冷却至-50℃的同时将氨气体导入至容器,使金属a溶解。于-50℃进行1小时搅拌后,恢复至室温。
[0080]
其后,将容器浸入油浴中,在搅拌的同时于100℃保持1小时后冷却至室温。将冷却后容器内残留的氨气体进行排气,得到上述金属酰胺。
[0081]
以下,以本发明的第一实施方式及第二实施方式为例,对本发明的氧氮氢化物、以其作为载体担载有过渡金属的金属担载物、担载金属催化剂、氨合成用催化剂、氨合成用催化剂进行详细地说明。此外,以本发明的第一实施方式及第二实施方式为例,对使用该氨合成用催化剂的氨的制造方法、本发明的氧氮氢化物的制造方法进行详细地说明。但是,本发明不限于上述实施方式,可以采用各种各样的形态。
[0082]“第一实施方式”[0083]
(包含第13族典型元素的氧氮氢化物)
[0084]
本实施方式的氧氮氢化物为将氮与氢掺杂于下述通式(6)表示的复合氧化物的氧位点的氧氮氢化物。本发明的氧氮氢化物为下述通式(7)表示的化合物。具有与未掺杂氮、氢的下述通式(6)表示的复合氧化物相同类型的晶体结构是优选的。也即,优选地,本实施方式的氧氮氢化物将氮与氢掺杂于下述通式(6)表示的复合氧化物的氧位点,并同时维持
该复合氧化物的晶体结构。
[0085]
ab2o4ꢀꢀꢀꢀꢀꢀꢀ
(6)
[0086]
ab2o
4-xnyhz
ꢀꢀꢀ
(7)
[0087]
(上述通式(7)中,b为选自al、ga及in组成的组中的至少1种,a、x、y、z与通式(1)中的a、x、y、z含义相同。)
[0088]
上述通式(7)表示的化合物为上述通式(1)表示的化合物中在n为1,m为2,l为4,b为选自al、ga、及in组成的组中的至少1种的情况下的化合物。
[0089]
掺杂于本实施方式的氧氮氢化物的氮的量及氢的量只要能够维持ab2o4的晶体结构就没有特别限定。
[0090]
就x、y、z的关系而言,例如,优选2x-(3y z)=0,以使得氧氮氢化物为电荷中性。
[0091]
上述通式(7)中,x表示0.5≤x≤1.6表示的数;y表示0.25≤y≤0.8表示的数;z表示0.25≤z≤0.8表示的数是优选的。
[0092]
上述通式(7)中,x表示1.0≤x≤1.4表示的数;y表示0.4≤y≤0.6表示的数;z表示0.3≤z≤0.5表示的数是更优选的。
[0093]
例如,通过后述的实施例合成了baal2o
4-xnyhz
表示的氧氮氢化物。
[0094]
就上述掺杂的氮和氢而言,只要不损害本发明的效果,其一部分可以进一步被氮和氢以外的原子取代,具体而言,也可以包含电子、碳、卤原子等。
[0095]
(氧氮氢化物的制造方法)
[0096]
本实施方式的上述通式(7)表示的氧氮氢化物的制造方法包括在氨气氛下对下述通式(8)表示的金属酰胺与下述通式(9)表示的第13族元素的氧化物进行加热的工序。详细的制造方法在后述的实施例中进行说明。本实施方式的氧氮氢化物ab2o
4-xnyhz
的制造方法例如包括将a(nh2)2与b2o3进行混合的混合工序、将通过混合工序得到的混合物在氨气氛下进行加热处理的加热处理工序。此外,混合工序前,还包括进一步将a(nh2)2进行脱水处理的前处理工序是优选的。在这种情况下,优选地,上述混合工序为在ar手套箱中等的稀有气体气氛下对脱水处理后的a(nh2)2和b2o3进行混合的工序。
[0097]
a(nh2)2ꢀꢀꢀ
(8)
[0098]
b2o3ꢀꢀꢀ
(9)
[0099]
上述通式(8)、(9)中,a、b与上述通式(7)中的a、b含义相同。
[0100]
作为上述脱水处理工序,可举出例如于300℃以上且低于900℃、优选地于400℃以上且低于800℃、更优选地于500℃以上且低于700℃进行真空加热处理的方法。
[0101]
作为上述加热处理,可举出例如在氨气流中于300℃以上且低于900℃加热处理1~50小时、优选地于400℃以上且低于900℃加热处理2~50小时、更优选地于500℃以上且低于900℃加热处理5~25小时的方法。
[0102]
使a与b的摩尔比(a:b)成为0.8:2.0~1.2:2.0(mol/mol)的方式进行混合是优选的,0.9:2.0~1.1:2.0是更优选的,0.95:2.0~1.05:2.0是进一步优选的。
[0103]
所使用的原料a(nh2)2的形态及原料b2o3的形态为粉末是优选的。例如,可举出市售的高纯度化学公司制的al2o3粉末(平均粒径:2~3μm)。ba(nh2)2可通过以下的步骤进行合成。将金属ba(aldrich公司制、纯度99.99%)放入耐压容器中,冷却至-50℃左右的同时将氨气体导入上述容器,由此使金属ba溶解。将所得的溶解物于-50℃进行1小时搅拌后,恢
复至室温。其后,将包含得到的溶解物的上述容器浸入油浴中,在搅拌的同时于100℃保持1小时后冷却至室温。接着,冷却后,将该容器内残留的氨气体进行排气,由此得到ba(nh2)2。
[0104]
本实施方式的氧氮氢化物优选为通过在氨气氛下对a(nh2)2与b2o3进行加热而得到的氧氮氢化物。通过包括将a(nh2)2与b2o3混合的混合工序、将通过混合工序得到的混合物在氨气氛中进行加热处理的加热处理工序的制造方法而得到的氧氮氢化物是更优选的。上述混合工序前,还包括进一步将b2o3进行脱水处理的前处理工序是优选的。在这种情况下,优选地,上述混合工序中,优选在ar手套箱中等的稀有气体气氛下对脱水处理后的b2o3与a(nh2)2进行混合。此外,优选的加热处理温度、优选的加热时间、优选的原料的加入量比等与上述制造方法相同。
[0105]
《氧氮氢化物中包含的氮及氢的定量>
[0106]
可以利用升温脱附分析装置(belcata)对合成的氧氮氢化物ab2o
4-xnyhz
进行分析,求出脱附的氮及氢的量。基于脱附的氮及氢的量的结果,可得到氧氮氢化物中包含的氮及氢的比例。例如,在之后的实施例中,基于通过升温脱附分析装置(belcata)分析而得到的结果,于800℃合成的baal2o
4-xnyhz
的组成为baal2o
3.7n0.17h0.09
。
[0107]
(金属担载物)
[0108]
本实施方式的金属担载物为将过渡金属(m)担载于载体而得的物质。上述载体为包含将氮与氢掺杂于ab2o4的氧位点而得的氧氮氢化物的组合物。本实施方式的金属担载物为将过渡金属(m)担载于载体而得的物质。上述载体为包含上述通式(7)表示的氧氮氢化物的组合物。上述载体为包含上述说明的本实施方式的氧氮氢化物的组合物是优选的。上述过渡金属(m)为选自由ru、co及fe组成的组中的至少1种是优选的。
[0109]
上述过渡金属的担载量没有特别限定,通常相对于上述载体100质量份而言为0.01质量份(0.01质量%)以上,优选地为0.5质量份(0.5质量%)以上,更优选地为1质量份(1质量%)以上,进一步优选地为2质量份(2质量%)以上,通常为50质量份(50质量%)以下,优选为30质量份(30质量%)以下,更优选为20质量份(20质量%)以下,进一步优选为10质量份(10质量%)以下。只要在上述下限值以上,即可实现本发明的效果,只要在上述上限值以下,即可实现担载量与成本相称的本发明的效果。
[0110]
本实施方式的金属担载物为将过渡金属(m)担载于载体的物质。上述载体优选为包含在氨气氛下对b2o3与a(nh2)2进行加热而得到的氧氮氢化物的组合物。更优选地为下述组合物,该组合物包含通过包括将b2o3与a(nh2)2混合的混合工序、将通过混合工序得到的混合物在氨气氛中进行加热处理的加热处理工序的制造方法而得到的氧氮氢化物。上述混合工序前,还包括进一步将b2o3进行脱水处理的前处理工序是优选的。在这种情况下,优选地,上述混合工序为在ar手套箱中等的稀有气体气氛下对脱水处理后的b2o3与a(nh2)2进行混合的工序。另外,优选的加热处理温度、优选的加热时间、优选的原料的加入量比等与上述制造方法相同。
[0111]
《过渡金属>
[0112]
本实施方式中使用的过渡金属没有特别限定,但通常为周期表第6族、7族、8族、9族或10族的过渡金属,优选为第6族、8族、或9族的过渡金属,更优选地为第8族或9族金属。
[0113]
此外,作为具体的金属元素,没有特别限定,但通常为cr、mo、mn、re、fe、ru、os、co、rh、ni、pd、pt,从在与氮的键能高的方面考虑,优选为mo、re、fe、ru、os、co,在将氨合成用催
化剂用作氨合成用催化剂时,从在具有氨合成活性的方面考虑,更优选为ru、co或fe,从在具有最高的催化剂活性的方面考虑,进一步优选为ru。
[0114]
上述的各元素可单独使用,也可以将2种类以上组合使用。此外可以使用这些元素的金属间化合物,例如,co3mo3n、fe3mo3n、ni2mo3n、mo2n等。优选地将各元素单独或2种类以上的组合进行使用,更优选地,从在成本的方面考虑单独使用是有利的。
[0115]
《过渡金属(m)向氧氮氢化物的担载方法>
[0116]
过渡金属(m)向氧氮氢化物的担载方法没有特别限定,例如,将通过上述方法得到的粉末状氧氮氢化物(ab2o
4-xnyhz
)、待担载的金属的化合物放入石英玻璃管内,在真空中于50℃以上90℃以下加热0.5~4小时,然后继续于100℃以上150℃以下加热0.5~4小时,由此将所要担载的金属化合物附着于粉末状ab2o
4-xnyhz
的表面。最后于150℃以上300℃以下加热0.5~5小时,将金属化合物热分解,由此可得到在ab2o
4-xnyhz
上固定有过渡金属(m)的担载物(以下,m/ab2o
4-xnyhz
)。
[0117]
例如,使用过渡金属(m)各自为ru、co、fe的过渡金属化合物ru3(co)
12
、co2(co)8、fe2(co)9,能够合成金属担载物ru担载ab2o
4-xnyhz
(简称为ru/ab2o
4-xnyhz
)、co担载ab2o
4-xnyhz
(简称为co/ab2o
4-xnyhz
)、fe担载ab2o
4-xnyhz
(简称为fe/ab2o
4-xnyhz
)。
[0118]
《金属担载物的形状>
[0119]
本实施方式的金属担载物的形状没有特别限定,具体而言可以为块状、粉末状、被膜状等任意的形状,但通常为粉末状。粉末状的金属担载物的粒径没有特别限定,通常为1nm以上、10μm以下。
[0120]
本实施方式的金属担载物中过渡金属粒径没有特别限定,但通常为1nm以上、100nm以下。优选地,在作为氨合成用催化剂使用时,从在作为氮解离的活性点的台阶位(step site)数变多的方面考虑,为20nm以下有利,更优选为10nm以下。
[0121]
(担载金属催化剂)
[0122]
本实施方式的担载金属催化剂包含上述金属担载物。本实施方式的担载金属催化剂包含过渡金属和对上述过渡金属进行担载的载体,上述载体为包含下述通式(7)表示的氧氮氢化物的组合物。
[0123]
ab2o
4-xnyhz
ꢀꢀ
(7)
[0124]
(上述通式(7)中,a为选自由ba及sr组成的组中的至少1种,b为选自由al、ga及in组成的组中的至少1种,x表示0.2≤x≤2.0表示的数;y表示0.1≤y≤1.0表示的数;z表示0.1≤z≤1.0表示的数。)
[0125]
(氨合成用催化剂)
[0126]
本发明的氨合成用催化剂为将过渡金属(m)担载于载体而得的物质。上述载体为包含将氮与氢掺杂于ab2o4的氧位点的氧氮氢化物的组合物。本实施方式的氨合成用催化剂包含过渡金属和对上述过渡金属进行担载的载体,上述载体为包含下述通式(7)表示的氧氮氢化物的组合物。上述载体为包含上述说明的本实施方式的氧氮氢化物的组合物是优选的。
[0127]
ab2o
4-xnyhz
ꢀꢀ
(7)
[0128]
(上述通式(7)中,a为选自由ba及sr组成的组中的至少1种,b为选自由al、ga及in组成的组中的至少1种,x表示0.2≤x≤2.0表示的数;y表示0.1≤y≤1.0表示的数;z表示
0.1≤z≤1.0表示的数。)
[0129]
就上述氮与氢而言,只要不损害本发明的效果,其一部分也可以进一步包含氮及氢这两者以外的原子,具体而言,可以包含电子、碳、卤原子等。
[0130]
《过渡金属>
[0131]
本实施方式中使用的过渡金属没有特别限定,但通常为周期表第6族、7族、8族、9族或10族的过渡金属,优选为第6族、8族、或9族的过渡金属,更优选地为第8族或9族金属。
[0132]
此外,作为具体的金属元素,没有特别限定,但通常为cr、mo、mn、re、fe、ru、os、co、rh、ni、pd、pt,从与氮的键能高的方面考虑,优选为mo、re、fe、ru、os、co,在将氨合成用催化剂用作氨合成用催化剂时,从具有氨合成活性的方面考虑,更优选为ru、co或fe,从具有最高的催化剂活性的方面考虑,进一步优选为ru。
[0133]
上述的各元素可单独使用,也可以将2种类以上组合使用。此外可以使用这些元素的金属间化合物,例如,co3mo3n、fe3mo3n、ni2mo3n、mo2n等。优选地将各元素单独或2种类以上的组合进行使用,更优选地,从成本的方面考虑单独使用是有利的。
[0134]
(氨合成用催化剂的制造方法)
[0135]
本发明的氨合成用催化剂为将过渡金属(m)担载于载体的物质。上述载体为包含将氮与氢掺杂于ab2o4的氧位点的氧氮氢化物的组合物。就本实施方式的氨合成用催化剂而言,将上述过渡金属担载于包含含有上述氧氮氢化物的组合物的上述载体来进行制造。制造方法没有特别限定,通常,使过渡金属、或成为过渡金属的前体的化合物(以下,过渡金属化合物)担载于上述载体来进行制造。
[0136]
成为本实施方式的氨合成用催化剂的原料的、上述氧氮氢化物的组合物可以使用市售的试剂、工业原料,也可以使用从对应的金属出发通过已知的方法而得到的物质。
[0137]
可在对本实施方式中使用的上述氧氮氢化物组合物在氢气氛中进行200~500℃左右、数小时(例如340℃2小时)加热的前处理后,通过后述的过渡金属担载工序担载上述过渡金属。
[0138]
在使用将上述载体预先在氢气氛下加热了的试样制造的催化剂中,例如,在用于氨合成反应的情况下,在反应开始后可立即得到高活性。
[0139]
将过渡金属(m)担载于本实施方式中使用的上述载体的方法没有特别限定,可以使用已知的方法。通常使用将作为担载的过渡金属的化合物且能够通过还原、热分解等转化为过渡金属的过渡金属化合物担载于上述载体后,转化为过渡金属的方法。
[0140]
上述过渡金属化合物没有特别限定,但可以使用容易热分解的过渡金属的无机化合物或有机过渡金属络合物等。具体而言,可以使用过渡金属的络合物、过渡金属的氧化物、硝酸盐、盐酸盐等的过渡金属盐等。
[0141]
例如,作为ru化合物,可举出十二羰基三钌[ru3(co)
12
]、四(三苯基膦)二氯化钌(ii)[rucl2(pph3)4]、三(三苯基膦)二氯化钌(ii)[rucl2(pph3)3]、三(乙酰丙酮)钌(iii)[ru(acac)3]、二茂钌[ru(c5h5)]、亚硝酰基硝酸钌[ru(no)(no3)3]、钌酸钾、氧化钌、硝酸钌、氯化钌等。三(乙酰丙酮)钌(iii)[ru(acac)3]是优选的。
[0142]
作为fe化合物,可举出五羰基铁[fe(co)5]、十二羰基三铁[fe3(co)
12
]、九羰基铁[fe2(co)9]、四羰基铁碘化物[fe(co)4i2]、三(乙酰丙酮)铁(iii)[fe(acac)3]、二茂铁[fe(c5h5)2]、氧化铁、硝酸铁、氯化铁(fecl3)等。
[0143]
作为co化合物,可举出例如八羰基钴[co2(co)8]、三(乙酰丙酮)钴(iii)[co(acac)3]、钴(ii)乙酰丙酮[co(acac)2]、二茂钴[co(c5h5)2]、氧化钴、硝酸钴、氯化钴等。
[0144]
这些过渡金属化合物中,[ru3(co)
12
]、[fe(co)5]、[fe3(co)
12
]、[fe2(co)9]、[co2(co)8]等过渡金属的羰基络合物通过在担载后进行加热来担载过渡金属,因此从制造本实施方式的氨合成用催化剂时能省略后述的还原处理的方面考虑是优选的。
[0145]
上述过渡金属化合物的使用量没有特别限定,可以适当地使用用于实现所期望的担载量的量,但通常相对于使用的上述载体100质量份,通常为0.01质量份(0.01质量%)以上,优选为2质量份(2质量%)以上,优选为10质量份(10质量%)以上,更优选为20质量份(20质量%)以上,通常为50质量份(50质量%)以下,优选为40质量份(40质量%)以下,更优选为30质量份(30质量%)以下。
[0146]
作为将上述过渡金属化合物担载于载体的方法,具体而言可使用例如物理混合法、cvd法(化学气相沉积法)、溅射法等的方法。
[0147]
物理混合法是将上述载体与上述过渡金属化合物进行固体混合后,在氮、氩、氦等的非活性气体气流中、或者在真空下进行加热的方法。此时的加热温度没有特别限定,通常为200℃以上、600℃以下。加热时间没有特别限定,通常期望其为2小时以上。
[0148]
在此,只要是通过热分解能够转化为过渡金属的过渡金属化合物,则过渡金属通常在上述阶段中被担载,成为本实施方式的氨合成用催化剂。
[0149]
在使用除了通过热分解转化为过渡金属的过渡金属化合物以外的物质的情况下,通常将过渡金属化合物还原而成为本实施方式的氨合成用催化剂。
[0150]
就将上述过渡金属化合物还原的方法(以下称为还原处理)而言,只要不妨碍本发明的目的则无特别限定,但可举出例如,在包含还原性气体的气氛下进行的方法、在包含上述过渡金属化合物的溶液中加入nabh4、nh2nh2或、福尔马林等还原剂来使其析出于上述金属氢化物的表面的方法,优选地在包含还原性气体的气氛下进行。作为上述还原性气体,可举出氢、氨、甲醇(蒸气)、乙醇(蒸气)、甲烷、乙烷等。
[0151]
此外,在上述还原处理时,不妨碍本发明的目的、特别是不妨碍氨合成反应的还原性气体以外的其他成分也可以共存于反应体系。具体而言,在还原处理时,除氢等还原性气体以外还可以使不妨碍反应的氩、氮这样的气体共存,使氮共存是优选的。
[0152]
在将上述还原处理在包含氢的气体中进行的情况下,通过使氢与氮一起共存,能够与后述的氨的制造同时进行。也即,在将本实施方式的氨合成用催化剂用作后述的氨合成用催化剂的情况下,也可以将上述过渡金属化合物担载于上述金属氢化物而成的物质置于氨合成反应的反应条件中,由此将上述过渡金属化合物还原,转化为过渡金属。
[0153]
上述还原处理时的温度没有特别限定,通常可于200℃以上,优选于300℃以上,优选于低于700℃进行。更优选可于400℃以上且低于700℃进行。通过于上述的还原处理温度范围内进行,充分地、或者在优选范围发生上述过渡金属的生长。
[0154]
上述还原处理时的压力没有特别限定,通常为0.01mpa以上、10mpa以下。若使还原处理时的压力与后述的氨合成条件为相同条件,则不再需要繁杂的操作,在制造效率的方面有利。
[0155]
上述还原处理的时间没有特别限定,在于常压实施的情况下,通常为1小时以上、2小时以上是优选的。
[0156]
此外,在于反应压力高的条件、例如1mpa以上进行的情况下,1小时以上是优选的。
[0157]
在使用除了通过热分解转化为过渡金属的过渡金属化合物以外的物质的情况下,与前述的还原处理方法同样地,将固体混合物中包含的过渡金属化合物利用通常的方法进行还原,由此成为本实施方式的氨合成用催化剂。
[0158]
作为除了上述氧氮氢化物及上述过渡金属以外的成分,可进一步包含sio2、al2o3、zro2、mgo、活性炭、石墨、sic等作为上述氧氮氢化物的载体。
[0159]
本实施方式的氨合成用催化剂能够使用通常的成型技术而作为成型体使用。具体而言,可举出粒状、球状、片状、环状、通心粉、四叶、骰子、蜂窝状等的形状。此外,也可以在适当的支承体上涂布后使用。
[0160]
在使用本实施方式的氨合成用催化剂时,其反应活性没有特别限定,在以反应温度300℃、反应压力0.9mpa时的氨的生成速率为例的情况下,1.0mmol/g
·
h以上是优选的,从适于实用的制造条件的方面出发,2.0mmol/g
·
h以上是更优选的,从适于高效率的制造条件的方面出发,3.0mmol/g
·
h以上是进一步优选的,从适于更高效率的制造条件的方面考虑,5.0mmol/g
·
h以上是进一步优选的。
[0161]
以下对使用本实施方式的氨合成用催化剂的氨制造方法进行记载。
[0162]
(氨的制造方法)
[0163]
本实施方式的氨的制造方法(以下有时称为本实施方式的制造方法)为将本实施方式的担载金属催化剂或本实施方式的氨合成用催化剂作为催化剂使用,使氢与氮于上述催化剂上反应而合成氨的方法。
[0164]
作为具体的制造方法,只要是使氢与氮于上述催化剂上接触而合成氨的方法,就没有特别限定,可以按照适当已知的制造方法进行制造。
[0165]
在本实施方式的氨制造方法中,通常,在使氢与氮于上述催化剂上接触时,加热催化剂,制造氨。
[0166]
本实施方式的制造方法中反应温度没有特别限定,但通常为200℃以上、优选为250℃以上,更优选为300℃以上,通常为600℃以下,优选为500℃以下,更优选为450℃以下。氨合成为放热反应,因此在低温区域在化学平衡理论上对氨生成更有利,但为了实现充分的氨生成速率,于上述的温度范围进行反应是优选的。
[0167]
在本实施方式的制造方法中,与上述催化剂接触的氮与氢的摩尔比率没有特别限定,通常,以氢相对氮的比率(h2/n2(体积/体积))计通常为于0.4以上,优选为于0.5以上,更优选为于1以上,通常为于10以下,优选为于5以下进行。
[0168]
本实施方式的制造方法中反应压力没有特别限定,以包含氮与氢的混合气体的压力计通常为0.01mpa以上,优选为0.1mpa以上,通常为20mpa以下,优选为15mpa以下,更优选为10mpa以下。此外,如果考虑实际应用,在大气压以上的加压条件下进行反应是优选的。
[0169]
在本实施方式的制造方法中,在使氮与氢与上述催化剂接触前,利用使用脱水材料的方法、进行深冷分离的方法、氢气体等除去附着于上述催化剂上的水分、氧化物是优选的。作为除去的方法可举出还原处理。
[0170]
本实施方式的制造方法中,为了实现更良好的氨收率,本实施方式的制造方法中使用的氮和氢中的水分含量少是优选的,没有特别限定,通常,氮与氢的混合气体中的总水分含量为100ppm以下,优选地,50ppm以下是优选的。
[0171]
在本实施方式的制造方法中,反应容器的形式没有特别限定,可以使用在氨合成反应中通常能够使用的反应容器。作为具体的反应形式,可以使用例如分批式反应形式、封闭循环系反应形式、流通系反应形式等。其中从实用的观点出发,流通系反应形式是优选的。此外也可以使用将填充有催化剂的一种反应器、或多个反应器连接的方法、在同一反应器内具有多个反应层的反应器的任意方法。
[0172]
从氢与氮出发合成氨的反应为伴随着体积收缩的放热反应,因此为了提高氨收率,在工业上除去反应热是优选的,可以使用具有通常使用的除热手段的已知的反应装置。例如具体而言,可以使用将填充有催化剂的反应器串联地多个连接、在各反应器的出口设置中间冷却器进行除热的方法等。
[0173]
在本实施方式的氨制造方法中,可以单独使用通过本实施方式的制造方法得到的氨合成用催化剂,可以与在氨合成中通常能够使用的其他已知的催化剂组合使用。
[0174]“第二实施方式”[0175]
(包含第14族典型元素的氧氮氢化物)
[0176]
本实施方式的氧氮氢化物为将氮与氢掺杂于下述通式(10)表示的复合氧化物的氧位点的氧氮氢化物。本技术发明的氧氮氢化物为下述通式(11)表示的化合物。具有与未掺杂氮、氢的下述通式(10)表示的复合氧化物相同类型的晶体结构是优选的。也即,优选地,本实施方式的氧氮氢化物为在将氮与氢掺杂于下述通式(10)表示的复合氧化物的氧位点的同时维持其复合氧化物的晶体结构。
[0177]
a3bo5ꢀꢀꢀꢀꢀꢀꢀ
(10)
[0178]
a3bo
5-xnyhz
ꢀꢀꢀ
(11)
[0179]
(上述通式(7)中,b为选自si、ge及sn组成的组中的至少1种,a、x、y、z与通式(1)中的a、x、y、z含义相同。)
[0180]
上述通式(11)表示的化合物为上述通式(1)表示的化合物中在n为3,m为1,l为5,b为选自si、ge及sn组成的组中的至少1种的情况下的化合物。
[0181]
掺杂于本实施方式的氧氮氢化物的氮的量及氢的量只要能够维持a3bo5的晶体结构就没有特别限定。
[0182]
就x、y、z的关系而言,例如,优选2x-(3y z)=0以使氧氮氢化物为电荷中性。
[0183]
上述通式(1)中,x表示由0.1≤x≤3.5表示的数;y表示由0.1≤y≤2.0表示的数;z表示由0.1≤z≤2.0表示的数是优选的。
[0184]
上述通式(1)中,x表示2.0≤x≤3.5表示的数;y表示1.0≤y≤2.0表示的数;z表示1.0≤z≤2.0表示的数是更优选的。
[0185]
例如,通过后述的实施例合成了ba3sio
5-xnyhz
表示的氧氮氢化物。
[0186]
就上述掺杂的氮和氢而言,只要不损害本发明的效果,其一部分可以进一步被氮和氢这两者以外的原子取代,具体而言,也可以包含电子、碳、卤原子等。
[0187]
(氧氮氢化物的制造方法)
[0188]
本实施方式的上述通式(11)表示的氧氮氢化物的制造方法包括在氨气氛下对下述通式(8)表示的金属酰胺与下述通式(12)表示的第14族元素的氧化物进行加热的工序。详细的制造方法在后述的实施例中进行说明。本实施方式的氧氮氢化物a3bo
5-xnyhz
的制造方法例如包括将a(nh2)2与bo2进行混合的混合工序、将通过混合工序得到的混合物在氨气
氛下进行加热处理的加热处理工序。此外,混合工序前,还包括进一步将a(nh2)2进行脱水处理的前处理工序是优选的。在这种情况下,优选地,在上述混合工序中,在ar手套箱中等的稀有气体气氛下对脱水处理后的a(nh2)2和bo2进行混合。
[0189]
a(nh2)2ꢀꢀꢀ
(8)
[0190]
bo2ꢀꢀꢀ
(12)
[0191]
上述通式(8)、(13)中,a、b与上述通式(11)中的a、b含义相同。
[0192]
作为上述脱水处理工序,可举出例如于300℃以上且低于900℃、优选地于400℃以上且低于800℃、更优选地于500℃以上且低于700℃进行真空加热处理的方法。
[0193]
作为上述加热处理,可举出例如在氨气流中于300℃以上且低于900℃加热处理1~24小时、优选地于300℃以上且低于700℃加热处理2~12小时、更优选地于400℃以上且低于700℃加热处理2~6小时的方法。
[0194]
使a与b的摩尔比(a:b)成为2.4:1~3.6:1(mol/mol)的方式进行混合是优选的,2.7:1~3.3:1是更优选的,2.85:1~3.15:1是进一步优选的。
[0195]
所使用的原料a(nh2)2的形态及原料bo2的形态为粉末是优选的。例如,可举出市售的aldrich公司制的sio2粉末(平均粒径:低于10nm)。ba(nh2)2可通过与第一实施方式同样的方法进行制造。
[0196]
本实施方式的氧氮氢化物优选为通过在氨气氛下对a(nh2)2与bo2进行加热而得到的氧氮氢化物。通过包括将a(nh2)2与bo2混合的混合工序、将通过混合工序得到的混合物在氨气氛中进行加热处理的加热处理工序的制造方法而得到的氧氮氢化物是更优选的。上述混合工序前,还包括进一步将bo2进行脱水处理的前处理工序是优选的。在这种情况下,优选地,上述混合工序中,在ar手套箱中等的稀有气体气氛下对脱水处理后的a(nh2)2与bo2进行混合。另外,优选的加热处理温度、优选的加热时间、优选的原料的加入量比等与上述制造方法相同。
[0197]
《氧氮氢化物中包含的氮及氢的定量>
[0198]
可以利用升温脱附分析装置(belcata)对合成的氧氮氢化物a3bo
5-xnyhz
进行分析,求出脱附的氮及氢的量。基于脱附的氮及氢的量的结果,可得到氧氮氢化物中包含的氮及氢的比例。例如,在之后的实施例中,基于升温脱附分析装置(belcata)分析而得到的结果,于600℃合成的a3bo
5-xnyhz
的组成为a3bo
1.62n1.66h1.77
。
[0199]
(金属担载物)
[0200]
本实施方式的金属担载物为将过渡金属(m)担载于载体的物质。上述载体为包含将氮与氢掺杂于a3bo5的氧位点的氧氮氢化物的组合物。上述载体为包含上述通式(11)表示的氧氮氢化物的组合物。上述载体为包含上述说明的本实施方式的氧氮氢化物的组合物是优选的。上述过渡金属(m)为选自由ru、co及fe组成的组中的至少1种是优选的。
[0201]
上述过渡金属的种类、过渡金属(m)向氧氮氢化物的担载方法、其担载量、金属担载物的制造方法、金属担载物的形状与第一实施方式相同。
[0202]
(担载金属催化剂)
[0203]
本实施方式的担载金属催化剂包含上述金属担载物。本实施方式的担载金属催化剂包含过渡金属和将上述过渡金属担载的载体,上述载体包含下述通式(11)表示的氧氮氢化物的组合物。
[0204]
a3bo
5-xnyhz
ꢀꢀ
(11)
[0205]
(上述通式(11)中,a为选自由ba及sr组成的组中的至少1种,b为选自si、ge及sn组成的组中的至少1种,x表示由0.1≤x≤3.5表示的数;y表示由0.1≤y≤2.0表示的数;z表示由0.1≤z≤2.0表示的数。)
[0206]
(氨合成用催化剂)
[0207]
本发明的氨合成用催化剂为将过渡金属(m)担载于载体的物质。上述载体为包含将氮与氢掺杂于a3bo5的氧位点而成的氧氮氢化物的组合物。本实施方式的氨合成用催化剂包含过渡金属和担载上述过渡金属的载体,上述载体为包含下述通式(11)表示的氧氮氢化物的组合物。上述载体为包含上述说明的本实施方式的氧氮氢化物的组合物是优选的。
[0208]
a3bo
5-xnyhz
ꢀꢀ
(11)
[0209]
(上述通式(11)中,a为选自由ba及sr组成的组中的至少1种,b为选自si、ge及sn组成的组中的至少1种,x表示由0.1≤x≤3.5表示的数;y表示由0.1≤y≤2.0表示的数;z表示由0.1≤z≤2.0表示的数。)
[0210]
就上述氮及氢而言,只要不损害本发明的效果,其一部分可以进一步包含氮及氢这两者以外的其他原子,具体而言,可以包含电子、碳、卤原子等。
[0211]
本实施方式中使用的过渡金属与第一实施方式的过渡金属相同。
[0212]
(氨合成用催化剂的制造方法)
[0213]
本实施方式的氨合成用催化剂为将过渡金属(m)担载于载体而成的物质。上述载体为包含将氮与氢掺杂于a3bo5的氧位点的氧氮氢化物的组合物。就本实施方式的氨合成用催化剂而言,将上述过渡金属担载于包含含有上述氧氮氢化物的组合物的上述载体来进行制造。制造方法没有特别限定,通常,使过渡金属、或成为过渡金属的前体的化合物(以下、过渡金属化合物)担载于上述载体来进行制造。
[0214]
本实施方式的氨合成用催化剂可以通过与第一实施方式相同的方法进行制造。
[0215]
作为除了上述氧氮氢化物及上述过渡金属以外的成分,可进一步包含sio2、al2o3、zro2、mgo、活性炭、石墨、sic等作为上述氧氮氢化物的载体。
[0216]
本实施方式的氨合成用催化剂能够使用通常的成型技术而作为成型体使用。具体而言,可举出粒状、球状、片状、环状、通心粉、四叶、骰子、蜂窝状状等的形状。此外,也可以在适当的支承体上涂布后使用。
[0217]
在使用本实施方式的氨合成用催化剂时,其反应活性没有特别限定,在以反应温度300℃、反应压力0.9mpa时的氨的生成速率为例的情况下,1.0mmol/g
·
h以上是优选的,从适于实用的制造条件的方面出发,2.0mmol/g
·
h以上是更优选的,从适于高效率的制造条件的方面出发,3.0mmol/g
·
h以上是进一步优选的,从适于更高效率的制造条件的方面考虑,5.0mmol/g
·
h以上是进一步优选的。
[0218]
(氨的制造方法)
[0219]
就本实施方式的氨的制造方法而言,除了使用本实施方式的氨合成用催化剂以外,与第一实施方式的氨的制造方法相同。
[0220]
本实施方式的氨的制造方法中,可以单独使用通过本实施方式的制造方法得到的氨合成用催化剂,也可以与在氨合成中通常能够使用的其他已知的催化剂组合使用。
[0221]
实施例
[0222]
以下,基于实施例,对本发明更详细地进行说明。利用气相色谱仪求出nh3的生成量、或者将生成的nh3溶解于硫酸水溶液中并利用离子色谱仪对该溶液进行定量而求出氨生成速率,由此进行氨合成活性的评价。
[0223]
(离子色谱分析)
[0224]
将从反应容器排出的氨气体于5mm硫酸水溶液中溶解,利用离子色谱仪对捕捉到的铵离子(nh
4
)进行分析。分析条件如下所述。
[0225]
[测定条件]
[0226]
装置:岛津制作所公司制prominence
[0227]
检测器:电导率检测器cdd-10avp(岛津制作所公司制)
[0228]
柱:离子色谱用柱ic-c4(岛津制作所公司制)
[0229]
洗脱液:3.0mm草酸 2.0mm 18-冠-6-醚水溶液流速:1.0ml/分钟
[0230]
柱温度:40℃
[0231]
(实施例1)
[0232]
(氨合成用催化剂的制备)
[0233]
[baal2o
4-xnyhz
粉末的合成]
[0234]
通过将al2o3于600℃进行真空加热处理将表面吸附的水等除去,在ar手套箱中使用玛瑙研钵将脱水处理后的al2o3和ba(nh2)2混合。此时,使ba与al的摩尔比成为1:2的方式混合。将得到的粉体在流量100ml/min的nh3气流中以2小时升温至800℃,于800℃进行20小时加热处理,由此得到本实施例的baal2o
4-xnyhz
粉末。
[0235]
如图1所示,在于600℃以6小时进行合成的情况下,没有得到目标的baal2o4相。另一方面,在于800℃以6小时进行合成的情况下,能够确认目标的baal2o4相的存在。进而,在于800℃以20小时进行合成的本实施例中,得到大致为单相的baal2o4相。
[0236]
《baal2o
4-xnyhz
中包含的氮及氢的定量>
[0237]
将利用升温脱附分析装置(belcata)对在本实施例中合成的baal2o
4-xnyhz
进行分析的结果示于图2中。
[0238]
如图2所示,氮和氢从550℃附近开始脱附,可以确认baal2o
4-xnyhz
中包含了氮和氢。氮即使于900℃也没有完全脱附,因此难以对baal2o
4-xnyhz
中全部的氮进行定量,估算大概的含量时,可知组成为baal2o
3.7n0.17h0.09
。
[0239]
[ru向baal2o
4-xnyhz
的担载]
[0240]
将本实施例中合成的粉末状baal2o
4-xnyhz 0.50g、ru3(co)
12
(aldrich公司制、99%)0.056g(相对于ba3sio
5-xnyhz
,作为被担载的金属ru相当于5质量%)放入石英玻璃管内,将其在真空中于70℃加热1小时,然后继续于120℃加热1小时,由此使ru3(co)
12
附着于粉末状baal2o
4-xnyhz
的表面。将这样得到的粉末状物质最后于250℃加热2小时,将ru3(co)
12
热分解,由此得到在baal2o
4-xnyhz
上固定有ru的担载物(以下,ru/baal2o
4-xnyhz
)。
[0241]
以下,使用上述氨合成用催化剂,进行氨合成。
[0242]
[使用ru担载baal2o
4-xnyhz
的氨合成]
[0243]
《氨合成反应>
[0244]
将上述ru/baal2o
4-xnyhz
作为催化剂,使该催化剂与氮与氢的混合气体接触,进行氨合成反应。将上述ru/baal2o
4-xnyhz 0.1g装入sus制反应管,使用具备其的固定床流通式
反应装置进行反应。原料的氮气体和氢气体的水分浓度分别在检测限以下。关于该反应时的原料气体的流量,氮气体为15ml/min,氢气体为45ml/min(共计60ml/min)。此外该反应时的反应压力为0.9mpa,反应温度为300℃,反应时间为30小时。
[0245]
《氨的生成速率>
[0246]
使从上述固定床流通式反应装置出来的气体在0.005m硫酸水溶液中鼓泡,使上述气体中的氨溶解,利用离子色谱仪将生成的铵离子通过上述的方法进行定量。利用离子色谱仪经时地对通过氨合成反应生成的氨的生成速率进行测定,其结果是,ru/baal2o
4-xnyhz
与现有的作为ru催化剂的cs-ru/mgo相比,于所有的反应温度条件显示出高得多的催化剂活性(如图3所示)。于300℃的氨生成速率为8.4mmol/g
·
hr。该值为远高于cs-ru/mgo(0.55mmol/g
·
hr)的值。结果示于表1中。
[0247]
(实施例2)
[0248]
(氨合成用催化剂的制备)
[0249]
[ba3sio
5-xnyhz
粉末的合成]
[0250]
通过将sio2于600℃进行真空加热处理将表面吸附的水等除去,在ar手套箱中使用玛瑙研钵将脱水处理后的sio2和ba(nh2)2混合。此时,使ba与si的摩尔比为3:1的方式混合。将得到的粉体在流量100ml/min的nh3气流中以2小时升温至600℃,于600℃进行6小时加热处理,由此得到ba3sio
5-xnyhz
粉末。
[0251]
[ru向ba3sio
5-xnyhz
的担载]
[0252]
将通过上述的方法得到的粉末状ba3sio
5-xnyhz 0.50g、ru3(co)
12
(aldrich公司制、99%)0.056g(相对于ba3sio
5-xnyhz
,作为被担载的金属ru相当于5质量%)放入石英玻璃管内,将其在真空中于70℃加热1小时,然后继续于120℃加热1小时,由此将ru3(co)
12
附着于粉末状ba3sio
5-xnyhz
的表面。将这样而得到的粉末状物质最后于250℃加热2小时,将ru3(co)
12
热分解,由此得到在ba3sio
5-xnyhz
上固定有ru的担载物(以下,ru/ba3sio
5-xnyhz
)。以下,使用上述氨合成用催化剂,进行氨合成。
[0253]
[使用ru担载ba3sio
5-xnyhz
的氨合成]
[0254]
《氨合成反应>
[0255]
将上述ru/ba3sio
5-xnyhz
作为催化剂,使该催化剂与氮与氢的混合气体接触,进行氨合成反应。将上述ru/ba3sio
5-xnyhz 0.1g装入sus制反应管,使用具备其的固定床流通式反应装置进行反应。原料的氮气体的水分浓度和氢气体的水分浓度分别在检测限以下。关于该反应时的原料气体的流量,氮气体为15ml/min,氢气体为45ml/min(共计60ml/min)。此外该反应时的反应压力为0.9mpa,反应温度为300℃,反应时间为30小时。
[0256]
《氨的生成速率>
[0257]
使从上述固定床流通式反应装置出来的气体在0.005m硫酸水溶液中鼓泡,使上述气体中的氨溶解,利用离子色谱仪将生成的铵离子通过上述的方法进行定量。利用离子色谱仪经时地对通过氨合成反应生成的氨的生成速率进行测定。300℃、0.9mpa时的氨的合成速率如表1所示,为6.34mmol/g
·
hr。结果示于表1中。
[0258]
此外,改变上述氨合成反应的反应温度,评价了氨生成速率的反应温度依赖性。结果示于图3中。
[0259]
(比较例1)
[0260]
[cs-ru/mgo粉末的合成]
[0261]
除了使用添加有cs的mgo(记为cs/mgo)代替实施例1的baal2o
4-xnyhz
以外,使用与实施例1同样的方法,制备了5质量%ru-cs/mgo催化剂(cs/ru元素比=1)。
[0262]
[使用cs-ru/mgo的氨合成]
[0263]
《氨合成反应>
[0264]
在与实施例1相同条件下实施氨合成反应。300℃、0.9mpa时的氨的合成速率如表1所示,为0.55mmol/g
·
hr。结果示于表1中。
[0265]
(比较例2)
[0266]
[ru/ceo2粉末的合成]
[0267]
除了使用ceo2代替实施例1的baal2o
4-xnyhz
以外,使用与实施例1同样的方法,制备了5质量%ru/ceo2催化剂。
[0268]
[使用ru/ceo2的氨合成]
[0269]
《氨合成反应>
[0270]
在与实施例1相同条件下实施氨合成反应。300℃、0.9mpa时的氨的合成速率如表1所示,为0.72mmol/g
·
hr。结果示于表1中。
[0271]
(比较例3)
[0272]
[ru/c12a7:e-粉末的合成]
[0273]
代替实施例1的baal2o
4-xnyhz
,通过与国际公开第2012/0077658号中记载的方法同样的方法,制备了2质量%ru/c12a7:e-催化剂。
[0274]
[使用ru/c12a7:e-的氨合成]
[0275]
《氨合成反应>
[0276]
在与实施例1相同条件下实施氨合成反应。300℃、0.9mpa时的氨的合成速率如表1所示,为0.76mmol/g
·
hr。结果示于表1中。
[0277]
(比较例4)
[0278]
[ca2nh粉末的合成]
[0279]
代替实施例1的baal2o
4-xnyhz
,通过与国际公开第2015/129471号中记载的方法同样的方法,制备了5质量%ru/ca2nh催化剂。
[0280]
[使用ru/ca2nh的氨的合成]
[0281]
《氨合成反应>
[0282]
在与实施例1相同条件下实施氨合成反应。300℃、0.9mpa时的氨的合成速率如表1所示,为1.74mmol/g
·
hr。结果示于表1中。
[0283]
《baal2o
4-xnyhz
粉末的xrd>
[0284]
通过上述的方法、在实施例1中合成的试样的xrd图案示于图1中。此外,为了对比,al2o3、bao、baal2o4的xrd图案也示于图1中。
[0285]
(实施例3~5)
[0286]
[于各种各样的加热处理温度合成的ba3sio
5-xnyhz
粉末的评价]
[0287]
除了分别使用表2所示出的加热处理温度代替实施例1的nh3气流中的加热处理温度600℃以外,使用与实施例1同样的方法制备了ba3sio
5-xnyhz
粉末。
[0288]
《ba3sio
5-xnyhz
粉末的xrd>
[0289]
通过上述方法、于各种各样的加热处理温度合成的试样的xrd图案示于图4中。此外,为了对比,ba3sio5的xrd图案也示于图4中。
[0290]
如图4所示,在使加热处理温度为400℃以上进行合成的情况下,能够确认ba3sio5结晶相的存在,得到大致为单相的材料。此外,即使使加热处理温度高至800℃,也同样能够确认ba3sio5结晶相的存在。进而,确认了与标准的氧化物ba3sio5比较,峰整体地向低角度侧偏移。认为这是因为离子半径比氧大的氮被以高浓度掺杂。
[0291]
[表1]
[0292][0293]
以下为表1的实施例及比较例的反应条件。
[0294]
催化剂量:0.1g,反应温度:300℃,反应气体流量:60ml/min,
[0295]
反应气体组成:n2/h2=1/3(v/v)、反应压力:0.9mpa。[表2]
[0296][0297]
(实施例6)
[0298]
[co向baal2o
4-xnyhz
的担载]
[0299]
将在实施例1中合成的粉末状baal2o
4-xnyhz 0.855g、co2(co)8(关东化学公司制、95%)0.145g(相对于ba3sio
5-xnyhz
,作为被担载的金属co相当于5质量%)放入石英玻璃反应管,使氮15ml/min和氢45ml/min流通于其中,以2小时升温至400℃、维持5小时,由此得到在baal2o
4-xnyhz
上固定有co的担载物(以下,co/baal2o
4-xnyhz
)。
[0300]
以下,使用上述氨合成用催化剂,进行氨合成。
[0301]
[使用co担载baal2o
4-xnyhz
的氨合成]
[0302]
《氨合成反应>
[0303]
将上述co/baal2o
4-xnyhz
作为催化剂,使该催化剂与氮与氢的混合气体接触,进行氨合成反应。将上述co/baal2o
4-xnyhz 0.1g装入sus制反应管,使用具备其的固定床流通式
反应装置进行反应。原料的氮气体和氢气体的水分浓度分别在检测限以下。关于该反应时的原料气体的流量,氮气体为15ml/min,氢气体为45ml/min(计60ml/min)。此外该反应时的反应压力为0.9mpa,反应温度为300℃,反应时间为30小时。
[0304]
《氨的生成速率>
[0305]
使从上述固定床流通式反应装置出来的气体在0.005m硫酸水溶液中鼓泡,使上述气体中的氨溶解,利用离子色谱仪将生成的铵离子通过上述的方法进行定量。利用离子色谱仪经时地对通过氨合成反应生成的氨的生成速率进行测定。300℃、0.9mpa时的氨的合成速率如表3中所示,为1.11mmol/g
·
hr。结果示于表3中。
[0306]
(实施例7)
[0307]
(氨合成用催化剂的制备)
[0308]
[co向ba3sio
5-xnyhz
的担载]
[0309]
将在实施例2中得到的粉末状ba3sio
5-xnyhz
、co2(co)8(关东化学公司制、95%)(相对于ba3sio
5-xnyhz
,作为被担载的金属co相当于5质量%)放入石英玻璃反应管内,使氮15ml/min和氢45ml/min流通于其中,以2小时升温至400℃,维持5小时,由此得到在ba3sio
5-xnyhz
上固定有co的担载物(以下,co/ba3sio
5-xnyhz
)。
[0310]
以下,使用上述氨合成用催化剂,进行氨合成。
[0311]
[使用co担载ba3sio
5-xnyhz
的氨合成]
[0312]
《氨合成反应>
[0313]
将上述co/ba3sio
5-xnyhz
作为催化剂,使该催化剂与氮与氢的混合气体接触,进行氨合成反应。将上述co/ba3sio
5-xnyhz 0.1g装入sus制反应管,使用具备其的固定床流通式反应装置进行反应。确认原料的氮气体的水分浓度和氢气体的水分浓度分别在检测限以下。关于该反应时的原料气体的流量,使氮气体为15ml/min,使氢气体为45ml/min(共计60ml/min)。此外,使该反应时的反应压力为0.9mpa,使反应温度为300℃,使反应时间为30小时。
[0314]
《氨的生成速率>
[0315]
使从上述固定床流通式反应装置出来的气体在0.005m硫酸水溶液中鼓泡,使上述气体中的氨溶解,利用离子色谱仪通过上述的方法将生成的铵离子进行定量。利用离子色谱仪经时地对通过氨合成反应生成的氨的生成速率进行测定。算出300℃、0.9mpa时的氨的合成速率(mmol/g
·
hr)。
[0316]
(实施例8)
[0317]
[fe向baal2o
4-xnyhz
的担载]
[0318]
将实施例1中合成的粉末状baal2o
4-xnyhz
、fe2(co)2(strem chemicals公司制、99%)(相对于ba3sio
5-xnyhz
,作为被担载的金属fe相当于5质量%)放入石英玻璃管内,对其流通氮15ml/min和氢45ml/min流通,以2小时升温至400℃,维持5小时,由此得到在baal2o
4-xnyhz
上固定有fe的担载物(以下,fe/baal2o
4-xnyhz
)。
[0319]
以下,使用上述氨合成用催化剂,进行氨合成。
[0320]
[使用fe担载baal2o
4-xnyhz
的氨合成]
[0321]
《氨合成反应>
[0322]
将上述fe/baal2o
4-xnyhz
作为催化剂,使该催化剂与氮与氢的混合气体接触,进行
氨合成反应。将上述fe/baal2o
4-xnyhz 0.1g装入sus制反应管,使用具备其的固定床流通式反应装置进行反应。确认原料的氮气体和氢气体的水分浓度分别在检测限以下。关于该反应时的原料气体的流量,使氮气体为15ml/min,使氢气体为45ml/min(共计60ml/min)。此外使该反应时的反应压力为0.9mpa,使反应温度为300℃,使反应时间为30小时。
[0323]
《氨的生成速率>
[0324]
使从上述固定床流通式反应装置出来的气体在0.005m硫酸水溶液中鼓泡,使上述气体中的氨溶解,利用离子色谱仪通过上述的方法将生成的铵离子进行定量。利用离子色谱仪经时地对通过氨合成反应生成的氨的生成速率进行测定。算出300℃、0.9mpa时的氨的合成速率(mmol/g
·
hr)。
[0325]
(实施例9)
[0326]
(氨合成用催化剂的制备)
[0327]
[fe向ba3sio
5-xnyhz
的担载]
[0328]
将实施例2中得到的粉末状ba3sio
5-xnyhz 0.837g、fe2(co)9(strem chemicals公司制、99%)0.163g(相对于ba3sio
5-xnyhz
,作为被担载的金属fe相当于5质量%)放入石英玻璃反应管内,对其流通氮15ml/min和氢45ml/min,以2小时升温至400℃,维持5小时,由此得到在ba3sio
5-xnyhz
上固定有fe的担载物(以下,fe/ba3sio
5-xnyhz
)。
[0329]
以下,使用上述氨合成用催化剂,进行氨合成。
[0330]
[使用fe担载ba3sio
5-xnyhz
的氨合成]
[0331]
《氨合成反应>
[0332]
将上述fe/ba3sio
5-xnyhz
作为催化剂,使该催化剂与氮与氢的混合气体接触,进行氨合成反应。将上述fe/ba3sio
5-xnyhz 0.1g装入sus制反应管,使用具备其的固定床流通式反应装置进行反应。原料的氮气体的水分浓度和氢气体的水分浓度分别在检测限以下。关于该反应时的原料气体的流量,氮气体为15ml/min,氢气体为45ml/min(共计60ml/min)。此外该反应时的反应压力为0.9mpa,反应温度为300℃,反应时间为30小时。
[0333]
《氨的生成速率>
[0334]
将从上述固定床流通式反应装置出来的气体在0.005m硫酸水溶液中鼓泡,使上述气体中的氨溶解、利用离子色谱仪将生成的铵离子通过上述的方法进行定量。利用离子色谱仪经时地对通过氨合成反应生成的氨的生成速率进行测定。300℃、0.9mpa时的氨的合成速率如表1所示,为0.916mmol/g
·
hr。结果示于表1中。
[0335]
(实施例10)
[0336]
[ni向baal2o
4-xnyhz
的担载]
[0337]
将实施例1中合成的粉末状baal2o
4-xnyhz 0.839g、ni(c5h5)2(东京化成公司制、98%)0.161g(相对于ba3sio
5-xnyhz
,作为被担载的金属ni相当于5质量%)放入石英玻璃反应管内,对其流通氮15ml/min和氢45ml/min,以2小时升温至400℃,维持5小时,由此得到在baal2o
4-xnyhz
上固定有ni的担载物(以下,ni/baal2o
4-xnyhz
)。
[0338]
以下,使用上述氨合成用催化剂,进行氨合成。
[0339]
[使用ni担载baal2o
4-xnyhz
的氨合成]
[0340]
《氨合成反应>
[0341]
将上述ni/baal2o
4-xnyhz
作为催化剂,使该催化剂与氮与氢的混合气体接触,进行
氨合成反应。将上述ni/baal2o
4-xnyhz 0.1g装入sus制反应管,使用具备其的固定床流通式反应装置进行反应。原料的氮气体和氢气体的水分浓度分别在检测限以下。关于该反应时的原料气体的流量,氮气体为15ml/min,氢气体为45ml/min(共计60ml/min)。此外该反应时的反应压力为0.9mpa,反应温度为400℃,反应时间为30小时。
[0342]
《氨的生成速率>
[0343]
使从上述固定床流通式反应装置出来的气体在0.005m硫酸水溶液中鼓泡,使上述气体中的氨溶解、利用离子色谱仪将生成的铵离子通过上述的方法进行定量。利用离子色谱仪经时地对通过氨合成反应生成的氨的生成速率进行测定。400℃、0.9mpa时的氨的合成速率如表3所示,为1.20mmol/g
·
hr。结果示于表3中。
[0344]
(实施例11)
[0345]
(氨合成用催化剂的制备)
[0346]
[ni向ba3sio
5-xnyhz
的担载]
[0347]
将实施例2中得到的粉末状ba3sio
5-xnyhz 0.839g、ni(c5h5)2(东京化成公司制、98%)0.161g(相对于ba3sio
5-xnyhz
,作为被担载的金属ni相当于5质量%)放入石英玻璃反应管内,对其流通氮15ml/min和氢45ml/min,以2小时升温至400℃,维持5小时,由此得到在ba3sio
5-xnyhz
上固定有ni的担载物(以下,ni/ba3sio
5-xnyhz
)。
[0348]
以下,使用上述氨合成用催化剂,进行氨合成。
[0349]
[使用ni担载ba3sio
5-xnyhz
的氨合成]
[0350]
《氨合成反应>
[0351]
将上述ni/ba3sio
5-xnyhz
作为催化剂,使该催化剂与氮与氢的混合气体接触,进行氨合成反应。将上述ni/ba3sio
5-xnyhz 0.1g装入sus制反应管,使用具备其的固定床流通式反应装置进行反应。原料的氮气体的水分浓度和氢气体的水分浓度分别在检测限以下。关于该反应时的原料气体的流量,氮气体为15ml/min,氢气体为45ml/min(共计60ml/min)。此外,该反应时的反应压力为0.9mpa,反应温度为400℃,反应时间为30小时。
[0352]
《氨的生成速率>
[0353]
使从上述固定床流通式反应装置出来的气体在0.005m硫酸水溶液中鼓泡,使上述气体中的氨溶解,利用离子色谱仪将生成的铵离子通过上述的方法进行定量。利用离子色谱仪经时地对通过氨合成反应生成的氨的生成速率进行测定。400℃、0.9mpa时的氨的合成速率如表1所示,为0.282mmol/g
·
hr。结果示于表1中。
[0354]
[表3]
[0355]
[0356]
以下为表3的实施例6及9的反应条件。
[0357]
催化剂量:0.1g,反应温度:300℃,反应气体流量:60ml/min,
[0358]
反应气体组成:n2/h2=1/3(v/v)、反应压力:0.9mpa。
[0359]
以下为表3的实施例10及11的反应条件。
[0360]
催化剂量:0.1g,反应温度:400℃,反应气体流量:60ml/min,
[0361]
反应气体组成:n2/h2=1/3(v/v)、反应压力:0.9mpa。
[0362]
本发明的氨合成用催化剂的效果在于下述方面:与在合成复合氧化物后将其氧位点用氮、氢取代的通常的方法不同,通过将包含a
nbmol
型复合氧化物的a位点元素的金属酰胺材料与包含b位点元素的过渡金属氧化物作为原料使用,可以在一步并且低温下合成氧氮氢化物。
[0363]
此外,在通常的氧化物上固定有ru等过渡金属纳米粒子的催化剂中,与通常的氨合成催化剂同样地,在过渡金属纳米粒子上发生氮分子及氢分子的解离,通过生成氨的langmuir-hinshelwood反应机理进行反应。然而,在本发明的催化剂中,通过在载体材料的骨架中掺杂的氮及氢直接参与反应的mars van krevelen机理进行氨合成,特别是即使在低温区域也显示了高的催化剂活性。其结果是,本发明的催化剂的相对于氨合成的活化能显示出与以往催化剂相比为其一半左右的值。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。