(1r,2s)-(3,4-二氟苯基)环丙胺的盐酸盐晶型的制备方法
技术领域
1.本发明涉及一种(1r,2s)-(3,4-二氟苯基)环丙胺的盐酸盐晶型的制备方法。
背景技术:
2.(1r,2s)-(3,4-二氟苯基)环丙胺盐酸盐是抗血小板药物替格瑞洛的重要中间体之一,且具有式a的化学结构:
[0003][0004]
替格瑞洛是由阿斯利康公司研发的一种新型、具有选择性的抗血小板药,该药能可逆性地作用于血管平滑肌细胞(vsmc)上的嘌呤2受体(purinoceptor2,p2)亚型p2y12,不需要代谢激活,对二磷酸腺苷(adp)引起的血小板聚集有明显的抑制作用,且口服使用后起效迅速,能有效改善急性冠心病患者的症状。与噻吩并吡啶类药物不同,替卡格雷对p2y12受体是可逆抑制剂,所以对于那些需在先期进行抗凝治疗后再行手术的病人尤为适用。
[0005]
中国专利申请cn104030930b公开了(1r,2s)-(3,4-二氟苯基)环丙胺盐酸盐的制备方法,但并未涉及晶型的方面。
[0006]
在(1r,2s)-(3,4-二氟苯基)环丙胺盐酸盐的制备中,发现cn104030930b中的制备方法得到的化合物纯度波动较大,且使用到了钯试剂,提高了反应成本,不利于大规模生产。
[0007]
本领域技术人员熟知在医药合成领域中需要高纯度的化合物。极高的纯度可改进长期储存的稳定性。另一方面,降低反应成本,更有利于工业化。
[0008]
现有技术中记载了杂质imp.1化合物作为参考标准控制(1r,2s)-(3,4-二氟苯基)环丙胺的质量,并具体记载了纯品(1r,2s)-(3,4-二氟苯基)环丙胺中杂质imp.1的含量为0.01%~0.6%,但现有技术并未记载相关纯化方法以使(1r,2s)-(3,4-二氟苯基)环丙胺中的imp.1杂质含量达到安全标准的方法。
技术实现要素:
[0009]
本发明所要解决的技术问题是现有的(1r,2s)-(3,4-二氟苯基)环丙胺盐酸盐的纯度有待提高,为此,本发明提供了一种(1r,2s)-(3,4-二氟苯基)环丙胺的盐酸盐晶型的制备方法。该方法能以高收率、高纯度制备得到如式a所示的化合物的晶型,其重现性好,易于规模化生产。
[0010]
本发明提供了一种如式a所示化合物的晶型的制备方法,其包括下述步骤:将反溶剂加入至溶液中,析晶,得到如式a所示的化合物的晶型,所述溶液为含如式a所示的化合物的粗品与溶剂形成的溶液,
[0011][0012]
如式a所示化合物的晶型的制备方法中,所述溶剂可为醇类溶剂和/或酮类溶剂;所述醇类溶剂可为甲醇、乙醇和异丙醇中的一种或多种,优选为甲醇和/或乙醇;所述酮类溶剂可为甲基异丁基酮。
[0013]
如式a所示化合物的晶型的制备方法中,所述反溶剂可为醚类溶剂、卤代烃类溶剂和酯类溶剂中的一种或多种;所述醚类溶剂优选为甲叔醚和/或异丙醚;所述卤代烃类溶剂优选为二氯甲烷;所述酯类溶剂优选为醋酸异丙酯、乙酸乙酯和醋酸叔丁酯中的一种或多种;较佳地,所述反溶剂优选醋酸异丙酯、异丙醚和乙酸乙酯中的一种或多种。
[0014]
如式a所示化合物的晶型的制备方法中,所述如式a所示化合物和反溶剂的质量体积比的比值可为0.2~0.3g/ml,例如0.25g/ml。
[0015]
如式a所示化合物的晶型的制备方法中,所述溶剂和反溶剂的体积比优选为1:(0.1~10),更优选1:(0.2~5),例如1:4.2或1:5。
[0016]
如式a所示化合物的晶型的制备方法中,所述溶剂和反溶剂的组合可为甲醇/甲叔醚、甲醇/异丙醚、甲醇/醋酸异丙酯、甲醇/乙酸乙酯、甲醇/醋酸叔丁酯、乙醇/甲叔醚、乙醇/异丙醚、乙醇/醋酸异丙酯、乙醇/乙酸乙酯、乙醇/醋酸叔丁酯、异丙醇/甲叔醚、异丙醇/异丙醚、异丙醇/醋酸异丙酯、异丙醇/乙酸乙酯、或异丙醇/醋酸叔丁酯,例如甲醇/醋酸异丙酯、甲醇/乙酸乙酯、甲醇/异丙醚、乙醇/异丙醚、或乙醇/乙酸乙酯。
[0017]
如式a所示化合物的晶型的制备方法中,所述含如式a所示的化合物的粗品还可包括杂质imp.1化合物,所述的杂质imp.1化合物的结构式为:所述含如式a所示的化合物的粗品中杂质imp.1化合物的含量可为0.6%~8%,例如4.9%。
[0018]
如式a所示化合物的晶型的制备方法中,所述溶液可为将所述如式a所示的化合物与所述溶剂升温至溶清,然后降温得到。
[0019]
如式a所示化合物的晶型的制备方法中,所述溶液为将所述如式a所示的化合物与所述溶剂升温至溶清,然后降温得到,所述溶液升温至溶清的温度为75℃~90℃,例如75℃~80℃。
[0020]
如式a所示化合物的晶型的制备方法中,所述溶液为将所述如式a所示的化合物与所述溶剂升温至溶清,然后降温得到,所述溶液的降温可为下述步骤:将所述溶液降温至70
±
5℃,保温1
±
0.5h,然后降温至60
±
5℃,保温1
±
0.5h,然后降温至50
±
5℃,保温1
±
0.5h;或者,所述溶液的降温还可通过持续降低温度或经由预定冷却梯度降低温度的形式进行控制,所述预定冷却梯度可为约60分钟内降至70
±
5℃,然后约60分钟内降至60
±
5℃,然后再约60分钟内降至50
±
5℃。
[0021]
如式a所示化合物的晶型的制备方法中,所述析晶的温度可为将所述反溶剂和所述溶液形成的混合溶液降温至-10℃~35℃,优选为0℃~35℃,更优选为10℃~30℃,例如25℃~30℃。
[0022]
如式a所示化合物的晶型的制备方法中,较佳地,所述反溶剂滴加至所述溶液中。
[0023]
如式a所示化合物的晶型的制备方法中,当所述反溶剂滴加至所述溶液中时,所述滴加的速度可为50
±
5ml/h。
[0024]
如式a所示化合物的晶型的制备方法中,当所述反溶剂滴加至所述溶液中时,所述滴加时的温度可为50
±
5℃。
[0025]
如式a所示化合物的晶型的制备方法还可包括后处理,所述后处理可包括过滤、洗涤、干燥。
[0026]
所述后处理中,所述洗涤使用的溶剂可为甲叔醚、异丙醚和醋酸异丙酯中的一种或多种,优选为甲叔醚。所述洗涤过程的温度可为0℃~5℃。
[0027]
所述后处理过程中,所述干燥过程的温度、压力和持续时间的设定值以可使一种或多种溶剂的含量降至给定值以下。例如:如式a所示化合物的晶型中的溶剂含量低于或等于5000ppm,优选低于2000ppm,更优选低于1000ppm。
[0028]
如式a所示化合物的晶型的制备方法中,所述如式a所示的化合物的晶型,使用cuk
α1
辐射,以2θ角表示的x-射线粉末衍射图,在15.59
±
0.2
°
,18.35
±
0.2
°
,24.84
±
0.2
°
,28.36
±
0.2
°
,29.14
±
0.2
°
,31.7
±
0.2
°
,33.04
±
0.2
°
和36.3
±
0.2
°
处具有衍射峰。
[0029]
如式a所示化合物的晶型的制备方法中,所述如式a所示的化合物的晶型的x-射线粉末衍射图,还可包括选自11.21
±
0.2
°
,16.75
±
0.2
°
,18.78
±
0.2
°
,21.31
±
0.2
°
,22.25
±
0.2
°
,23.58
±
0.2
°
,25.44
±
0.2
°
,26.76
±
0.2
°
,31.38
±
0.2
°
,34.81
±
0.2
°
,37.02
±
0.2
°
,38.09
±
0.2
°
,38.88
±
0.2
°
,39.62
±
0.2
°
,40.95
±
0.2
°
,41.62
±
0.2
°
,42.50
±
0.2
°
,43.09
±
0.2
°
,43.40
±
0.2
°
中的至少一个2θ特征吸收峰。
[0030]
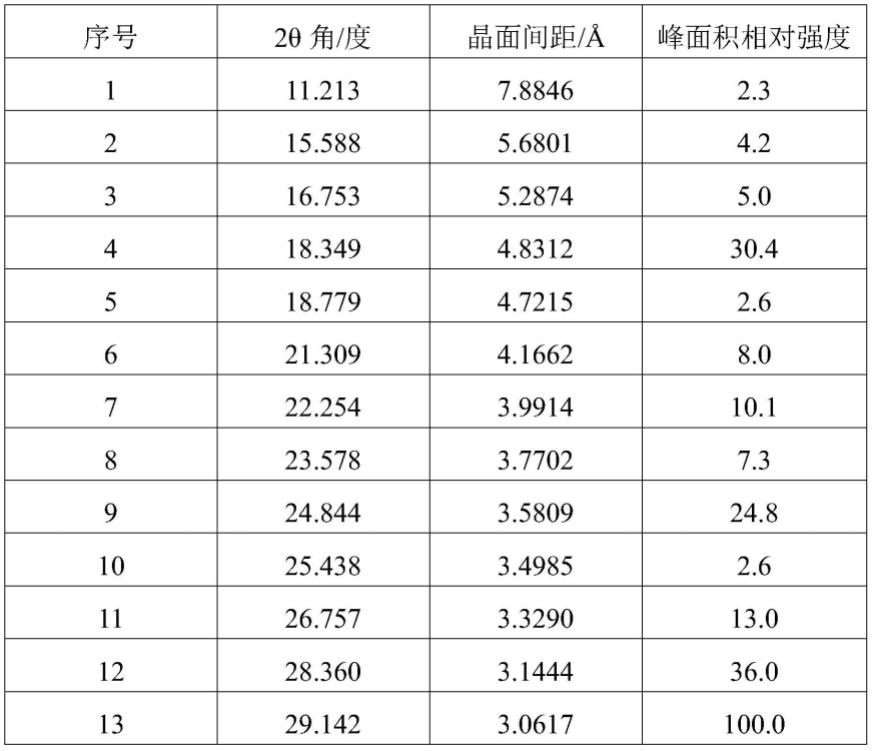
如式a所示化合物的晶型的制备方法中,较佳地,所述如式a所示的化合物的晶型以2θ角表示的x-射线粉末衍射图,其衍射峰和相对强度如下表所示:
[0031]
[0032][0033]
;更佳地,所述如式a所示的化合物的晶型以2θ角表示的x-射线粉末衍射图基本上如图1所示。
[0034]
如式a所示的化合物的晶型的制备方法中,还可包括以下步骤:在溶剂中,化合物sm5和氯化氢反应,得到所述含如式a所示的化合物的粗品,即可,所述溶剂同如前所述的任一项制备方法中包含的溶剂,
[0035][0036]
如式a所示的化合物的晶型的制备方法中,较佳地,所述化合物sm5以sm5溶于所述溶剂形成的sm5溶液的形式参与反应;更佳地,所述sm5溶液中化合物sm5和溶剂的质量体积比的比值为1.0g/ml~1.5g/ml,例如1.0g/ml。
[0037]
如式a所示的化合物的晶型的制备方法中,较佳地,所述氯化氢以氯化氢溶于所述溶剂形成的氯化氢溶液的形式参与反应。所述氯化氢溶液中氯化氢的质量浓度可为20%。
[0038]
如式a所示的化合物的晶型的制备方法中,所述化合物sm5和氯化氢的摩尔比可为1:(1~1.5),例如1:1。
[0039]
如式a所示的化合物的晶型的制备方法中,所述反应的温度可为本领常规,例如75℃~90℃,再例如75℃或80℃。
[0040]
如式a所示的化合物的晶型的制备方法中,所述化合物sm5相对于溶剂与反溶剂形成混合溶剂的质量比可为1:(2~7),例如1:(3~5),再例如1:(4~5)。
[0041]
如式a所示的化合物的晶型的制备方法中,还可进一步包括以下步骤:化合物sm4在次氯酸钠的碱性溶液作用下,发生hofmann降解反应,得到所述化合物sm5即可;
[0042][0043]
所述的hofmann降解反应的步骤和条件可以参照本领域常规的hofmann降解反应的步骤和条件进行选择。
[0044]
如式a所示的化合物的晶型的制备方法中,还可进一步包括以下步骤:溶剂中,碱性条件下,化合物sm3与氨甲醇溶液发生酯交换反应,得到所述的化合物sm4,即可,
[0045]
[0046]
所述的化合物sm4的制备方法的步骤和条件可以参照本领域常规的酯交换反应的步骤和条件进行选择。
[0047]
如式a所示的化合物的晶型的制备方法中,还可进一步包括以下两个步骤,步骤一:溶剂中,化合物sm1在碱的作用下反应得到化合物sm2;步骤二:溶剂中,在碱的存在下,化合物sm2和膦酰基乙酸三乙酯反应,得到所述的化合物sm3,即可,
[0048][0049]
所述化合物sm3的制备方法的步骤和条件可以参照本领域常规的类似反应的步骤和条件进行选择。
[0050]
本发明还提供了一种如式a所示的化合物的晶型,所述晶型的x-射线粉末衍射图的2θ特征吸收峰为:15.59
±
0.2
°
,18.35
±
0.2
°
,24.84
±
0.2
°
,28.36
±
0.2
°
,29.14
±
0.2
°
,31.7
±
0.2
°
,33.04
±
0.2
°
和36.3
±
0.2
°
,
[0051][0052]
在某一实施方案中,所述如式a所示的化合物的晶型的x-射线粉末衍射图,还可包括选自11.21
±
0.2
°
,16.75
±
0.2
°
,18.78
±
0.2
°
,21.31
±
0.2
°
,22.25
±
0.2
°
,23.58
±
0.2
°
,25.44
±
0.2
°
,26.76
±
0.2
°
,31.38
±
0.2
°
,34.81
±
0.2
°
,37.02
±
0.2
°
,38.09
±
0.2
°
,38.88
±
0.2
°
,39.62
±
0.2
°
,40.95
±
0.2
°
,41.62
±
0.2
°
,42.50
±
0.2
°
,43.09
±
0.2
°
,43.40
±
0.2
°
中的至少一个2θ特征吸收峰。
[0053]
在某一实施方案中,所述如式a所示的化合物的晶型的x-射线粉末衍射图的数据还可如下表所示:
[0054]
[0055][0056]
在某一实施方案中,所述如式a所示的化合物的晶型以2θ角表示的x-射线粉末衍射图基本上还可如图1所示。
[0057]
在某一实施方案中,所述如式a所示的化合物的晶型的制备方法中,制备得到的化合物sm5中,通过hplc测定杂质imp.1化合物的含量可为0.6%~8%,例如4.9%,
[0058][0059]
在某一实施方案中,所述如式a所示的化合物的晶型在通过hplc测定时杂质imp.1化合物的含量可等于或小于0.5%,例如等于或小于0.1%,再例如等于或小于0.05%。
[0060]
在某一实施方案中,所述hplc测定方法的色谱条件如下:
[0061]
(a)色谱柱:十八烷基硅烷键合硅胶为填充剂的色谱柱;例如phenomenex gemini c18 4.6
×
250mm,5μm;
[0062]
(b)流动相:(a)0.01mol/l磷酸二氢钾溶液和0.05mol/l的高氯酸钠用磷酸调节ph值至2.5;(b)乙腈;
[0063]
(c)洗脱条件:
[0064]
时间(分钟)流动相a(%)流动相b(%)08020284060324060
32.18020408020
[0065]
(d)检测波长:210nm;流速:1.0ml/min;柱温40℃;
[0066]
(e)称取供试液,取适量于自动进样瓶,进样量10ul,所述供试液为如式a所述的晶型溶于溶剂中经离心形成的离心液,浓度为1mg/ml;所述的溶剂同如前所述的任一项中所述的溶剂。
[0067]
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0068]
本发明所用试剂和原料均市售可得。
[0069]
本发明的积极进步效果在于:本发明提供了一种(1r,2s)-(3,4-二氟苯基)环丙胺的盐酸盐晶型的制备方法。该方法能以高收率、高纯度制备得到如式a所示的化合物的晶型,该方法重现性好,易于规模化生产。
附图说明
[0070]
图1为实施例4的(1r,2s)-(3,4-二氟苯基)环丙胺盐酸盐晶型粉末x-衍射图
具体实施方式
[0071]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0072]
以下合成实施例用于制备化合物a及其所述的晶型方法,即将其视为举例说明的可能方法,而不将本发明限制在其内容中。合称路线如下:
[0073][0074]
实施例1
[0075][0076]
取1l四颈瓶中加入sm1(100g,0.52mol),甲苯(500ml)。机械搅拌,冰浴下滴加氢氧化钠水溶液(41.3g氢氧化钠溶解于300ml水,1.04mol),约30min滴毕。继续反应2h,hplc监测原料<1%。加入饱和食盐水(200ml),静置分层,有机层用无水硫酸钠干燥,减压蒸除部分溶剂,得sm2甲苯溶液。氮气保护下,2l四颈瓶中加入上述sm2甲苯溶液,叔丁醇钠(55g,0.57mol),机械搅拌,冰浴下缓慢滴加膦酰基乙酸三乙酯(127.95g,0.57mol),约30min滴毕。升温至内温60℃,继续反应14h,hplc监测sm2<2%。冰浴下,加入水(500ml)进行淬灭,静置分层,水层用甲苯(250ml)萃取,合并有机相,用饱和氯化钠水溶液洗涤,减压蒸除溶剂,得棕色油状物sm3(118.0g,101.1%)。
[0077]
esi-ms(m/z):227.0[m h]
。
[0078]1h nmr(cdcl3)δ:1.19~1.23(m,1h),1.27~1.29(t,3h,j=7.0hz),1.57~1.59(m,1h),1.83~1.85(m,1h),2.46~2.49(m,1h),4.16~4.21(q,2h,j1=9.0hz,j2=4.3hz),6.80~6.90(m,2h),7.0~7.26(m,1h)。
[0079]
实施例2
[0080][0081]
取350ml厚壁耐压瓶中加入上述油状物sm3(118.0g),甲醇(250ml),冰浴下通氨气至饱和,加入甲酸甲酯(37.4g,0.62mol),甲醇钠甲醇溶液(102.8g,质量分数为30%),密闭加热至60℃,反应4h,hplc监测原料<1%,将反应液冷却至40℃,吹扫氨气,蒸除2/3体积甲醇,升温至60℃,缓慢滴加水(800ml),有固体析出,搅拌2h,自然降至室温,搅拌1h,过滤,滤饼用水洗涤(100ml
×
2),置45℃恒温烘箱中干燥,得白色固体sm4(77.1g,收率75.1%)。
[0082]
esi-ms(m/z):198.0[m h]
。
[0083]1h nmr(dmso-d6)δ:1.16~1.23(m,1h),1.29~1.34(m,1h),1.79~1.85(m,1h),2.24~2.26(m,1h),5.73(s,2h),6.79~6.87(m,2h),6.89~7.01(m,1h)。
[0084]
实施例3
[0085][0086]
四颈瓶中加入30%的氢氧化钠水溶液(243.6g,1.80mol),机械搅拌,次氯酸钠水溶液(176.7g,0.66mol,有效率含量13.3%),30℃下,分批加入sm4(60g,0.30mol),加完继续反应12h,hplc监测原料<2%,将反应液冷却至0~5℃,控温不超过10℃,滴加浓盐酸调
ph 8~9,加入二氯甲烷(500ml),分液,水层用二氯甲烷(200ml
×
2)萃取,合并有机层,水洗(200ml
×
2),减压蒸除溶剂,得黄色油状物sm5(52.1g),通过hplc测定油状物sm5中杂质imp.1化合物的含量为4.9%。
[0087]
实施例4
[0088]
上述制得的sm5油状物50.0g(杂质imp.1化合物的含量为4.9%),加入无水甲醇50ml溶清,滴加20%氯化氢甲醇溶液60g。升温至t=75℃溶清,缓慢降温至70℃,保温1h,缓慢降温至60℃,保温1h,缓慢降温至50℃,保温1h,t=50℃缓慢滴加醋酸异丙酯250ml(约五小时滴加完毕,控制滴加速度)滴加完毕,t=50℃保温反应1h,缓慢降温至25-30℃保温反应4h,离心得白色片状结晶固体,冰甲叔醚50ml洗涤,烘干,称重得化合物a的晶型58.0g,摩尔收率95.3%,纯度99.6%。
[0089]
经由hplc测定纯度和杂质。色谱分析方法如下:
[0090]
(a)色谱柱:十八烷基硅烷键合硅胶为填充剂的色谱柱(phenomenex gemini c18 4.6
×
250mm,5μm);
[0091]
(b)流动相:(a)0.01mol/l磷酸二氢钾溶液和0.05mol/l的高氯酸钠用磷酸调节ph值至2.5;(b)乙腈;
[0092]
(c)洗脱条件:
[0093]
时间(分钟)流动相a(%)流动相b(%)0802028406032406032.18020408020
[0094]
(d)检测波长:210nm、流速:1.0ml/min;柱温40℃;
[0095]
(e)称取供试液(此处供试液为如式a所述的晶型溶于溶剂中经离心形成的离心液,其浓度为1mg/ml),取适量于自动进样瓶,进样量10ul。
[0096]
化合物a的晶型的纯度为99.4%;杂质imp.1化合物检测不到。
[0097]
如图1所示xrpd检测证明为化合物a的晶型。化合物a的晶型的xrpd衍射峰如表1所示。
[0098]
表1
[0099]
[0100][0101]
实施例5
[0102]
上述制得的sm5油状物50.0g(杂质imp.1化合物的含量为4.9%),加入无水甲醇50ml溶清,滴加20%氯化氢甲醇溶液60g。升温至t=75℃溶清,缓慢降温至70℃,保温1h,缓慢降温至60℃,保温1h,缓慢降温至50℃,保温1h,t=50℃缓慢滴加乙酸乙酯250ml(约五小时滴加完毕,控制滴加速度)滴加完毕,t=50℃保温反应1h,缓慢降温至25-30℃保温反应4h,离心得白色片状结晶固体,冰甲叔醚50ml洗涤,烘干,称重得化合物a的晶型55.0g,摩尔收率90.4%,纯度99.5%,杂质imp.1化合物检测不到。
[0103]
实施例6
[0104]
上述制得的sm5油状物50.0g(杂质imp.1化合物的含量为4.9%),加入无水甲醇50ml溶清,滴加20%氯化氢甲醇溶液60g。升温至t=75℃溶清,缓慢降温至70℃,保温1h,缓慢降温至60℃,保温1h,缓慢降温至50℃,保温1h,t=50℃缓慢滴加异丙醚250ml(约五小时滴加完毕,控制滴加速度)滴加完毕,t=50℃保温反应1h,缓慢降温至25-30℃保温反应4h,离心得白色片状结晶固体,冰甲叔醚50ml洗涤,烘干,称重得化合物a的晶型56.0g,摩尔收率92.0%,纯度99.5%,杂质imp.1化合物检测不到。
[0105]
实施例7
[0106]
上述制得的sm5油状物50.0g(杂质imp.1化合物的含量为4.9%),加入无水乙醇60ml溶清,滴加20%氯化氢乙醇溶液60g。升温至t=80℃溶清,缓慢降温至70℃,保温1h,缓慢降温至60℃,保温1h,缓慢降温至50℃,保温1h,t=50℃缓慢滴加异丙醚250ml(约五小时滴加完毕,控制滴加速度)滴加完毕,t=50℃保温反应1h,缓慢降温至25-30℃保温反应4h,离心得白色片状结晶固体,冰甲叔醚50ml洗涤,烘干,称重得化合物a的晶型54.7g,摩尔收率90.0%,纯度99.4%,杂质imp.1化合物检测不到。
[0107]
实施例8
[0108]
上述制得的sm5油状物50.0g(杂质imp.1化合物的含量为4.9%),加入无水乙醇60ml溶清,滴加20%氯化氢乙醇溶液60g。升温至t=80℃溶清,缓慢降温至70℃,保温1h,缓慢降温至60℃,保温1h,缓慢降温至50℃,保温1h,t=50℃缓慢滴加乙酸乙酯250ml(约五小
时滴加完毕,控制滴加速度)滴加完毕,t=50℃保温反应1h,缓慢降温至25-30℃保温反应4h,离心得白色片状结晶固体,冰甲叔醚50ml洗涤,烘干,称重得化合物a的晶型54.0g,摩尔收率88.8%,纯度99.4%,杂质imp.1化合物检测不到。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。