1.本发明涉及用于检测含有减毒病毒的样品中至少一种回复病毒(revertant virus)的存在的方法。本发明还涉及用于定量分析病毒样品中病毒单倍型的混合物的方法。本发明还涉及可用于执行所述方法的试剂盒。
2.发明背景
3.登革热病毒是黄病毒科(flaviviridae)中的虫媒病毒。它是一种具有立方型衣壳和线性正义rna基因组的包膜病毒。登革热病毒在大部分热带地区流行,并且由传染性蚊子、通常是埃及伊蚊(aedesaegypti)传播,从而导致有症状的感染或无症状的血清转化。有症状的登革热感染是一种全身性的动态性疾病。它具有包括严重和非严重的临床表现在内的广泛的临床谱(clinical spectrum)。有四种不同的登革热血清型(denv 1-4),它们在遗传上与黄热病和类似的蜱传脑炎病毒有关。
4.目前在临床试验中测试的一种四价登革热疫苗(tdv)由所有四种登革热血清型tdv1、tdv2、tdv3和tdv4组成。tdv2是一种源自pdk-53的活减毒重组病毒,该pdk-53在den-2病毒在原代犬肾(pdk)细胞中连续传代后变得非致病性。对于tdv2病毒的减毒表型是必需且足够的突变已被遗传鉴定(butrapet等人,j.virol.,第74卷,第7期(2000),3011-3019)。这些突变分别是5’非编码区中基因组核苷酸位置57处的c-至-t,以及非结构蛋白(ns)1和3中氨基酸位置53和250处的gly-至-asp和glu-至-val突变。这对应于基因组核苷酸位置57处的c至t突变、基因组核苷酸位置2579处的g-至a突变和基因组核苷酸位置5270处的a至t突变。这三个减毒遗传决定子位于tdv2病毒基因组的结构基因区之外。tdv1、tdv3和tdv4分别是具有登革热病毒2pdk53骨架及登革热病毒1、登革热病毒3和登革热病毒4的prm和e蛋白的嵌合变体,该嵌合变体维持了tdv-2的减毒表型。
5.当被施用于人中时,tdv由四种病毒组成。由于疫苗病毒复制,宿主免疫响应被触发。病毒的扩增可能会产生突变,导致回复为野生型。在疫苗接种后的前30天内出现发热病例的情况下,重要的是要确认这些患病不是由可能已回复为野生型并且成为致病性的疫苗病毒引起的,但可能是由于感染其他密切相关的黄病毒或任何其他微生物剂导致。在如此复杂的情况下,辨认和鉴定可疑样品中存在的基因组变体非常重要。这需要可准确捕获rna病毒基因组的遗传多样性的测序方法。
6.wo 2017/179017 a1描述了使用基于taqman的错配扩增突变测定(taq-mama)来灵敏检测含有减毒病毒的样品中回复体(revertant)的存在,该测定不基于测序。
7.huang等人,plos negl.trop.diseases 7(2013),e2243,1-11涉及制造四价登革热疫苗(denvax)种子的遗传和表型特征。对于病毒的测序,使用了自动测序仪上的sanger测序。为了测试疫苗的位置5’nc-57基因座处的回复,使用了基于taqman的错配扩增突变测定(taq-mama)。这代表了改进的定量snp测定。根据作者的说法,检测灵敏度可以降低到检测0.01-0.07%回复。作者进一步报告说,通过共有基因组序列分析可检测到的回复灵敏度限值为10%至30%。因此,本领域技术人员会认为基因组序列分析不适用于灵敏检测含有减毒病毒的样品中的回复体。
8.此外,已经在鼠神经毒力模型中测试了可能的回复体。
9.因此,现有技术所经历的缺点是不能提供完整的单病毒单倍型诸如回复体的序列信息。由于缺乏对完整单病毒单倍型的鉴定,现有技术无法量化复杂混合物诸如患者样品或活减毒疫苗中完整单病毒单倍型的量。
10.ardui等人,nucl.acids res.46(2018),2159-2168描述了smrt测序已在乙型肝炎病毒、丙型肝炎病毒和hiv-1中执行。那些研究都没有涉及在含有减毒病毒的样品中检测回复体。
11.dilernia等人,nucl.acids res.43(2015),e129,第1页至第13页(由ardui等人引用)描述了用于smrt测序方法纠错的复杂算法的应用。从表1可以看出,根本无法检测到预期频率低于1.56%的基因组变体。对于更高的频率,观察值至少在某种程度上与预期值有显著差异。研究的基因组彼此之间至少有五个核苷酸不同。
12.鉴于此,需要提供用于检测含有活减毒病毒(特别是登革热病毒)的复杂样品中的回复体的改进方法。进一步地,需要对复杂样品(特别是含有登革热病毒的样品)中的单病毒单倍型进行定量。
技术实现要素:
13.本发明根本的技术问题通过提供如权利要求中定义的主题来解决。
14.根据第一方面,用于检测含有减毒病毒的样品中至少一种回复病毒的存在的方法包括以下步骤:
15.a)从所述样品纯化病毒核酸;
16.b)任选地,从所述病毒核酸制备双链dna模板;
17.c)通过聚合酶链式反应(pcr)扩增a)的所述病毒核酸或b)的所述双链dna模板,以使得生成具有至少2kb的长度并且包含所述减毒病毒的至少一个减毒基因座的pcr产物;
18.d)通过单分子实时(smrt)测序对所述pcr产物进行测序;以及
19.e)将所述pcr产物中所述至少一个减毒基因座处的至少一个核苷酸与野生型病毒中的相应核苷酸进行比较,其中所述野生型核苷酸在所述至少一种减毒基因座处的存在指示所述样品中回复病毒的存在。
20.发明人发现smrt测序(pacbio测序)可用于活减毒病毒疫苗样品中的回复体的定性和定量确定。已知例如与传统sanger方法的0.001%的低测序错误率相比,smrt测序表现出13%的高测序错误率。由于使用高数量或读段计数以及额外高数量的umi计数,pacbio固有的高序列错误显著降低。成功使用smrt测序来确定长度超过5kb的序列中的单个或多个回复体,其中回复体在活减毒疫苗样品中以低量出现可以被认为是令人惊讶的有利效果。smrt测序的使用提供了高读段长度的优势,这对于覆盖整个登革热病毒基因组很有用。对活减毒疫苗样品的smrt测序相对于已知方法(诸如三个单独的pcr测序反应)的进一步优势是,当使用常规pcr方法来确定减毒基因座处的序列时,与“平均”单倍型相比,可以确定单个单倍型的完整序列。平均单倍型意指序列不是通过对单个核酸分子的测序来确定的,而是通过对覆盖完整核酸分子的不同部分的不同核酸分子的测序来确定的。因此,所确定的位于第二或第三减毒基因座处的核酸序列可能不源自用于确定第一减毒基因座处的序列的核酸分子相同的核酸分子。虽然sanger测序可用于确定包含不同核酸分子的样品中回复
体的存在,但它不能在一次测序反应中确定回复体的完整核酸序列。
21.已知smrt测序与第二代测序技术相比展现出较低的通量。这被认为是定量分析(诸如基因/同种型丰度估计)的障碍。为了克服这一障碍,诸如smrt的第三代测序技术可以被评价为混合测序,即第二代和第三代测序技术数据的组合。因此,令人惊讶的是,单独使用smrt测序足以对复杂的病毒混合物(诸如登革热病毒)进行定量分析。本发明人在优选实施方案中提供了一种用于检测含有减毒病毒的样品中的回复体的高度灵敏的方法。基于样品中病毒的总量,可检测到的回复病毒的量可能低至0.2%。这明显优于针对hiv病毒的现有技术方法。
22.此外,在本发明的优选实施方案中,样品中存在的至少两种不同的单倍型可以被彼此区分开来,它们可以仅在单个核苷酸上彼此不同。这明显优于用于hiv病毒的现有技术方法,后者只能区分至少五个核苷酸彼此不同的单倍型。
23.根据第二方面,提供了一种用于定量分析病毒样品中的病毒单倍型混合物的方法,其包括以下步骤:
24.a)从所述样品纯化病毒核酸;
25.b)任选地,通过使用包含唯一分子标识符(umi)序列的引物从所述病毒核酸制备双链dna模板以使得所述双链dna模板包含umi序列;
26.c)通过聚合酶链式反应(pcr)扩增所述双链dna模板以使得具有至少2kb的长度并且包含唯一分子标识符(umi)序列的pcr产物;
27.d)通过单分子实时(smrt)测序对所述pcr产物进行测序;以及
28.e)确定所述pcr产物的相对量,并从而确定样品中包含的病毒单倍型。
29.令人惊讶的是,发明人发现使用上述方法的umi工作流程鉴定出8到11个单倍型每个回复体,即与基于相同样品仅鉴定出2到5个单倍型每个回复体的blasr或quiver工作流程相比更灵敏。从疾病控制和预防中心(center of disease control and prevention,cdc)获得分别用于在位置ns1-2579(p1)、ns3-5270(p3)和5'nc-57(p5和p51)处回复的登革热病毒参考单回复体样品。在所述参考样品中,与blasr或quiver工作流程相比,umi工作流程鉴定出显著更多的单独单倍型。
30.根据第三方面,提供了根据第一方面的方法在含有活减毒病毒的疫苗的质量控制中的用途。
31.根据第四方面,提供了根据第二方面的方法在诊断患者样品中的病毒感染中的用途。
32.根据第五方面,提供了一种用于检测含有减毒登革热病毒的样品中至少一个回复病毒的存在的试剂盒,该试剂盒包括包含seq id no:9(5f)的核苷酸序列的第一引物对和包含seq id no:6(3r)的核苷酸序列的第二引物。
33.根据第六方面,提供了一种用于定量分析登革热病毒样品中的病毒单倍型混合物的试剂盒,该试剂盒包括包含seq id no:21的核苷酸序列的第一引物(引物iia)和包含seq id no:9(5f)的核苷酸序列的第二引物。
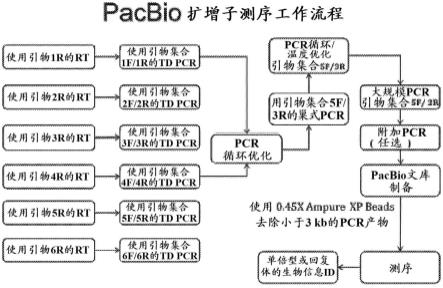
34.附图简述
35.图1是示出了smrt(pacbio)测序工作流程的实例的图表。pcr意指聚合酶链式反应;rt意指逆转录pcr;td pcr意指着陆pcr(touchdown pcr);单倍型id意指单倍型的标识;
回复体id意指回复体的标识。
36.图2示出了tdv2基因组。
37.图3示出了用于sanger测序的tdv2基因组上的引物的位置。
38.图4示出了插入物长度的映射的读段。
39.发明详述
40.在本说明书和权利要求书中使用术语“包含/包括(comprise)”或“包含/包括(comprising)”的情况下,它不排除其他要素或步骤。对于本发明的目的,术语“由
……
组成”被认为是术语“包括”的任选实施方案。如果在下文中组被定义为包括至少一定数量的实施方案,这也应理解为公开了任选地仅由这些实施方案组成的组。
41.在指代单数名词时使用不定冠词或定冠词,如“一个”或“一种”、“该/所述”的情况下,除非特别说明,否则这包括该名词的复数形式。
42.反之亦然,当使用名词的复数形式时,它也指单数形式。
43.此外,说明书和权利要求书中的术语第一、第二、第三或(a)、(b)、(c)等被用于区分相似的要素,而不一定用于描述顺序次序或时间次序。应当理解,如此使用的术语在适当的情况下是可互换的,并且本文描述的本发明的实施方案能够以除了本文描述或例示的顺序之外的其他顺序操作。
44.在本发明的上下文中,所指示的任何数值通常与将被本领域技术人员理解为仍然确保所讨论的特征的技术效果的精度区间相关联。如本文所用,与所指示数值的偏差在
±
10%的范围内,并且优选在
±
5%的范围内。前面提及的与所指示数值区间的
±
10%并且优选
±
5%的偏差也由本文使用的关于数值的术语“约”和“大约”指示。
45.根据本发明的第一方面,提供了一种用于检测含有减毒病毒的样品中至少一种回复病毒的存在的方法,其包括以下步骤:
46.a)从所述样品纯化病毒核酸;
47.b)任选地,从所述病毒核酸制备双链dna模板;
48.c)通过聚合酶链式反应(pcr)扩增a)的所述病毒核酸或b)的所述双链dna模板以使得生成具有至少2kb的长度并且包含所述减毒病毒的至少一个减毒基因座的pcr产物;
49.d)通过单分子实时(smrt)测序对所述pcr产物进行测序;以及
50.e)将所述pcr产物中所述至少一个减毒基因座处的至少一个核苷酸与野生型病毒序列中的相应核苷酸进行比较,其中所述野生型核苷酸在所述至少一种减毒基因座处的存在指示所述样品中回复病毒的存在。
51.本文中的“病毒”意指任何病毒,包括双链和单链dna病毒,以及双链和单链rna病毒。
[0052]“减毒病毒”在本文中意指与野生型病毒相比,该病毒在宿主细胞中具有降低的复制能力和/或降低的感染性。复制能力和/或感染性可以在合适的细胞系统中体外或在合适的动物模型中体内确定。减毒可以通过使病毒在外来宿主中,诸如在组织培养物、含胚胎卵子或活体动物中连续传代来实现。可替代地,减毒可以通过化学剂执行。
[0053]
活减毒病毒对于在病毒疫苗中的使用很重要,因为与灭活病毒相比,活减毒病毒产生更强的免疫响应。活减毒病毒可以是rna病毒或dna病毒。合适的活减毒rna病毒可以选自登革热病毒、脊髓灰质炎病毒、风疹病毒、麻疹病毒、黄热病病毒、腮腺炎病毒和流感病
毒。优选地,活减毒rna病毒是登革热病毒。合适的活减毒dna病毒包括水痘带状疱疹病毒、牛痘病毒、天花病毒、单纯疱疹病毒和基孔肯雅病毒。活减毒病毒与野生型病毒的不同之处可能在于病毒核酸序列中的一个或多个突变。例如,对于一种四价登革热疫苗(tdv),已知新生小鼠的神经毒力减弱是由突变5’ncr-57c-》u、ns1-53gly-》asp(nt-2579g-》a)和ns3-250 glu-》val(nt-5270a-》u)决定的(butrapet等人,j.virol.74(2000),3011-3019)。上述核苷酸序列编号涉及seq id no:23所示的四价登革热病毒血清型2(tdv2)的核苷酸序列。
[0054]“回复病毒”在本文中意指源自减毒病毒的病毒,其在一个或多个减毒基因座处的核苷酸回复为野生型序列。回复病毒的检测对于基于活减毒病毒的疫苗质量控制特别重要。在tdv的情况下,回复体将具有位于位置57处的核苷酸c、位于位置2579处的核苷酸g和/或位于位置5270处的核苷酸a。
[0055]
本文中的“样品”意指来自接种疫苗的个体或患者的任何样品,或在所制造疫苗的质量控制阶段进行测试的样品。样品可以来自全血或血清,最优选样品来自血清。样品也可以是疫苗组合物或施用前的储备溶液。这对于质量控制和安全原因可能很重要。
[0056]
优选地,待根据本发明分析的疫苗组合物包含血清型1至4中的每一种血清型的登革热抗原,该血清型1至4各自独立地选自由以下组成的组:(a)活减毒登革热病毒和(b)活减毒嵌合病毒。
[0057]
优选地,待根据本发明分析的疫苗组合物包含血清型1至4中的每一种血清型的登革热抗原,其中血清型1、3和4的所述登革热抗原各自是活减毒嵌合登革热病毒并且血清型2的所述登革热抗原选自由活减毒登革热病毒和活减毒嵌合登革热病毒组成的组。例如,本发明的待分析的疫苗组合物可包含血清型1至4中的每一种血清型的登革热抗原,其中血清型1、3和4的所述登革热抗原各自为活减毒嵌合登革热/登革热病毒并且血清型2的所述登革热抗原是活减毒登革热病毒。例如,待根据本发明分析的疫苗组合物可以是huang等人,plos negl trop dis 7(5):e2243(2013)公开的血清型1至4中每一种血清型的登革热抗原的四价混合物(称为denvax)。可替代地,待根据本发明分析的疫苗组合物可包含血清型1至4中的每一种血清型的登革热抗原,其中血清型1、3和4的所述登革热抗原各自为活减毒嵌合yf/登革热病毒并且血清型2的所述登革热抗原是活减毒登革热病毒。
[0058]
优选地,待根据本发明分析的疫苗组合物包含血清型1至4中的每一种血清型的登革热抗原,其中血清型1、3和4的所述登革热抗原各自独立地选自由以下组成的组:(a)活减毒登革热病毒和(b)活减毒嵌合登革热病毒,并且血清型2的所述登革热抗原是活减毒嵌合登革热病毒。例如,待根据本发明分析的疫苗组合物可包含血清型1至4中的每一种血清型的登革热抗原,其中血清型1、3和4的所述登革热抗原各自为活减毒登革热病毒并且血清型2的所述登革热抗原是活减毒嵌合登革热/登革热病毒。例如,根据本发明的疫苗组合物可以是durbin等人,journal of infectious diseases(2013),207,957-965公开的血清型1至4中每一种血清型的登革热抗原的四价混合物(称为tv001、tv002、tv003和tv004')中的任一种。优选地,待根据本发明的这个实施方案分析的疫苗组合物是tv003。
[0059]
优选地,待根据本发明分析的疫苗组合物包含血清型1至4中的每一种血清型的登革热抗原,其中所述登革热抗原中的每一种均是活减毒嵌合登革热病毒,优选嵌合yf/登革热病毒,更优选包含其中的prm-e序列已经用登革热病毒的prm-e序列取代的减毒yf基因组骨架的嵌合yf/登革热病毒。
[0060]
优选地,待根据本发明分析的活减毒嵌合登革热病毒包含来自登革热病毒的一种或多种蛋白和来自不同的黄病毒的一种或多种蛋白。优选地,该不同的黄病毒是黄热病病毒(即嵌合yf/登革热病毒)。优选地,待根据本发明分析的活减毒嵌合登革热病毒包含其中的prm-e序列已经被登革热病毒的prm-e序列取代的减毒黄热病病毒基因组。可替代地,待根据本发明分析的活减毒嵌合登革热病毒包含来自第一登革热病毒的一种或多种蛋白和来自第二登革热病毒的一种或多种蛋白(即嵌合登革热/登革热病毒)。优选地,所述第一登革热病毒和所述第二登革热病毒属于不同血清型。当所述第一登革热病毒和所述第二登革热病毒属于相同血清型时,所述第一和第二登革热病毒是不同的菌株。
[0061]
如有必要,可以通过使病毒在合适的宿主细胞中生长来增加样品中的病毒浓度。合适的宿主细胞包括哺乳动物细胞,诸如如维罗细胞(vero cell)。优选将维罗细胞以1.5e6个细胞/烧瓶接种在t25 cm2烧瓶中,并通过将该细胞在37℃和5%co2张力下孵育来使该细胞过夜生长至80-90%汇合度。优选地,将该病毒在含有1%fbs和1%青霉素/链霉素的测定培养基dmem中稀释。优选地,以0.1moi用tdv感染维罗细胞并且让病毒在37℃5%co2孵育箱中吸附1小时。为了获得高质量的病毒rna,实施例部分描述了优选的方法。
[0062]“从样品中纯化病毒核酸”可以使用本领域已知的任何dna或rna分离方法来执行。本领域已知的任何方法都可用于dna提取或纯化。合适的方法尤其包括诸如以下的步骤:离心步骤、沉淀步骤、色谱步骤、透析步骤、加热步骤、冷却步骤和/或变性步骤。
[0063]
本文中“制备双链dna模板”意指如果病毒核酸是rna,则该方法进一步包括下面的步骤:利用逆转录酶、反向引物和核苷酸混合物进行逆转录从而生成rna/dna杂合体,随后将该rna/dna杂合转化为双链cdna模板。该反应可以在自动热循环仪中进行。优选地,对于从登革热病毒中纯化的rna,逆转录使用具有seq id no:6的核苷酸序列的寡核苷酸作为反向引物在约45℃至约65℃范围内的温度下、优选在约50℃至约55℃范围内的温度下执行。
[0064]
如果病毒核酸是单链dna,则双链dna-模板由模板依赖性dna聚合酶的活性生成。如果从病毒中纯化出的病毒核酸已经是双链核酸,则不需要该任选步骤。
[0065]“扩增步骤a)或步骤b)的所述病毒核酸以使得生成具有至少2kb的长度并且包含至少一个减毒基因座的pcr产物”在本文中意指将a)的病毒核酸或步骤b)的双链dna模板与合适的引物(正向和反向引物)、核苷酸和模板依赖性dna聚合酶一起孵育以使得生成具有至少2kb的长度的pcr产物,其中该pcr产物包含该病毒的减毒基因座中的一个或多个减毒基因座。该反应可以在自动热循环仪中进行。
[0066]
如本文所用的术语“引物”表示在诱导与核酸链互补的引物延伸产物的合成的条件下充当核苷酸合成的起始点的寡核苷酸。该引物可以被生成为使得它们能够结合至待逆转录或扩增的序列。该引物可包含5至50个核苷酸,优选8至30个核苷酸;更优选,该引物包含10至20个核苷酸。该引物序列与待逆转录或扩增的序列的至少一部分互补。优选地,该引物序列与待逆转录或扩增的序列内的序列完全互补。用于设计序列特异性引物和探针的方法在本领域中是已知的。
[0067]
优选地,所述pcr产物具有至少3kb的长度,并且更优选5kb长度。优选地,所述pcr产物包含两个减毒基因座。更优选地,所述pcr产物包含三个减毒基因座。对于tdv,所述pcr产物优选包含tdv2序列(seq id no:23)的位置57、2579和/或5270。
[0068]
优选地,步骤c)的pcr扩增反应包括至少10个使用dna聚合酶、合适的正向和反向
引物和核苷酸的pcr循环。更优选地,使用10至50个循环,甚至更优选10至30个循环。对于登革热病毒pcr产物扩增,pcr扩增反应包括使用具有seq id no:9(5f)的核苷酸序列的第一引物对和具有seq id no:6(3r)的核苷酸序列的第二引物。还优选的是pcr扩增反应包括在约90℃至约99℃的温度下的变性步骤、在约45℃至约75℃的温度下的退火步骤和在约60℃至约75℃的温度下的延伸步骤,更优选变性在约95℃下执行,退火在68℃下被执行10个循环(其中每个循环下降1℃),并且延伸在68℃下执行。
[0069]
优选地,在扩增步骤c)之后并且在测序步骤d)之前,该方法另外包括以下步骤:
[0070]
i)对pcr产物进行末端修复;
[0071]
ii)任选地,纯化经末端修复的pcr产物;以及
[0072]
iii)将步骤c)中制备的pcr产物的两条互补链用连接寡核苷酸联接,其中将一条互补链的3’端经由每个双链核酸片段中的连接寡核苷酸与另一条互补链的5'端连接,由此所述连接寡核苷酸提供所得经连接的核酸片段的单链部分。
[0073]
对pcr产物进行末端修复意在生成平末端pcr产物以用于随后连结衔接子以进行smrt测序。末端修复可以通过使用合适的dna聚合酶诸如t4 dna聚合酶(来自thermofischer的快速dna末端修复试剂盒)和核苷酸来实现。
[0074]“将步骤c)中制备的pcr产物的两条互补链用连接寡核苷酸联接”在本文中意指通过将发夹衔接子连结到上述双链dna产物的两端,形成闭合的单链环状dna,所谓的smrtbell。合适的发夹衔接子可从pacific biosciences(template prep kit)获得。使用合适的连结酶将发夹衔接子连接到平末端pcr产物。
[0075]
优选地,在步骤d)前,纯化经扩增的pcr产物。用于纯化pcr产物的方法是本领域已知的。合适的方法包括在琼脂糖凝胶上分离或通过柱或珠粒纯化。
[0076]
本文的步骤d)的“单分子实时(smrt)测序”包括由pacificbiosciences公司提供的测序技术和平台。smrt测序优选包括pacbiors和rsii平台以及pacbio sequel平台,最优选使用pacbio rsii平台执行smrt测序。当将具有smrtbell结构的样品加载到称为smrtcell的芯片上时,smrtbell会扩散到称为零模波导(zmw)的测序单元中,该单元为光检测提供最小的可用体积。zmw是一种这样的结构,该结构产生了一个经照亮的观察体积,该观察体积小到足以仅观察被dna聚合酶掺入的dna的单个核苷酸。
[0077]
为了对该smrt细胞进行smrt测序,在合适的条件下将与发夹衔接子互补的引物与单核苷酸混合物一起添加。四种单核苷酸碱基中的每一种都附接到四种不同荧光染料中的一种。当核苷酸被dna聚合酶掺入时,荧光标签被切割掉并扩散到zmw的观察区域之外,在该区域不再观察到其荧光。检测器检测核苷酸掺入的荧光信号,并根据染料的相应荧光进行碱基调用(base call),即碱基与荧光信号的相关性。
[0078]
本文的smrt测序还包括由pacific biosciences公司开发的技术。单分子实时(smrt)测序是一种并行化的单分子dna测序方法。单分子实时测序利用零模波导(zmw)。将单个dna聚合酶酶与作为模板的单个dna分子一起固定在zmw的底部。zmw是一种这样的结构,该结构产生了一个经照亮的观察体积,该观察体积小到足以仅观察被dna聚合酶掺入的dna的单个核苷酸。四种dna碱基中的每一种都附接至四种不同的荧光染料中的一种。当核苷酸被dna聚合酶掺入时,荧光标签被切割掉并扩散到zmw的观察区域之外,在该区域不再观察到其荧光。检测器检测核苷酸掺入的荧光信号,并且根据染料相应的荧光进行碱基调
用;参见eid等人,science 323(2009),133-138。
[0079]
优选地,使用pacbio rs和rsii设备以及pacbio sequel设备进行smrt测序,最优选地使用pacbio rsii设备执行smrt测序。pacific biosciences最近推出了sequel设备的进一步改进版本,称为sequel ii,其也可用于实施本发明。对于数据分析,pacific biosciences提供了可用于执行本发明的smrt分析软件。
[0080]
对于附接至荧光染料的dna碱基,可商购获得不同的化学物质。最初pacific biosciences提供了所谓的c1化学物质,随后提供了所谓的p6-c4化学物质。所有这些都涵盖在本发明的范围内。dna聚合酶随着时间的推移已经被优化用于pacbio系统。所有这些都涵盖在本发明中。
[0081]
为了降低smrt测序的序列错误率,优选使用高数量的序列读段进行数据分析。更优选使用至少103读段/轮,更优选104读段/轮,最优选5x104个读段/轮。确认特定序列的读段数越多,则该给定序列的统计可靠性就越高。低于10的读段被认为是不可靠的。
[0082]
smrt测序不同于第二代测序(sgs)技术,诸如公司illumina(miniseq,miseq、nextseq或hiseq)或ion torrent(pgm,s5,proton)和roche(454,焦磷酸测序)提供的技术。sgs技术的特点是与smrt测序相比,读段长度短得多,最高可达400bp,同时序列错误率低得多。使用sgs技术,特别是miseq system(illumina)或hiseq 1500system(illumina)对口服脊髓灰质炎病毒疫苗中的低频回复体进行量化,并且结果发现精确度与现有技术maprec(通过聚合酶链式反应和限制酶切割进行的突变体分析)测定相当(sarcey等人,j.virol.methods 246(2017,75-80)。鉴于读段长度短,sgs技术不提供大插入物(诸如1kb或更长)的单个序列,而是针对此类插入物,sgs技术提供了不同多核苷酸的平均序列。
[0083]
不同病毒的野生型病毒序列可从如由ncbi主持的公共数据库,诸如entrez(ww.ncbi.nlm.nih.gov)获得。登革热病毒血清型2的野生型病毒序列与seq id no:23所示的tdv2核苷酸序列的不同之处在于野生型序列具有位于位置57处的核苷酸c、位于位置2579处的核苷酸g和位于位置5270处的核苷酸a。
[0084]“将所述pcr产物中所述至少一个减毒基因座处的至少一个核苷酸与野生型病毒序列中的相应核苷酸进行比较”在本文中意指将所获得的pcr产物序列与所述一个或多个减毒基因座处的野生型序列进行比较。该比较可以手动或使用计算机程序进行。如果获得的pcr产物序列在所述减毒基因座处具有与野生型相同的核苷酸,则这表明样品中存在回复体。如果获得的pcr产物序列在所述减毒基因座处具有与减毒病毒相同的核苷酸,则这表明所述样品中不存在回复病毒。
[0085]
在一个实施方案中,如果减毒病毒是tdv,则将所述pcr产物中所述至少一个减毒基因座处的至少一个核苷酸与野生型病毒序列中的相应核苷酸进行比较,优选地,将所述pcr产物中两个减毒基因座处的至少一个核苷酸与野生型病毒序列中的相应核苷酸进行比较,并且更优选将所述pcr产物中三个减毒基因座处的至少一个核苷酸与野生型病毒序列中的相应核苷酸进行比较。
[0086]
在一个实施方案中,如果减毒病毒是tdv,则将所述pcr产物中的选自对应于根据seq id no.23的核酸序列中的核苷酸57、2579和5270的核苷酸的至少一个减毒基因座处的至少一个核苷酸与野生型病毒序列中的相应核苷酸进行比较。优选地,将所述pcr产物中的选自对应于根据seq id no.23的核酸序列中的核苷酸57、2579和5270的核苷酸的至少两个
减毒基因座处的至少一个核苷酸与野生型病毒序列中的相应核苷酸进行比较。
[0087]
更优选地,将所述pcr产物中的来自对应于根据seq id no.23的核酸序列中的核苷酸57、2579和5270的核苷酸的至少三个减毒基因座处的至少一个核苷酸与野生型病毒序列中的相应核苷酸进行比较。
[0088]
根据本发明的另一方面,提供了一种用于定量分析病毒样品中病毒单倍型混合物的方法,其包括以下步骤:
[0089]
a)从所述样品纯化病毒核酸;
[0090]
b)通过使用包含唯一分子标识符(umi)序列的引物从所述病毒核酸制备双链dna模板以使得所述双链dna模板包含umi序列;
[0091]
c)通过聚合酶链式反应(pcr)扩增所述双链dna模板以使得生成具有至少2kb的长度并且包含唯一分子标识符(umi)序列的pcr产物;
[0092]
d)通过单分子实时(smrt)测序对所述pcr产物进行测序;以及
[0093]
e)通过分析umi计数和读段计数来确定所述pcr产物的相对量,从而确定样品中包含的病毒单倍型。
[0094]
该病毒和该样品的实施方案如上所定义。“病毒单倍型”涉及给定病毒的完整核苷酸序列。病毒单倍型可以被定义为特定基因座(诸如减毒基因座)处的核苷酸组合。例如,登革热病毒单倍型t:57-a:2579-t:5270与单倍型t:57-a:2579-c:5270的不同之处在于核苷酸5270的差异。
[0095]“唯一分子标识符(umi)”是施加到dna分子中或在dna分子中鉴定的可用于将单个dna分子彼此区分开来的核苷酸序列(参见例如kivioja,nature methods 9,72-74(2012))。可以将umi连同与它们相关的dna分子一起测序,以确定读段序列是一个源dna分子的序列还是另一个源分子的序列。术语“umi”在本文中用于指多核苷酸的序列信息和物理多核苷酸本身。术语“umi”包括通常用于将一个样品的读段与其他样品的读段区分开来的条形码。
[0096]
通常,对单个源分子的多个拷贝进行测序。
[0097]
可以在将用于测序的源分子递送到流动池之前对它进行pcr扩增。pcr扩增簇中的每个分子均源自同一源dna分子,但被单独地测序。出于纠错和其他目的,确定来自单个簇的所有读段都被鉴定为源自同一源分子可能很重要。umi允许这种分组。通过扩增或以其他方式复制从而产生dna分子的多个拷贝的dna分子称为源dna分子。
[0098]
umi可被施加于单个dna分子中或在单个dna分子中鉴定。在一些实施方案中,可通过如经由通过聚合酶、核酸内切酶、转座酶等的连结或转座将umi物理地连接或结合至dna分子的方法将umi施加到dna分子中。因此,这些“施加的”umi也是称为物理umi。在某些情况下,它们也可以称为外生umi。在源dna分子中鉴定的umi称为虚拟umi。在某些情况下,虚拟umi也可以称为内生umi。
[0099]
可以通过多种方式定义物理umi。例如,它们可以是被插入在衔接子中或以其他方式插入在待测序的源dna分子中的随机的、伪随机的或部分随机的或非随机的核苷酸序列。在一些实施方案中,物理umi可能是如此独特,以致于它们中的每一个都有望唯一地鉴定出样品中存在的任何给定源dna分子。生成衔接子的集合体,每个衔接子都具有物理umi,并且使那些衔接子附接至待测序的片段或其他源dna分子,并且每个单独的经测序分子都具有
有助于将其与所有其他片段区分开来的umi。在此类实施方案中,可以使用非常大的量的不同物理umi(如,数千到数百万)来唯一地鉴定样品中的dna片段。
[0100]
物理umi必须具有足够的长度来确保每个和每一个源dna分子的这种唯一性。在一些实施方案中,可结合其他鉴定技术使用不太独特的分子标识符,以确保在测序过程中唯一地鉴定每个源dna分子。在此类实施方案中,多个片段或衔接子可以具有相同的物理umi。可将其他信息诸如比对位置或虚拟umi与物理umi组合在一起,以唯一地将读段鉴定为源自单一源dna分子/片段。在一些实施方案中,衔接子包括物理umi,其限于相对少量的非随机序列,如96个非随机序列。此类物理umi也称为非随机umi。在一些实施方案中,可将非随机umi与序列位置信息和/或虚拟umi组合在一起以鉴定可归属于相同源dna分子的读段。可以折叠所鉴定的读段以获得反映如本文所述的源dna分子的序列的共有序列。
[0101]“随机umi”可以被认为是从由给定一个或多个序列长度的所有可能的不同寡核苷酸序列组成的umi集合中选择的作为随机样品的物理umi。例如,如果umi集合中的每个umi均具有n个核苷酸,则该集合包括4n个具有彼此不同的序列的umi。选自该4n个umi的随机样品构成随机umi。
[0102]
相反,如本文使用的“非随机umi”指的是不是随机umi的物理umi。在一些实施方案中,为特定实验或应用预定义可用的非随机umi。在某些实施方案中,使用规则来生成序列以获得非随机umi。例如,可以生成集合的序列,以使该序列具有一个或多个特定模式。在一些实施方案中,每个序列与该集合中的每个其他序列的不同之处在于特定数目的(如,2、3或4)个核苷酸。也就是说,通过替换少于该特定数目的核苷酸,不能将任何非随机umi序列转换为任何其他可用的非随机umi序列。在一些实施方案中,从包括少于给定特定序列长度的所有可能的umi的umi集合中选择非随机umi。例如,可以从总共96个不同的序列(而不是总共46=4096个可能的不同序列)中选择具有6个核苷酸的非随机umi。在其他实施方案中,序列不是从集合中随机选择的。相反,一些序列被选择的概率比其他序列更高。
[0103]
umi的附接可以通过将umi序列(诸如随机序列)添加到待在pcr扩增反应中使用的正向引物的5'端或反向引物的3'端来进行。由此,将umi序列掺入到pcr扩增产物中。
[0104]
如果病毒核酸是经分离的病毒rna,则步骤b)包括在逆转录酶存在下使用在5'端另外包含umi序列的反向引物对该经分离的病毒rna进行逆转录的步骤。优选地,在登革热病毒的情况下,使用具有seq id no:22(umi 3r)的核苷酸序列的寡核苷酸作为反向引物在约45℃至约65℃范围内的温度下、优选在约50℃至约55℃范围内的温度下执行逆转录。
[0105]
可替代地,umi序列可以在步骤c)的pcr扩增被掺入到正向或反向引物中。
[0106]
优选地,步骤c)的pcr扩增包括pcr扩增反应,该pcr扩增反应包括至少10个使用dna聚合酶和正向引物和反向引物的pcr循环。更优选地,执行10至50个pcr循环,甚至更优选10至30个pcr循环。在登革热病毒的情况下,pcr扩增反应包括使用具有seq id no:9(5f)的核苷酸序列的第一引物和具有seq id no:21的核苷酸序列的第二引物(引物iia)。
[0107]
优选地,步骤c)的pcr扩增反应包括在约90℃至约99℃的温度下的变性步骤、在约45℃至约75℃的温度下的退火步骤和在约60℃至约75℃的温度下的延伸步骤。
[0108]
优选地,在扩增步骤c)之后并且在测序步骤d)之前,该方法另外包括以下步骤:
[0109]
i)对pcr产物进行末端修复;
[0110]
ii)任选地,纯化经末端修复的pcr产物;以及
[0111]
iii)将步骤c)中制备的pcr产物的两条互补链用连接寡核苷酸联接,其中将一条互补链的3’端经由每个双链核酸片段中的连接寡核苷酸与另一条互补链的5'端连接,由此所述连接寡核苷酸提供所得经连接的核酸片段的单链部分。
[0112]
步骤c)和d)的实施方案如上所定义。
[0113]“读段计数”代表读段数目。它是单分子测序中常用的一类量化方案。尽管基于读段计数的方案类似于用于批量rna测序的常用方法,但从单细胞中捕获的极少量转录物需要cdna扩增来构建文库;这不可避免地会导致较大的扩增偏差。因此,如果生成与所用引物对更强地结合的经截短或错误的cdna片段,则与正确的dna片段相比,该经截短或错误的cdna片段将以更高的量扩增。“umi计数”仅与唯一映射的读段数有关。为了减轻读段计数的偏差,最近的几个scrna测序方案已采用了一个额外的步骤,其中在扩增之前用唯一分子标识符(umi)修饰单个转录物,从而导致转录物计数被更准确地量化。umi计数是通过仅考虑含有唯一umi序列的序列的读段计数来确定的。
[0114]
根据一个优选的实施方案,本发明的方法在步骤d)之前包括以下步骤:
[0115]
i)通过sanger测序单独地确定pcr产物的在至少一个减毒基因座处的序列;
[0116]
ii)将所述至少一个减毒基因座处的至少一个核苷酸与野生型病毒序列进行比较;以及
[0117]
iii)如果确定所述野生型序列位于至少一个减毒基因座处,则执行步骤d)的通过smrt测序对所述pcr产物进行的测序。
[0118]
与smrt测序相比,sanger测序,即一种基于dna聚合酶在体外dna复制过程中选择性地掺入链终止双脱氧核苷酸的dna测序方法,成本要低得多。如果单个减毒基因座处的sanger测序指示存在回复病毒,则对整个病毒核酸分子进行smrt测序,以区分减毒基因座处的单回复体、双回复体和三回复体。
[0119]
用于seq id no:23所示的tdv2核苷酸序列的位置57处的第一减毒基因座的sanger测序的合适引物对是具有seq id no:13的核苷酸序列的正向引物和seq id no:17的反向引物。用于位置2579处的第二减毒基因座的sanger测序的合适引物对是具有seq id no:14的核苷酸序列的正向引物和seq id no:18的反向引物。用于位置5270处的第三减毒基因座的sanger测序的合适引物对是具有seq idno:15或16的核苷酸序列的正向引物,和具有seq id no:19或20的核苷酸序列的反向引物。
[0120]
根据进一步的方面,本发明提供了根据第一方面的方法在含有活减毒病毒的疫苗的质量控制中的用途。对于疫苗样品的质量控制,检测疫苗样品中回复体的存在尤为重要。很明显,在三个减毒基因座中的每一个处都具有回复的登革热病毒(三回复体)具有野生型复制能力,并因此可能具有毒性。然而,鉴于在其中的两个减毒基因座处具有回复的双回复体和在第三减毒基因座处具有回复的单回复体之间可能发生可能会导致毒力增加的重组事件,包含双回复体和单回复体的疫苗组合物也可能会造成风险。
[0121]
根据进一步的方面,本发明提供了根据第二方面的方法在诊断患者样品中的病毒感染中的用途。根据第二方面的方法可以用于诊断受病毒感染的患者的病毒感染。该方法还能够检测与不同病毒亚型(诸如不同的登革热血清型)的共感染。这可为受感染患者的个性化治疗提供优势。优选地,用于诊断受病毒感染的患者中的病毒感染的方法在体外对从患者取出的生物样品进行。优选地,该样品是血液样品,更优选地,该样品是血清样品。
[0122]
根据本发明的进一步方面,提供了一种用于检测含有减毒登革热病毒的样品中至少一种回复病毒的存在的试剂盒,其包括选自具有seq id no:1、seq id no:3、seq id no:5、seq id no:7、seq idno:9和seq id no:11的核苷酸序列的寡核苷酸的第一引物,并且包括选自具有seq id no:2、seq id no:4、seq id no:6、seq idno:8、seq id no:10和seq id no:12的核苷酸序列的寡核苷酸的第二引物。优选地,第一引物包含seq id no:9(5f)的核苷酸序列并且第二引物包含seq id no:6(3r)的核苷酸序列。更优选地,第一引物由seq id no:9的核苷酸序列组成并且第二引物由seq id no:6的核苷酸序列组成。
[0123]
该试剂盒可进一步包括本领域已知的合适的逆转录酶、核苷酸和合适的缓冲液。
[0124]
根据本发明的另一方面,提供了用于定量分析登革热病毒样品中的病毒单倍型混合物的试剂盒,该试剂盒包括包含seq id no:9(5f)的核苷酸序列的第一引物和包含seq id no:21的核苷酸序列的第二引物(引物iia)。优选地,第一引物由seq id no:9的核苷酸序列组成并且第二引物由seq id no:21的核苷酸序列组成。
[0125]
该试剂盒可进一步包括本领域已知的合适的逆转录酶、核苷酸和合适的缓冲液。
[0126]
本发明在以下实施例中进一步描述,这些实施例仅用于说明本发明的具体实施方案并且也不应被解读为以任何方式限制本发明的范围。
[0127]
实施方案列表
[0128]
1.一种用于检测含有减毒病毒的样品中至少一种回复病毒的存在的方法,其包括以下步骤:
[0129]
a)从所述样品纯化病毒核酸;
[0130]
b)任选地,从所述病毒核酸制备双链dna模板;
[0131]
c)通过聚合酶链式反应(pcr)扩增a)的所述病毒核酸或b)的所述双链dna模板以使得生成具有至少2kb的长度并且包含所述减毒病毒的至少一个减毒基因座的pcr产物;
[0132]
d)通过单分子实时(smrt)测序对所述pcr产物进行测序;以及
[0133]
e)将所述pcr产物中所述至少一个减毒基因座处的至少一个核苷酸与野生型病毒序列中的相应核苷酸进行比较,其中所述野生型核苷酸在所述至少一个减毒基因座处的存在指示所述样品中回复病毒的存在。
[0134]
2.如实施方案1所述的方法,其中所述病毒是rna病毒,优选地其中所述rna病毒选自登革热病毒、脊髓灰质炎病毒、风疹病毒、麻疹病毒、黄热病病毒、腮腺炎病毒和流感病毒,更优选所述rna病毒是登革热病毒。
[0135]
3.如实施方案1或2所述的方法,其中所述至少一种回复病毒以基于含有减毒病毒的样品中病毒的总量的小于1%、优选小于0.2%的量存在于所述样品中。
[0136]
4.如实施方案1至3中任一项所述的方法,其中所述至少一种回复病毒与减毒病毒的差异小于10个核苷酸,优选小于5个核苷酸,更优选小于3个核苷酸,并且最优选1个核苷酸。
[0137]
5.如实施方案1所述的方法,其中所述病毒是dna病毒,优选其中所述dna病毒是水痘带状疱疹病毒、牛痘病毒、天花病毒、单纯疱疹病毒和基孔肯雅病毒。
[0138]
6.如实施方案1至5中任一项所述的方法,其中所述pcr产物具有至少3kb,优选至少5kb的长度。
[0139]
7.如实施方案1至6中任一项所述的方法,其中所述pcr产物包含至少两个减毒基
因座,优选至少三个减毒基因座。
[0140]
8.如实施方案1、2、6或7中任一项所述的方法,其中所述减毒病毒是活减毒登革热病毒并且所述至少一个减毒基因座选自根据seq id no:23的登革热病毒2型的核苷酸序列的位置57、2579和/或5270。
[0141]
9.如实施方案1至8中任一项所述的方法,其中在步骤a)之前,使所述活减毒病毒在维罗细胞中生长。
[0142]
10.如实施方案1、2或6至9中任一项所述的方法,其中所述病毒核酸是经分离的病毒rna,并且步骤b)包括在逆转录酶存在下使用反向引物对所述经分离的病毒rna进行逆转录的步骤。
[0143]
11.如实施方案10所述的方法,其中所述逆转录使用包含seqid no:6(3r)的核苷酸序列的寡核苷酸作为反向引物在约45℃至约65℃范围内的温度下、优选在约50℃至约55℃范围内的温度下执行,优选所述寡核苷酸由seq id no:6的核苷酸序列组成。
[0144]
12.如实施方案1至11中任一项所述的方法,其中步骤c)包括pcr扩增反应,所述pcr扩增反应包括至少10个使用dna聚合酶和正向引物和反向引物的pcr循环。
[0145]
13.如实施方案12所述的方法,其中所述pcr扩增反应包括使用包含seq id no:6(3r)的核苷酸序列的第一引物和包含seq idno:9(5f)的核苷酸序列的第二引物,优选地所述第一引物由seq idno:6的核苷酸序列组成并且所述第二引物由seq id no:9的核苷酸序列组成。
[0146]
14.如实施方案12或13所述的方法,其中所述pcr扩增反应包括在约90℃至约99℃的温度下的变性步骤、在约45℃至约75℃的温度下的退火步骤和在约60℃至约75℃的温度下的延伸步骤。
[0147]
15.如实施方案1至14中任一项所述的方法,其中步骤c)中获得的经扩增的pcr产物在步骤d)之前被纯化。
[0148]
16.如实施方案1至15中任一项所述的方法,其中在步骤d)之前,将所述经扩增的pcr产物作为适用于smrt测序的文库提供。
[0149]
17.如实施方案16所述的方法,其中在步骤d)之前所述方法另外包括:
[0150]
i)对pcr产物进行末端修复;
[0151]
ii)任选地,纯化经末端修复的pcr产物;以及
[0152]
iii)将步骤c)中制备的所述pcr产物的两条互补链用连接寡核苷酸联接,其中将一条互补链的3’端经由每个双链核酸片段中的连接寡核苷酸与另一条互补链的5'端连接,由此所述连接寡核苷酸提供所得经连接的核酸片段的单链部分。
[0153]
18.一种用于定量分析病毒样品中病毒单倍型的混合物的方法,其包括以下步骤:
[0154]
a)从所述样品纯化病毒核酸;
[0155]
b)通过使用包含唯一分子标识符(umi)序列的引物从所述病毒核酸制备双链dna模板以使得所述双链dna模板包含所述umi序列;
[0156]
c)通过聚合酶链式反应(pcr)扩增所述双链dna模板以使得生成具有至少2kb的长度并且包含唯一分子标识符(umi)序列的pcr产物;
[0157]
d)通过单分子实时(smrt)测序对所述pcr产物进行测序;以及
[0158]
e)通过分析umi计数和读段计数来确定所述pcr产物的相对量,从而量化所述样品
中包含的所述病毒单倍型。
[0159]
19.如实施方案18所述的方法,其中所述病毒单倍型中的任一种以基于所述病毒样品中病毒的总量的小于10%、优选小于1%、更优选小于0.2%的量存在于所述样品中。
[0160]
20.如实施方案18所述的方法,其中所述病毒是rna病毒,优选地其中所述rna病毒选自登革热病毒、脊髓灰质炎病毒、风疹病毒、麻疹病毒、黄热病病毒、腮腺炎病毒和流感病毒,更优选所述rna病毒是登革热病毒。
[0161]
21.如实施方案18至20中任一项所述的方法,其中所述病毒单倍型中的至少两种彼此之间的差异小于10个核苷酸,优选小于5个核苷酸,更优选小于3个核苷酸,并且最优选1个核苷酸。
[0162]
22.如实施方案18所述的方法,其中所述病毒是dna病毒,优选其中所述dna病毒是水痘带状疱疹病毒。
[0163]
23.如实施方案18至21中任一项所述的方法,其中所述病毒是活减毒登革热病毒,优选包含选自根据seq id no:23的登革热病毒2型的核苷酸序列的位置57、2579和/或5270的至少一个减毒基因座的登革热病毒。
[0164]
24.如实施方案18至23中任一项所述的方法,其中所述pcr产物具有至少3kb,优选至少5kb的长度。
[0165]
25.如实施方案18、19或23中任一项所述的方法,其中所述病毒核酸是经分离的病毒rna并且步骤b)包括在逆转录酶存在下使用在5'端另外包含umi序列的反向引物对所述经分离的病毒rna进行逆转录的步骤。
[0166]
26.如实施方案25所述的方法,其中所述逆转录使用包含seqid no:22(umi 3r)的核苷酸序列的寡核苷酸作为反向引物在约45℃至约65℃的温度范围内执行,优选所述寡核苷酸由seq id no:22的核苷酸序列组成并且所述温度在约50℃至约55℃的范围内。
[0167]
27.如实施方案18至26中任一项所述的方法,其中步骤c)包括pcr扩增反应,所述pcr扩增反应包括至少10个使用dna聚合酶和正向引物和反向引物的pcr循环。
[0168]
28.如实施方案27所述的方法,其中所述pcr扩增反应包括使用包含seq id no:21的核苷酸序列的第一引物(引物iia)和包含seqid no:9(5f)的核苷酸序列的第二引物,优选地所述第一引物由seqid no:21的核苷酸序列组成并且所述第二引物由seq id no:9的核苷酸序列组成。
[0169]
29.如实施方案27或28所述的方法,其中所述pcr扩增反应包括在约90℃至约99℃的温度下的变性步骤、在约45℃至约75℃的温度下的退火步骤和在约60℃至约75℃的温度下的延伸步骤。
[0170]
30.如实施方案18至29中任一项所述的方法,其中步骤c)中获得的经扩增的pcr产物在步骤d)之前被纯化。
[0171]
31.如实施方案18至30中任一项所述的方法,其中在步骤d)之前,将所述扩增的pcr产物作为适用于smrt测序的文库提供。
[0172]
32.如实施方案31所述的方法,其中在所述扩增步骤c)之后并且在所述测序步骤d)之前,所述方法另外包括以下:
[0173]
i)对pcr产物进行末端修复;
[0174]
ii)任选地,纯化经末端修复的pcr产物;以及
[0175]
iii)将步骤(b)中制备的双链核酸片段的两条互补链用连接寡核苷酸联接,其中将一条互补链的3’端经由每个双链核酸片段中的连接寡核苷酸与另一条互补链的5'端连接,由此所述连接寡核苷酸提供所得经连接的核酸片段的单链部分。
[0176]
33.根据实施方案1至32中任一项所述的方法,其中所述样品是包含减毒病毒的组合物,或来自患者的样品。
[0177]
34.如实施方案1至33中任一项所述的方法,其中在步骤d)之前,所述方法包括:
[0178]
i)通过sanger测序单独地确定所述pcr产物的在所述减毒基因座中的至少一个处的序列;
[0179]
ii)将所述至少一个减毒基因座处的至少一个核苷酸与所述野生型病毒序列进行比较;以及
[0180]
iii)如果确定所述野生型序列位于至少一个减毒基因座处,则执行步骤d)的通过smrt测序对所述pcr产物进行的测序。
[0181]
35.根据实施方案1至17中任一项所述的方法在含有活减毒病毒的疫苗的质量控制中的用途。
[0182]
36.根据实施方案18至34中任一项所述的方法在诊断患者样品中的病毒感染中的用途。
[0183]
37.一种用于检测含有减毒登革热病毒的样品中至少一种回复病毒的存在的试剂盒,其包括选自具有seq id no:1、seq id no:3、seq id no:5、seq id no:7、seq id no:9和seq id no:11的核苷酸序列的寡核苷酸的第一引物,并且包括选自具有seq id no:2、seq id no:4、seq id no:6、seq id no:8、seq id no:10和seqid no:12的核苷酸序列的寡核苷酸的第二引物,优选所述第一引物包含seq id no:9(5f)的核苷酸序列并且所述第二引物包含seq idno:6(3r)的核苷酸序列,更优选所述第一引物由seq id no:9的核苷酸序列组成并且所述第二引物由seq id no:6的核苷酸序列组成。
[0184]
38.一种用于定量分析登革热病毒样品中的病毒单倍型混合物的试剂盒,其包括包含seq id no:21的核苷酸序列的第一引物(引物iia)和包含seq id no:9(5f)的核苷酸序列的第二引物,优选所述第一引物由seq id no:21的核苷酸序列组成并且所述第二引物由seq id no:9的核苷酸序列组成。
实施例
[0185]
材料和方法
[0186]
维罗细胞的生长和感染
[0187]
将维罗who细胞以1.5e6个细胞/烧瓶接种在t25 cm2烧瓶中,并通过将该细胞在37℃和5%co2张力下孵育来使该细胞过夜生长至80-90%汇合度。在感染实验开始之前,在倒置显微镜下观察细胞的健康和汇合度。
[0188]
将tdv-2病毒在含有1%fbs和1%青霉素/链霉素的测定培养基dmem(hyclone,cat#sv30010)中稀释。用tdv-2以0.1moi感染维罗细胞,并让该细胞在37℃5%co2孵育箱中吸附病毒1小时。孵育完成后,吸移出接种物(inoculum)并且给细胞补充6ml新鲜测定培养基。再让烧瓶在该温度下继续孵育5-7天。第5天更换培养基,并且在第7天通过收集并在4℃冷却的离心机中以10,000x g旋转细胞培养上清液10分钟来收获病毒。
[0189]
病毒rna提取
[0190]
使用3种不同的商业试剂盒对140μl tdv2/vero who病毒细胞培养上清液进行rna提取。根据试剂盒随附的说明书进行提取。qiagen qiaamp病毒rna微型试剂盒(批号145051122),omega病毒rna提取试剂盒和roche高度纯rna分离试剂盒按照制造商说明书使用而不添加载体rna。在nanodrop分光光度计上测量纯化的rna产量。
[0191]
用于tdv分析的pacbio测序优化
[0192]
工艺总结:流程图
[0193]
pacbio测序工作流程已在图1中进行了描述。首先使用选定的引物进行逆转录(rt),然后进行pcr。然后优化pcr条件。通过巢式pcr和进一步扩增富集pcr产物。用用于产生pacbio文库的试剂盒对该pcr产物进行处理。使用ampure珠粒去除较小的产物。然后执行smrt测序,并使用生物信息学工具分析数据。
[0194]
引物选择
[0195]
使用rna折叠软件(http://unafold.rna.albany.edu/?q=mfold)分析位置57和5270处变体周围的区域以鉴定具有最小二级结构的区域。选择具有未配对区域的区段,并使用ncbi的primer3服务器在这些区段中选取六个引物对。由于在5'端只有57bp可用(图2),因此这些引物中的一些相互重叠(表1)。
[0196]
用于rt-pcr优化的引物列表
[0197][0198]
[0199]
表1:引物序列和特征。注意反向引物也是用于逆转录的引物。
[0200]
该引物是成对的。
[0201]
序列名称aagacagattctttgagggagcta1f(seq id no:1)ggtctgtgaaatgggcttcg1r(seq id no:2)gacagattctttgagggagctaa2f(seq id no:3)gggtctgtgaaatgggcttc2r(seq id no:4)acagattctttgagggagctaag3f(seq id no:5)gtctgtgaaatgggcttcgtc3r(seq id no:6)agacagattctttgagggagctaag4f(seq id no:7)gtctgtgaaatgggcttcgt4r(seq id no:8)gacagattctttgagggagctaag5f(seq id no:9)cttgctgggtctgtgaaatgg5r(seq id no:10)aagacagattctttgagggagctaa6f(seq id no:11)tgggtctgtgaaatgggcttc6r(seq id no:12)
[0202]
表2:引物序列与seq id编号的相关性.
[0203]
引物获自idt dna (www.idtdna.com)。每种情况下的反向引物都用作第一链cdna反应中逆转录酶的引物。然后将该对用于pcr。
[0204]
rt-pcr优化
[0205]
使用反向引物(表1)对6.5μl(139.1ng)的tdv2病毒rna进行逆转录。将六个引物对用于着陆pcr反应。着陆pcr如下那样进行。在95℃持续30秒;每个循环下降1度持续10个循环的66℃下退火20秒(即循环1
–
66℃,循环2
–
65℃,循环3
–
64℃等);以及68℃下延伸6分钟。再运行25个在95℃下持续30秒的循环,在53℃下退火20秒并在68c下延伸6分钟。将rt-pcr产物在1x tae缓冲液中在1%琼脂糖凝胶上运行。
[0206]
然后将两个性能最佳的引物对(1和4)与巢式pcr引物(5f和3r,来自表1中的同一集合)一起使用,并针对循环数进行优化。将具有较少非特异性条带的引物对1(1f/r)转送用于退火温度优化。在一系列pcr循环中测试五个温度。选取11个循环的最佳值,并使用0.45xampure xp珠粒纯化去除小于3千碱基的非特异性扩增产物。
[0207]
pacbio测序和变体分析
[0208]
将来自优化的rt-pcr反应的材料用于构建pacbio测序文库。对一个smrtcell进行测序,从而产生366mbp的数据和12,467个插入物读段。使用blasr对该读段与登革热病毒参考基因组(genbank登录号u87411)进行比对,并使用quiver调用变体。数据汇总在结果部分中。
[0209]
tdv2回复体的鉴定
[0210]
sanger测序以确认回复体的身份
[0211]
在维罗who细胞中扩增四种回复体(见下表3),并使用omega试剂盒提取病毒rna。进行sanger测序以确认这些tdv回复体的身份。
[0212][0213]
表3:tdv2回复体列表
[0214]
rt-pcr优化
[0215]
将titan one tube rt-pcr试剂盒(roche)连同多个引物集合(此处未全部示出)和tdv2病毒rna一起用于优化rt和pcr条件,以产生覆盖包括nt57、nt2579和nt5270在内的三个减毒基因座的约5.3kb尺寸的单个大产物。最后,选择不会产生非特异性产物的最佳引物集合(如下所示)。
[0216]
d2-1(正向):agttgttagtctacgtggaccgac(seq id no:13)
[0217]
cd2-5358(反向):gaaatgggcttcgtccatgataatcagg(seq id no:20)
[0218]
将经优化的rt-pcr条件应用于四个tdv2回复体,以在每个反应管中使用100ng病毒rna扩增5.3kb产物。rt-pcr如下那样进行:50℃持续30秒,94℃持续2分钟(1个循环),在94℃下变性15秒,在60℃下退火30秒,并在68℃下延伸5分钟(30个循环)和在68℃下最终延伸5分钟(1个循环)。我们成功产生了5.3kb长的产物,对该产物经过sanger测序以鉴定tdv2回复体。
[0219]
pcr产物的纯化
[0220]
优化rt-pcr反应,并且成功扩增出5.35kb的pcr产物。来自qiagen的两个纯化试剂盒被用于纯化pcr产物;凝胶提取和pcr纯化试剂盒。凝胶提取试剂盒提供了更清洁的单条带产物并且被选择用于pcr产物的纯化。
[0221]
sanger测序和分析
[0222]
通过sanger测序对经纯化的pcr产物进行检查,并且对测序数据文件进行编辑,并且将该测序数据文件与takeda的tdv2参考测序进行比对。sanger测序反应中使用的引物和这些引物在覆盖三个目标减毒基因座的tdv2基因组上的位置分别见表4和图3的表和图。
[0223]
用于sanger测序的引物列表
[0224][0225]
[0226]
表4:用于sanger测序的引物
[0227]
pacbio测序和变体分析
[0228]
rt-pcr以扩增pcr产物
[0229]
采用经优化的rt-pcr程序使用基因特异性引物(gsp)从四种tdv2回复病毒rna中扩增cdna。用3r反向引物对6.5μl病毒rna进行逆转录,随后以25、30、35和40个pcr循环使用两个性能最佳的引物对(5f和3r)进行巢式pcr。选取了30个循环的最佳值,并使用0.45x ampure xp珠粒纯化来去除小于3千碱基的非特异性扩增产物。
[0230]
pacbio测序和变体分析
[0231]
使用来自rt-pcr反应的材料构建pacbio测序文库。
[0232]
pacbio的扩增子模板制备和测序方案的材料和方法:
[0233]
材料:
[0234]
·
smrtbell
tm
模板制备试剂盒.pacbio 100-259-100
[0235]
·
dna/聚合酶结合试剂盒.pacbio 100-372-700)
[0236]
·
magbead试剂盒.pacbio目录号100-676-500
[0237]
·
dna测序试剂.pacbio 100-612-400
[0238]
·
dna内部对照复合物.pacbio 100-364-600
[0239]
·
cells.pacbio 100-171-800
[0240]
·
预洗涤的ampure xp珠粒.beckman coulter a63881
[0241]
·
qiagen缓冲液eb.qiagen 19086
[0242]
·
磁架(magnetic rack).thermofisher scientific 12321
[0243]
·
dynamag
tm-96底磁(bottom magnet).thermofisher scientific 12332d
[0244]
·
eppendorf
tm
dna lobind微量离心管,eppendorf 022431021
[0245]
·
pcr 8-孔管条(pcr 8-well tube strips),vwr 20170-004
[0246]
·
qubit
tm
dsdna br测定试剂盒,thermofisher scientific q32850
[0247]
·
agilent dna 12000试剂盒,agilent 5067-1508
[0248]
·
硬壳pcr板96-孔,薄壁(hard-shell pcr plates 96-well,thin wall),biorad hsp9655
[0249]
预洗涤ampure xp珠粒:(在所有文库制备步骤中都使用洗涤过的ampure xp珠粒)
[0250]
1.彻底重新悬浮ampure xp珠粒。
[0251]
2.将500μl的ampure xp珠粒吸移动到1.5ml离心管中。
[0252]
3.在台式微量离心机中以最大速度旋转1分钟。
[0253]
4.将该管放在磁架上,直到上清液变澄清。
[0254]
5.将上清液转移到另一个1.5ml离心管,并留待以后使用。
[0255]
6.向该珠粒中加入1ml分子生物学级水并且涡旋以完全重新悬浮珠粒。
[0256]
7.在台式微量离心机中以最大速度将珠粒离心1分钟。
[0257]
8.将该管放在磁架上,直到上清液变澄清。
[0258]
9.丢弃水。
[0259]
10.再重复水洗4次(步骤6-9)。
[0260]
11.将1ml qiagen缓冲液eb添加到珠粒沉淀中并涡旋以完全重新悬浮珠粒。
[0261]
12.在台式微量离心机中以最大速度离心1分钟。
[0262]
13.将珠粒管放在磁架上,直到上清液变澄清。
[0263]
14.丢弃eb缓冲液。
[0264]
15.通过涡旋将步骤5中收集的原始ampure xp上清液中的珠粒完全重新悬浮。
[0265]
16.将洗过的珠粒在4℃下储存长达3个月。
[0266]
纯化pcr产物:
[0267]
1.确定您的大规模pcr反应的样品量。在这种情况下,3x pcr反应为每个样品产生150μl产物。将pcr产物移至新的1.5ml lobind微量离心管。向每个样品中加入0.45x体积(67.5μl)的ampure xp珠粒。
[0268]
2.彻底混合珠粒/dna溶液。
[0269]
3.快速向下旋转管以收集珠粒。
[0270]
4.通过在室温下旋转管10分钟来使dna与珠粒结合。请注意,珠粒/dna混合对产量至关重要。旋转后,珠粒/dna混合物应该看起来是均匀的。未能将dna与珠粒试剂彻底混合将导致dna结合效率低下和样品回收率降低。
[0271]
5.离心该管1-5秒。
[0272]
6.将该管放在磁珠粒架上,直到珠粒聚集该到管的一侧并且溶液看起来很澄清。将珠粒收集到一侧所需的实际时间取决于添加的珠粒的体积。
[0273]
7.在该管仍在磁珠粒架上时,慢慢吸移出变澄清的上清液。避免干扰珠粒沉淀。
[0274]
8.通过倚着该管的与珠粒相对的一侧缓慢浇注1.5ml新鲜制备的70%乙醇来清洗珠粒。使该管静置30秒。不要扰动珠粒沉淀。30秒后,吸取并丢弃乙醇。
[0275]
9.重复以上步骤8。
[0276]
10.去除残留的70%乙醇并干燥珠粒沉淀。从磁珠粒架上取下管并将其旋转以沉淀珠粒。珠粒和任何残留的70%乙醇都将位于管的底部。将管放回磁珠粒架上。吸移出任何剩余的70%乙醇。
[0277]
11.检查管中是否有任何剩余的液滴。如果液滴存在,则重复步骤10。
[0278]
12.从磁珠粒架上取下管,并让珠粒在管盖打开的情况下风干30至60秒。
[0279]
13.向珠粒中加入39μl qiagen缓冲液eb。通过以2000rpm涡旋1至2分钟来混合直至均匀。向下旋转管以沉淀珠粒,然后将管放在磁架上,直到上清液变澄清澄清。收集37μl洗脱样品并放入新的200μl pcr管中。将样品放置在冰上。
[0280]
修复dna损伤:
[0281]
1.将来自smrtbell
tm
模板制备试剂盒的以下试剂添加到每个样品中。
[0282]
[0283]
*当制备超过1个样品时,制作dna损伤修复缓冲液、nad 、atp高和dntp的主混合物。向每个样品中加入11μl主混合物和2uldna损伤修复混合物。
[0284]
2.通过轻弹管充分混合反应物。
[0285]
3.快速离心以收集管底部的反应物。
[0286]
4.将样品管置于热循环仪中,在37℃下孵育1小时,然后在4℃下孵育1分钟。将样品放置在冰上。
[0287]
修复末端:
[0288]
1.将来自smrtbell
tm
模板制备试剂盒的以下试剂添加到每个样品中。
[0289]
末端修复混合物2.5μl
[0290]
2.通过轻弹样品管充分混合反应物。
[0291]
3.短暂离心以收集样品。
[0292]
4.将样品放入热循环仪中并在25℃下孵育5分钟,然后将样品放回冰上。立即进行下一步,并且不要让反应物孵育超过5分钟。
[0293]
纯化经末端修复的产物:
[0294]
1.添加0.45x体积(23.6μl)的ampure xp珠粒至每个样品中。
[0295]
2.通过轻弹管彻底混合珠粒/dna溶液。
[0296]
3.快速向下旋转管以收集珠粒。
[0297]
4.通过在室温下旋转管10分钟来使dna与珠粒结合。请注意,珠粒/dna混合对产量至关重要。旋转后,珠粒/dna混合物应该看起来是均匀的。未能将dna与珠粒试剂彻底混合将导致dna结合效率低下和样品回收率降低。
[0298]
5.离心管1-5秒。
[0299]
6.将管置于dynamag
tm-96底部磁珠粒架中,直到珠粒聚集到试管的一侧并且溶液变得澄清。
[0300]
7.在该管仍在磁珠粒架上,慢慢吸移出变澄清的上清液。避免干扰珠粒沉淀。
[0301]
8.通过倚着该管的与珠粒相对的一侧缓慢浇注200μl新鲜制备的70%乙醇来清洗珠粒。使该管静置30秒。不要扰动珠粒沉淀。30秒后,吸取并丢弃乙醇200μl。
[0302]
9.重复以上步骤8。
[0303]
10.去除残留的70%乙醇并干燥珠粒沉淀。从磁珠粒架上取下管并将其旋转以沉淀珠粒。珠粒和任何残留的70%乙醇都将位于管底部。将管放回磁珠粒架上。吸移出任何剩余的70%乙醇。
[0304]
11.检查管中是否有任何剩余的液滴。如果液滴存在,则重复步骤10。
[0305]
12.从磁珠粒架上取下管,并让珠粒在管盖打开的情况下风干30至60秒。
[0306]
13.向珠粒中加入33μl qiagen缓冲液eb。通过以2000rpm涡旋1至2分钟混合直至均匀。向下旋转管以沉淀珠粒,然后将管放在磁架上,直到上清液变澄清。收集31μl每种洗脱样品并放入新的200μl pcr管中。
[0307]
14.将样品放置在冰上。
[0308]
平端连结:
[0309]
1.将来自smrtbell
tm
模板制备试剂盒的以下试剂添加到每个样品中。如果制备主混合物,则组合以下所有组分,但平端衔接子除外。将7μl主混合物等分到每个样品,然后将
衔接子直接添加到混合物中。
[0310][0311]
2.通过轻弹管来混合反应物。
[0312]
3.短暂离心管以收集样品
[0313]
4.将样品置于热循环仪中并在25℃下孵育1小时,然后在65℃下孵育10分钟以灭活连结酶。将样品放回在冰上。
[0314]
5.向每个样品中加入0.5μl exo iii和0.5μl exovii,并在37℃下孵育样品1小时。
[0315]
6.您必须进行到下一步。
[0316]
纯化smrtbell文库
[0317]
1.添加0.45x体积(18.45μl)的ampure xp珠粒至每个样品中。
[0318]
2.通过轻弹管彻底混合珠粒/dna溶液。
[0319]
3.快速向下旋转管以收集珠粒。
[0320]
4.通过在室温下旋转管10分钟来使dna与珠粒结合。请注意,珠粒/dna混合对产量至关重要。旋转后,珠粒/dna混合物应该看起来是均匀的。未能将dna与珠粒试剂彻底混合将导致dna结合效率低下和样品回收率降低。
[0321]
5.离心管1-5秒。
[0322]
6.将管置于dynamag
tm-96底部磁珠粒架中,直到珠粒聚集到管的一侧并且溶液变澄清。
[0323]
7.在该管仍在磁珠粒架上,慢慢吸移出变澄清的上清液。避免干扰珠粒沉淀。
[0324]
8.通过倚着该管的与珠粒相对的一侧缓慢浇注200μl新鲜制备的70%乙醇来清洗珠粒。使该管静置30秒。不要扰动珠粒沉淀。30秒后,吸取并丢弃乙醇。
[0325]
9.重复以上步骤8。
[0326]
10.去除残留的70%乙醇并干燥珠粒沉淀。从磁珠粒架上取下管并将其旋转以沉淀珠粒。珠粒和任何残留的70%乙醇都将位于管底部。将管放回磁珠粒架上。吸移出任何剩余的70%乙醇。
[0327]
11.检查管中是否有任何剩余的液滴。如果液滴存在,则重复步骤10。
[0328]
12.从磁珠粒架上取下管,并让珠粒在管盖打开的情况下风干30至60秒。
[0329]
13.向珠粒中加入52μl qiagen缓冲液eb。通过以2000rpm涡旋1至2分钟混合直至均匀。向下旋转管以沉淀珠粒,然后将管放在磁架上,直到上清液变澄清。收集50μl每种洗脱样品并放入新的200μl pcr管中。
[0330]
14.向洗脱的样品中加入0.45x体积(22.5μl)的ampure xp珠粒。重复步骤2
–
12。
[0331]
15.向珠粒中加入12μl qiagen缓冲液eb。通过以2000rpm涡旋1至2分钟混合直至均匀。向下旋转管以沉淀珠粒,然后将管放在磁架上,直到上清液变澄清。收集10μl的经洗
脱的文库。
[0332]
16.使用qubit
tm
dsdna br测定试剂盒和qubit 3.0荧光计量化1μl样品文库
[0333]
17.通过使用agilent dna 12000试剂盒在2100生物分析仪上运行1μl(0.5-50ng/μl)样品文库来确定该文库的大小分布。
[0334]
退火和结合smrtbell文库
[0335]
此步骤需要dna/聚合酶结合和磁珠粒试剂盒。使用pacbio p6c4结合计算器2.3.1.1版本来确定用于使测序引物退火和使聚合酶结合到smrtbell文库的条件。请参阅下面的结合计算器参数。
[0336][0337]
1.通过将以下试剂添加到新的200μl pcr管中来稀释测序引物。
[0338]
a.测序引物1ul
[0339]
b.洗脱缓冲液32.3μl
[0340]
2.在80℃下孵育2分钟,然后保持在4℃。条件化测序引物可在-20℃下储存并且可使用长达30天。
[0341]
3.通过按下列顺序添加适当量的试剂到200μl pcr管中来将稀释的测序引物退火。
[0342][0343]
4.在20℃下孵育30分钟。
[0344]
5.将样品放置在冰上。
[0345]
6.通过添加以下试剂至新的200μl pcr管中来修饰sa-dna聚合酶。
[0346]
a.sa-dna聚合酶p63μl
[0347]
b.结合缓冲液v227μl
[0348]
c.总体积(50nm最终)30μl
[0349]
7.将以下组分添加到引物退火文库中,并通过轻弹管进行混合。向下旋转以收集样品。
[0350][0351]
8.封闭pcr管并在30℃下孵育30分钟,然后将其放回冰上。聚合酶结合的smrtbell文库可在4℃下储存约7天,并应在10天内进行测序。7天后测序产量和质量开始下降。
[0352]
9.通过在新的200μl pcr管中为每个样本混合以下物质来制备smrtbell文库以进行测序。
[0353]
a.magbead结合缓冲液8.8μl
[0354]
b.聚合酶结合的文库0.22μl
[0355]
10.将稀释的文库放回冰上。
[0356]
11.通过涡旋混合magbeads,直到它们均匀。
[0357]
12.对于每个样品,将35μl magbeads添加到空的1.5ml lobind管中。对于多于一个的样品,制备主混合物并增添10%以解决移液损失。在这种情况下,我们有四个样品,因此4.1x35μl=143.5μl。
[0358]
13.离心几秒钟以收集magbeads并将它们放在磁珠粒架上,直到上清液变澄清。
[0359]
14.弃去上清液。
[0360]
15.向磁珠粒中加入143.5μl magbead洗涤缓冲液并通过吹打10次混合直至均匀。
[0361]
16.将珠粒离心几秒钟以收集珠粒并放回磁架上,直到上清液变澄清。
[0362]
17.弃去上清液。
[0363]
18.将143.5ul magbead结合缓冲液添加到珠粒中并通过吹打10次混合。
[0364]
19.将珠粒离心几秒钟以收集珠粒。
[0365]
20.将35μl珠粒等分到四个单独的1.5ml lobind管中,并置于冰上。
[0366]
21.将珠粒放在磁珠粒架上直到上清液变澄清,然后丢弃上清液。
[0367]
22.将来自步骤10的9μl稀释的文库添加到相应的珠粒管中。盖上珠粒管并通过轻弹管混合珠粒直至均匀。离心几秒钟以收集珠粒,然后将珠粒放回冰上。
[0368]
23.在4℃下在旋转器上孵育珠粒30分钟,然后将其放回冰上。
[0369]
24.当正在珠粒孵育时,在新的200μl pcr管中稀释dna内部对照复合物。混合以下。
[0370]
a.对照1mix
[0371]
i.magbead结合缓冲液99μl
[0372]
ii.dna内部对照复合物1μl
[0373]
b.对照2mix
[0374]
i.magbead结合缓冲液49μl
[0375]
ii.对照1混合物1μl
[0376]
25.将来自步骤23的珠粒离心几秒,然后置于磁珠粒架上直至上清液变澄清。丢弃上清液并将珠粒放回冰上。
[0377]
26.向珠粒中加入18μl magbead结合缓冲液并轻弹管进行混合。
[0378]
27.将珠粒放在磁珠粒架上,直到上清液变澄清,然后丢弃上清液。
[0379]
28.向珠粒中加入18μl magbead洗涤缓冲液并轻弹管进行混合。
[0380]
29.将珠粒放在磁珠粒架上,直到上清液变澄清,然后丢弃上清液。
[0381]
30.向珠粒中加入3μl稀释的对照2混合物和42μl magbead结合缓冲液,并轻弹管进行混合。离心几秒钟以收集珠粒混合物并将其置于冰上直至使用。
[0382]
31.将新的96孔pcr板放在冰上。
[0383]
32.通过吸移混合来自步骤30的珠粒直至均匀,并且将45μl的四种混合物中的每一种混合物等分到位于冰上的96孔pcr板的同一列(如,第1列,a-d行)中的单独的孔中。
[0384]
pacbio加载和数据收集时间
[0385]
1.使用pacbio rs remote软件2.3版配置pacbio运行。使用以下设置:
[0386]
a.采集方案=magbead onecellperwell v1
[0387]
b.acquisitiontime(movie time)=360
[0388]
c.insertsize=5000
[0389]
d.stagehs=真
[0390]
e.sizeselectionenabled=假
[0391]
f.dnacontrolcomplex=2kb_对照
[0392]
g.use2ndlook=假
[0393]
h.numberofcollections=1
[0394]
2.将含有测序文库、dna测序试剂和smrtcell的pcr板加载到pacbio rs ii中。对文库测序360分钟。
[0395]
变体检测和量化
[0396]
将原始测序数据和登革热病毒参考基因组(genbank登录号u87411;登革热病毒2型,菌株16681)输入到smrtanalysis软件2.3.0.140936.p5.167094版中,该软件由daemon 2.3.0.139497、smrtpipe 2.3.0.139497、smrt portal 2.3.0.140893和smrt view 2.3.0.140836组成。使用smrtanalysis软件的rs_readsofinsert_mapping.1方案,使用默认映射参数和以下过滤参数对p1、p3、p5和p51读段与登革热病毒参考基因组(genbank登录号u87411;登革热病毒2型,菌株16681)进行比对:最小完整通过=5,最小预测精确度=90,插入物的最小读段长度=5000,插入物的最大读段长度=7000。将由rs_readsofinsert_mapping.1方案生成的插入物fasta(fasta和fastq文件可以互换使用)文件的读段与含bwa的登革热病毒(genbank登录号u87411登革热病毒2型,菌株16681)参考基因组进行比对,并且使用sam2tsv(http://lindenb.github.io/jvarkit/sam2tsv.html)和自定义bash脚本从经比对的sam文件中鉴定出每个减毒位点处的变体。使用自定义perl脚本来量化三个目标减毒位点处的变体。
[0397]
对四个smrtcell进行测序,从而产生平均约8.7kb的过滤后插入物读段。使用blasr对该读段与野生型登革热病毒参考基因组(genbank登录号u87411)进行比对,并且用quiver调用变体。数据汇总在结果部分中。
[0398]
进一步优化rt-pcr以使用tdv2产生单一pcr产物(不含非特异性条带)
[0399]
来自第一阶段的现有方案没有产生单一且干净的扩增子pcr产物。因此,尝试了额外的方案优化和方法来产生具有单个扩增子的pcr产物。这些优化包括使用先前设计的用于逆转录和pcr的引物的不同组合,增加rt和着陆pcr反应温度。这些各种优化步骤在下文有描述:
[0400]
逆转录和着陆pcr优化
[0401]
为了确定用于优化的新pcr引物组合,使用unipro ugene软件完成使用先前设计的引物集合进行的计算机模拟(in silico)pcr或epcr,从而允许30%的引物结合位点错配。基于展现出低脱靶结合的epcr分析和我们使用巢式pcr的结果,选择引物5f和3r进行优化。引物序列在表5中。
[0402]
用于巢式pcr反应中的rt-pcr优化的两对引物
[0403][0404][0405]
表5:引物序列和特征.请注意,1r反向引物也是用于逆转录的引物。该引物是成对的。
[0406]
首先,使用6.5μl tdv2病毒rna(约108个病毒基因组拷贝/μl)和引物3r在两种不同的反应温度50℃和55℃下进行逆转录。然后,用引物5f和3r如下那样执行着陆pcr:95℃持续30秒,每个循环下降1度持续10个循环的68℃下退火20秒(即循环1
–
68℃,循环2
–
67℃,循环3
–
65℃等),以及在68℃下延伸6分钟。再运行5个在95℃下持续30秒的循环,在53、55.4、57.2、59.4、61.9、65.7或68℃下退火20秒并在68℃下延伸6分钟,得到总共15个循环。将pcr产物在1%tae琼脂糖凝胶中运行,并且结果表明产生了预期大小的单个pcr扩增子。
[0407]
唯一分子标识符的引入
[0408]
唯一分子标识符(umi)的优势在于它们可以纠正由文库制备,诸如某些变体的pcr复制和优先扩增产生的伪像(artifact)。此外,umi已被证明可以通过纠正测序错误来改进对罕见变体的检测。umi的概念是每个rna或dna分子都附接有分子标签或条形码。显示出相同umi的扩增产物是相同dna分子的复制物。具有不同umi的序列读段是原始分子。umi有可能通过消除假阳性来提高检测1%或更低频率的突变的精确度。目标量化可以通过对读段中的umi的数目而不是对总读段的数目进行计数来更好地实现,因为总读段由于不均匀扩增而出现偏差。在优化引物和rt-pcr条件后,下一步骤是将umi掺入到3r逆转录引物中,并且从而掺入到每个病毒分子中,以提高对观察到的变体的量化。
[0409]
将完全随机的16-核苷酸umi和5’pcr引物iia结合位点添加到3r引物的5'末端。引物序列和特征示于表6中。
[0410]
在掺入umi期间使用的引物和寡核苷酸序列
[0411][0412]
x=基于来自clontech laboratories,inc的专有序列的未公开核苷酸。
[0413]
表6:引物和寡核苷酸序列和特征。5f和umi 3r被用于使用先前建立的使用5f和3r的成功扩增来引入umi。当尝试模板切换方案时,smarter iia和5’pcr引物iia与umi 3r一起使用。
[0414][0415]
表7:引物和seq id编号
[0416]
接下来,使用6.5μl tdv2病毒rna(约108个病毒基因组拷贝/μl)在优化条件下执行rt-pcr。将pcr产物在1%tae琼脂糖凝胶中运行。随后,执行大规模pcr。使用0.45x ampure xp珠粒纯化来浓缩pcr产物并去除过量的引物。大规模pcr和ampure xp珠粒纯化得到了预期大小的单条带的强扩增。
[0417]
pacbio测序和变体分析
[0418]
采用来自优化的rt-pcr反应的材料使用如上所述的pacbio扩增子文库制备方案构建pacbio测序文库。
[0419]
使用基于读段和基于umi的方法量化tdv2基因组
[0420]
变体没有像在阶段i中那样用quiver调用。原因是典型的变体调用算法不维护单个读段id信息,因为变体是基于变体位点处的读段一致性和覆盖率调用的。因此,不需要单独的读段信息。然而,为了利用基于umi的量化方法,含有相同umi和序列的多个读段被折叠成该读段的单个例示(instance)。因此,在使用基于umi的量化方法时,有必要维护单个读
段id信息,并且因此典型的变体调用器不便于使用umi进行分析。因此,开发了一种用于促进umi的使用的替代变体鉴定工作流程。简而言之,该工作流程由以下组成:修剪掉smarter umi 3r衔接子序列,提取umi序列并将其附加到插入物fastq文件的tdv2读段的读段id上,使用bwa-mem对经修剪的fastq文件与登革热病毒参考基因组(genbank登录号u87411;登革热病毒2型,菌株16681)进行比对以及对每个减毒位点处的回复体进行计数。
[0421]
将优化的文库制备和变体检测方案应用于tdv2单回复体
[0422]
用大规模pcr将umi掺入到单回复体中
[0423]
如上文“引入唯一分子标识符至每个病毒分子中”部分中所描述的那样执行大规模pcr。将pcr产物在1%tae琼脂糖凝胶中运行。结果示出了每个回复体的预期大小的单个扩增子。
[0424]
pacbio测序和变体分析
[0425]
采用如上所概述的pacbio扩增子文库制备方案使用所述材料构建pacbio测序文库。针对每个回复体,对一个smrtcell进行测序。
[0426]
接下来,如“使用基于读段和基于umi的方法量化tdv2变体”部分中所述的那样进行变体分析。
[0427]
将优化的文库制备和变体检测方案应用于四种复杂的回复体混合物
[0428]
复杂回复体混合物的制备
[0429]
基于如通过qrt-pcr确定的每个单独回复体中的基因组拷贝来制备四个复杂的回复体混合物汇集物,并将其运送到分析组,而没有公开任何有关组成的信息。结果部分中描述了该汇集物的详细信息。
[0430]
用大规模pcr将umi掺入到复杂回复体混合物中
[0431]
使用6.5μl病毒rna(约2.8x 107个病毒基因组拷贝/μl)和smarter umi 3r引物,如以上“引入唯一分子标识符至每个病毒分子中”部分所述的那样对四种复杂回复体混合物(复杂混合物1-4)执行rt。接着,如上所述的那样执行大规模着陆pcr,并且将pcr产物在1%tae琼脂糖凝胶中运行。结果示出了复杂混合物1-4的预期大小的单个扩增子。
[0432]
pacbio测序和变体分析
[0433]
采用如上所述的pacbio扩增子文库制备方案使用所述材料构建pacbio测序文库。针对每个复杂回复体混合物,对一个smrtcell进行测序。
[0434]
接下来,如“使用基于读段和基于umi的方法量化tdv2变体”部分中所述的那样进行变体分析。
[0435]
结果
[0436]
用于tdv2分析的pacbio测序的优化
[0437]
rt-pcr优化
[0438]
在着陆pcr反应中,在预期大小范围内的所有泳道中都存在条带,外加几个非特异性扩增产物。
[0439]
针对巢式pcr中的循环数进行了优化的两个表现最佳的引物对(1和4)给出了预期大小的条带的强扩增。引物对1具有比引物对4少的非特异性产物,并被转送用于退火温度优化。选取了11个循环的最佳值,并使用0.45x ampure xp珠粒纯化来去除小于3千碱基的非特异性扩增产物。
[0440]
pacbio测序和变体分析
[0441]
如上所述的那样使用所述材料构建pacbio测序文库。对一个smrtcell进行测序,产生366mbp的数据和12,467个插入物读段。每个插入物读段的平均通过次数为6.15,得到0.9828的平均读段质量。平均插入物长度读段为5321个核苷酸。
[0442]
注意:平均通过次数是聚合酶读取同一分子的平均次数。pacbio测序模板源自pacbio分析软件,是单链环。聚合酶继续运行,直到它脱落、被激光灭活或运行结束。
[0443]
读段与登革热病毒参考基因组(genbank登录号u87411;登革热病毒2型,菌株16681)的比对最初鉴定出了11个变体。插入物长度的映射的读段如图4所示。
[0444]
这些变体中的前三个变体因覆盖率低而没有得到良好的支持,从分析中被丢弃。其余变体得到了调用每个变体的读段百分比的充分支持。数据汇总在表8中。
[0445][0446][0447]
表8:观察到的变体以及支持读段数目的概述.
[0448]
检查了位置57、2579和5270处的三种减毒变体的组合,并将其概述在表9中。
[0449]
计数百分比57257952701047296.703tat280.259cat150.139aat120.111gat2011.856tgt50.046tct480.443taa210.194tac170.157tag20.018cgt10.009agt40.037tga10.009tgg10.009cac10.009aac00cga
[0450]
表9:三个减毒基因座的单倍型分析.
[0451]
读段计数示出在第一列中。该数据被排列成使得观察到的变体呈位置57、2579和5270的顺序。尽管pcr限制了推断有关输入rna的定量信息的能力,但tdv2单倍型tat是迄今为止最丰富的。没有发现具有参考单倍型cga的读段。其余的有一个或两个就tdv2单倍型而言的变体。
[0452]
从设盲样品中鉴定单回复体
[0453]
进行sanger测序以鉴定单回复体
[0454]
选择以下条件作为最终优化条件:
[0455]
rt在55℃下持续30分钟
[0456]
退火温度:60℃
[0457]
pcr循环数:30
[0458]
使用来自qiagen的凝胶提取试剂盒纯化所有tdv2回复体的所有pcr产物。sanger测序分析证实了tdv2回复体的身份(表10)。
[0459][0460]
表10:sanger测序结果概述
[0461]
进行pac bio测序以鉴定回复体
[0462]
单回复体分析
[0463]
30个循环被确定为最佳的,因为pcr产物在35-40个循环时开始在凝胶上向上拖尾,这指示了pcr嵌合体。
[0464]
pacbio测序和变体分析
[0465]
pacbio测序统计数据显示于表11中。使用blasr对p1、p3、p5和p51读段与登革热病毒参考基因组(genbank登录号u87411;登革热病毒2型,菌株16681)进行比对。映射的读段的数目示出在表11中。变体用quiver进行调用。单倍型t:57-g:2579-t:5270、t:57-a:2579-a:5270,c:57-a:2579-t:5270和c:57-g:2579-t:5270分别是p1、p3、p5和p51中最丰富的单倍型,占群体的》99%(表12)。
[0466][0467]
表11:单回复体p1、p3、p5和p51的pacbio测序和映射统计数据.
[0468][0469][0470][0471][0472]
表12:在三个减毒位点处的变体计数.
[0473][0474]
表13.使用常规rt-pcr和pacbio测序的回复体分析的概述
[0475]
进一步优化rt-pcr以产生单个pcr产物(使用tdv2病毒rna)
[0476]
逆转录和着陆pcr优化
[0477]
rt和着陆pcr反应的进一步优化产生了预期大小的单个pcr扩增子。结果表明产生了预期大小的单个pcr扩增子。基于这些结果,55℃被确定为最佳rt温度,因为存在5kbp以下存在较少的条带化(banding)和拖尾。由于扩增子丰度在57.2℃以上开始下降,因此57℃被确定为pcr的最后五个循环的最佳退火温度。
[0478]
唯一分子标识符的引入
[0479]
将umi序列添加到基因特异性反向3r引物中。添加umi后,使用6.5μl tdv2病毒rna(约108个病毒基因组拷贝/μl)在优化条件下执行rt-pcr。将pcr产物在1%tae琼脂糖凝胶中电泳。随后,执行大规模pcr。使用0.45x ampure xp珠粒纯化来浓缩pcr产物并去除过量的引物。大规模pcr和ampure xp珠粒纯化给出了单条带的强扩增。
[0480]
pacbio测序和变体分析
[0481]
对一个smrtcell进行测序,并产生了42.9mbp的数据和8038个插入物读段。在每个插入物读段周围的平均通过次数为4.99,得到的平均读段质量为0.9838。平均插入物长度读段为5346个核苷酸。
[0482]
使用基于读段和基于umi的方法量化tdv2基因组
[0483]
使用读段和umi的变体计数如表14所示。结果表明t:57-a:2579-t:5270tdv2单倍型是迄今为止最常见的,而未观察到野生型单倍型(c:57-g:2579-a:5270)。然而,存在一些回复体单倍型,包括p1(t:57-g:2579-t:5270)、p3(t:57-a:2579-a:5270)和p5(c:57-a:2579-t:5270)。
[0484]
[0485][0486]
表14:在三个减毒位点处使用读段或umi的tdv2变体计数.
[0487]
将优化的文库制备和变体检测方案应用于tdv2单回复体
[0488]
大规模pcr将umi掺入到单回复体中
[0489]
在这个阶段,优化了rt-pcr和文库制备方案。因此,我们跳过pcr优化步骤,并且直接转到进行如上文“引入唯一分子标识符至每个病毒分子中”部分所述的大规模pcr。将pcr产物在1%tae琼脂糖凝胶中运行。结果示出了每个回复体的预期大小的单个扩增子。
[0490]
pacbio测序和变体分析
[0491]
针对每个回复体,对一个smrtcell进行测序。接下来,如“使用基于读段和基于umi的方法量化tdv2变体”部分中所述的那样进行变体分析。结果示于表15的下表(1/4至4/4)。
[0492]
表15:tdv2单回复体(p1、p3、p5和p51)的三个减毒基因座处使用读段或umi的变体计数表
[0493][0494][0495]
表15(1/4):在三个减毒位点处使用读段或umi的p1变体计数。
[0496][0497]
表15(2/4):在三个减毒位点处使用读段或umi的p3变体计数。
[0498][0499][0500]
表15(3/4):在三个减毒位点处使用读段或umi的p5变体计数。
[0501][0502]
表15(4/4):在三个减毒位点处使用读段或umi的p51变体计数。
[0503]
pacbio测序统计数据示于表16中。使用blasr对p1、p3、p5和p51读段与登革热病毒参考基因组(genbank登录号u87411;登革热病毒2型,菌株16681)进行比对。映射的读段的数量示于表16中。
[0504]
与使用blasr和quiver工作流程的回复体分析类似,单倍型t:57-g:2579-t:5270、t:57-a:2579-a:5270、c:57-a:2579-t:5270和c:57-g:2579-t:5270分别在p1、p3、p5和p51中最丰富。但与鉴定出2-5个单倍型每个回复体的blasr和quiver工作流程相反,最近的umi工作流程鉴定出了8-11个单倍型每个回复体(参见表15)。
[0505][0506]
表16:单回复体p1、p3、p5和p51的pacbio测序统计数据.
[0507]
结果分析显示,在对这些回复体进行rt-pcr和测序后,三个主要减毒位点处的核苷酸发生了预期变化。在30个循环的pcr反应中使用6500个病毒rna拷贝时获得了过滤后8.7k的总读段(大小5.4kb)。
[0508]
复杂混合物中回复体百分比的定量
[0509]
复杂混合物的制备:
[0510]
对单个回复体进行qrt-pcr,并根据下表中给出的基因组拷贝制备4种单独的混合物(表17)。
[0511][0512]
表17:复杂样品的组合
[0513]
大规模pcr将umi掺入到复杂回复体混合物中
[0514]
将pcr产物在1%tae琼脂糖凝胶中运行。结果示出了复杂混合物1-4的预期大小的单个扩增子。大规模rt-pcr将umi掺入到复杂回复体混合物,复杂混合物1-4中。使用smarter iia寡核苷酸进行rt,然后使用5f正向引物和5'pcr引物iia反向引物进行着陆pcr。
[0515]
pacbio测序和变体分析
[0516]
针对复杂回复体混合物,对一个smrtcell进行测序。pacbio测序统计数据示于表18中。
[0517][0518]
表18:四个复杂回复体汇合物的pacbio测序统计数据。
[0519]
表19:tdv2单回复体的复杂混合物的三个减毒基因座处使用读段或umi的变体计数表.
[0520][0521][0522]
表19(1/4):在三个减毒位点处使用读段或umi的复杂混合物1(p#1)变体计数。该混合物中单倍型的估计百分比为23.27(p1)、57.76(p3)、6.89(p5)和11.20(tdv2)。
[0523][0524][0525]
表19(2/4):在三个减毒位点处使用读段或umi的复杂混合物2(p#2)变体计数。该混合物中单倍型的估计百分比为10.84(p3)、21.56(p51)、37.16(p513)和29.59(tdv-2)。
[0526][0527]
表19(3/4):在三个减毒位点处使用读段或umi的复杂混合物3(p#3)变体计数。该混合物中单倍型的估计百分比为9.62(p3)、25.12(p51)、37.16(p513)和27.72(wt denv-2)。
[0528][0529][0530]
表19(4/4):在三个减毒位点处使用读段或umi的复杂混合物4(p#4)变体计数。该混合物中单倍型的估计百分比为65.57(wtdenv-2)和33.70(tdv-2)。
[0531]
对于复杂混合物1,鉴定了13个单倍型,其中t:57-a:2579-a:5270是最丰富的,其次是t:57-g:2579-t:5270、t:57-a:2579-t:5270和c:57-a:2579-t:5270(表19;1/4);其余的单倍型占群体的百分比非常小,为约0.77%。复杂混合物2含有的单倍型的数目最大,为18个,其中c:57-g:2579-a:5270最丰富,其次是t:57-a:2579-t:5270、c:57-g:2579-t:5270和t:57-a:2579-a:5270;其余的单倍型占群体的约0.85%(表19;2/4)。复杂混合物3含有的单倍型的数目最少,为11个。单倍型c:57-g:2579-a:5270最丰富,其次是c:57-g:2579-t:5270和t:57-a:2579-a:5270,其余单倍型占群体的约0.38%(表19;3/4)。复杂混合物4含有第二多的单倍型,为14个。然而,只有两种单倍型,c:57-g:2579-a:5270和t:57-a:2579-t:5270,占群体的99%以上,其余的单倍型仅占群体的约约0.7%(表19;4/4)。
[0532]
减毒位点读段_计数读段_百分比umi_计数umi_百分比p#1
ꢀꢀꢀꢀ
t:57-a:2579-a:5270526157.78495557.76t:57-g:2579-t:5270211723.25199623.26t:57-a:2579-t:5270102511.2596111.20c:57-a:2579-t:52706256.865916.88p#2
ꢀꢀꢀꢀ
c:57-g:2579-a:5270266337.14255837.16t:57-a:2579-t:5270211929.55203729.59c:57-g:2579-t:5270154721.57148421.56t:57-a:2579-a:527078010.8774610.83p#3
ꢀꢀꢀꢀ
c:57-g:2579-a:5270574064.75542864.88c:57-g:2579-t:5270223725.23210125.11t:57-a:2579-a:52708539.628059.62p#4
ꢀꢀꢀꢀ
c:57-g:2579-a:5270515965.64492665.57t:57-a:2579-t:5270264533.65253233.70
[0533]
野生型:c:57-g:2579-a:5270
[0534]
疫苗:t:57-a:2579-t:5270
[0535]
表20.复杂样品的减毒位点处的读段和umi计数。
[0536]
pacbio结果与设盲复杂样品的比较
[0537]
将通过pacbio测序确定的复杂混合物中每种回复体的百分比与通过takeda制备的设盲混合物进行比较。从表21可以看出,汇集物p#1、p#2、p#3和p#4中每种单倍型的量与通过takeda配制的原始量相当。
[0538][0539]
表21:通过pacbio测序对复杂汇集物中的单倍型进行的定量
[0540]
通过e基因序列的rt-pcr扩增确定的身份
[0541]
逆转录聚合酶链式反应(rt-pcr)是一种灵敏的rna检测方法,它涉及两步反应。首先使rna被逆转录酶逆转录为cdna。然后将该cdna用作后续pcr步骤的模板。分离出扩增的dna片段并且使其在2%琼脂糖凝胶中可视化;方法条件列于表23中。每种单价原料药的身份均由对适当的登革热血清型特异的e基因序列的rt-pcr扩增的阳性结果和对其他三种登革热血清型特异的e基因序列的rt-pcr扩增的阴性结果来确定。设计了四个引物集合来区分登革热疫苗病毒的四种血清型。血清型特异性引物集合被设计成使得每个引物均在相应登革热血清型的基因组内仅具有一个特异性结合位点,并且每个引物集合均产生特定大小的dna片段。根据制造商的说明,使用病原体试剂盒从测试样品中分离出病毒rna。使用一步式rt-pcr试剂盒(highqu gmbh)对经分离的病毒rna执行rt-pcr。四种疫苗病毒株的扩增子对于tdv-1、tdv-2、tdv-3和tdv-4分别为419、390、731和226个碱基对,并且在2%琼脂糖凝胶上易于区分(表22)。
[0542]
表22.用于基于rt-pcr的身份测试的引物序列和扩增子大小
[0543][0544]
上述引物序列已被分配了以下序列标识符。
[0545]
tdv-1正向seq id no:24 反向seq id no:25tdv-2正向seq id no:26 反向seq id no:27tdv-3正向seq id no:28 反向seq id no:29tdv-4正向seq id no:30 反向seq id no:31
[0546]
表23:rt-pcr条件
[0547][0548]
通过qpcr确定的残留宿主细胞
[0549]
基于qpcr的方法被用于检测和估计vero dna的残留宿主细胞dna。
[0550]
该方法利用寡磁珠粒捕获来改进dna检测,并可在life technologies inc制造的试剂盒中获得。该方法被用于确保可以在散装原料药中检测到低水平的残留维罗宿主细胞dna。
[0551]
对于dna分离和纯化,残留dna样品制备试剂盒从在维罗细胞中产生的产物中提取宿主细胞dna。该试剂盒使用化学裂解和磁珠粒从包括含有高蛋白质和低dna浓度的样品在内的多式多样的样品类型中有效提取基因组dna。每个测试样品一式三份地被提取,并且对每次提取执行一次pcr反应。
[0552]
对于定量pcr,定量dna试剂盒被用于量化来自用于生产散装原料药的维罗细胞系的残留dna。定量dna试剂盒使用定量pcr通过
对试剂盒中提供的标准曲线插值来对测试样品中的低水平残留宿主细胞dna进行快速、特异性定量。
[0553]
有4个对照用于dna提取和qpcr。提取回收对照(extraction recovery control,erc)是掺有500pg维罗对照dna的原料药样品。阴性对照(nc)是分子生物学级的水,其用作从dna提取到qpcr的整个程序的阴性对照。无模板对照(ntc)是作为qpcr中的阴性对照的分子生物学级水。qpcr试剂盒中提供了内部阳性对照(ipc)作为qpcr的阳性对照。
[0554]
为使测试有效,生成的标准曲线必须满足r2大于0.99、效率为90%至110%且y截距在24至29之间的条件。ntc和se的一式三份阴性对照dna量平均值必须小于30fg。erc的一式三份回收率百分比平均值必须在50%和150%之间。ipc在所有测试样品和对照中的ct值都必须小于40.00。为了使样品结果有效,测试样品重复的可重复性必须小于或等于25%cv。
[0555]
来自qpcr的有效测试样品的ct值用于确定结果。如果ct未被确定,则结果报告为“阴性”。如果3次重复中至少2次重复的ct值落在标准曲线内,则结果报告为“阳性”。dna浓度将通过从标准曲线中插入ct值来计算。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
