cpi-613线粒体靶向小分子前药及其自组装纳米粒、制备方法和应用
技术领域
1.本发明属于药物制剂医药领域,具体涉及cpi-613线粒体靶向小分子前药及其自组装纳米粒的构建,以及在药物递送中的应用。
背景技术:
2.化疗仍然是目前治疗癌症最普遍的治疗方法,但传统的化疗药物如紫杉醇,多柔比星,顺铂等由于其安全性和有效性等问题,在临床上仍有诸多限制。一方面通过制剂手段为传统化疗药物减毒增效,另一方面也在寻找新的有效的抗肿瘤药物。抑制肿瘤细胞的代谢通路是一种有效的抗肿瘤机制。
3.cpi-613是一种合成的α-硫辛酸衍生物,是一种新型有效的丙酮酸脱氢酶(pdh)和α-酮戊二酸脱氢酶(α-kgdh)抑制剂,可以导致肿瘤细胞的代谢失调。不同于抑制糖酵解的pdk抑制剂,cpi-613反而激活了pdk,但是它的抗肿瘤作用正是来自它对pdk的过度激活,同时它还能抑制α-kgdh的活性,在阻断肿瘤细胞糖酵解通路的同时,也阻断了线粒体呼吸。多种代谢通路的阻断可能是cpi-613发挥抗肿瘤效果的原因。
4.线粒体是细胞内的重要细胞器,也是肿瘤细胞的代谢中心。将抗肿瘤药物特异性的靶向到线粒体可以降低对正常细胞的毒副作用,增加药效。线粒体是一种双层脂质膜包被的细胞器,在线粒体内膜的质子泵可以将线粒体基质中的质子送到膜间隙,从而形成跨线粒体内膜的跨膜电位,简称线粒体膜电位。在肿瘤细胞中,为了适应肿瘤独特的代谢方式,肿瘤细胞的线粒体膜电位变高,因此,当带正电的药物或制剂进入细胞后会在高的膜电位的驱动下靶向到线粒体,达到药物聚集以产生线粒体毒性,诱发肿瘤细胞的凋亡。离域亲脂性阳离子是典型的线粒体靶向剂,最常见的就是tpp(三苯基膦),可以通过共价连接把药物特异性地传递到线粒体。目前,以tpp为靶头,将药物聚集在线粒体提高效率的策略已得到广泛应用。cpi-613的羧基是唯一能与tpp共价连接的活性位点,为了提高cpi-613的药物释放速率,引入含单硫键的连接臂作为ros智能响应的断键系统。
5.另一方面,自组装纳米粒由于,稳定性好,与辅料相关的毒性问题小的优点,已成为近几年化疗药物递送研究的热点。因此,将两种策略结合起来,以期达到提高药物递送效率,提高药物的抗肿瘤效果的目的。
技术实现要素:
6.本发明的目的是提供一种具有氧化响应敏感性的有线粒体靶向功能的cpi-613小分子前药,并制备该前药的自组装纳米粒,探讨前药与纳米粒制剂的稳定性,药物释放、细胞毒性、药效学方面的研究,为开发肿瘤微环境智能响应型药物递送系统提供新的策略和更多的选择,满足临床中对高效化疗制剂的迫切需求。
7.本发明通过以下技术方案实现上述目的:
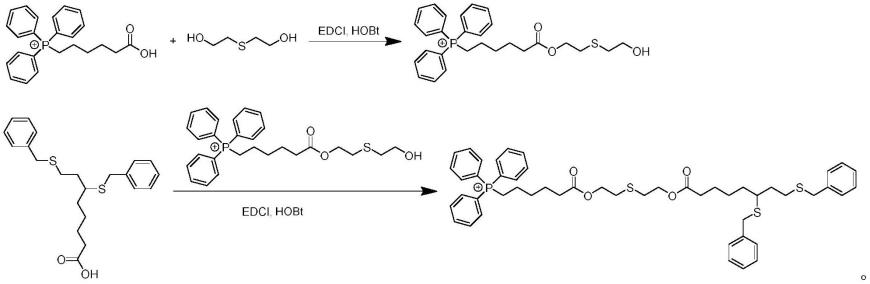
8.本发明所述的cpi-613线粒体靶向小分子前药,是以亲脂性药物cpi-613为模拟药
物,以含亲水基团的5-羧戊基三苯基溴化磷为靶头,以ros响应的硫二甘醇为连接臂连接制得,具有氧化响应敏感性,其结构如下所示:
[0009][0010]
进一步地,所述靶头还可以为其他末端为羧基的三苯基膦盐,选自3-丙基三苯基溴化磷、4-羧丁基三苯基溴化磷、6-己基三苯基溴化磷,或为亲脂性杂环阳离子化合物,选自地喹氯铵(dqa)、苯并噻唑盐、吲哚乙烯喹啉盐f16类化合物、小檗碱、胍阳离子或双胍阳离子、罗丹明。
[0011]
所述ros响应的连接臂还可以为苯硼酸酯类、二硫键类、单硒键类、二硒键类、脯氨酸类和芳基草酸类化合物。
[0012]
所述模拟药物还可以为其他含有羧基基团的作用于线粒体的抗肿瘤活性化合物,选自α-硫辛酸、α-生育酚琥珀酸酯。
[0013]
本发明还提供了所述cpi-613线粒体靶向小分子前药的合成方法,包括如下步骤:
[0014]
将5-羧戊基三苯基溴化磷与硫二甘醇以酯键相连接得到中间产物,再将该中间产物与cpi-613以酯键相连接,得到终产物cpi-613线粒体靶向小分子前药。
[0015][0016]
一种药物组合物,包含所述cpi-613线粒体靶向小分子前药和药学上可接受的载体和赋形剂。
[0017]
本发明还提供所述cpi-613线粒体靶向小分子前药自组装纳米粒的制备方法,包括如下步骤:
[0018]
将一定量的cpi-613线粒体靶向小分子前药与peg修饰剂溶解在乙醇溶液中,在搅拌的状态下,将上述乙醇溶液缓慢滴加到水中,从而自发形成纳米粒,搅拌直至乙醇挥发完全,加水定容,得到自组装纳米粒溶液。
[0019]
所述的peg修饰剂选自tpgs、dspe-peg、plga-peg、pe-peg和dspe-peg-aa,优选的peg修饰剂为tpgs。所述peg修饰剂的分子量为1000-5000,具体为1000、2000和5000,优选的分子量为2000。cpi-613线粒体靶向小分子前药与peg修饰剂的重量比为:(100:5)~(100:30),在此范围条件下发挥最好的抗肿瘤效果。
[0020]
本发明考察了所述cpi-613线粒体靶向小分子前药自组装纳米粒制剂(简称tsc自组装纳米制剂)的稳定性、氧化响应敏感性、细胞毒性、体内药效。
[0021]
具体实施方式结果表明,tsc自组装纳米制剂粒径范围在160~230nm之间,化学稳定性良好,应保存在4℃条件下,对ros有浓度依赖性的响应释放。在细胞水平上有显著增强的抗肿瘤效果,对不同的肿瘤细胞株有不同的抗肿瘤敏感性,而对正常小鼠成纤维细胞来说毒性与cpi-613相当;并且较为显著地影响了线粒体膜电位的变化,进一步促进肿瘤细胞的凋亡诱导。在小鼠人源胰腺癌模型中,低剂量下纳米粒具有显著的抗肿瘤效果,但高剂量下抗肿瘤效果与母药cpi-613相当,均具有显著的抑制肿瘤生长的效果,没有明显的差异。在提高小鼠生存期方面,tsc自组装纳米粒在高低剂量组中都表现出显著的优势。
[0022]
本发明所述的cpi-613线粒体靶向小分子前药或其药物组合物或其自组装纳米粒以静脉注射的方式给药,用于肿瘤微环境智能响应型药物传递系统。
[0023]
本发明所述的cpi-613线粒体靶向小分子前药或其药物组合物或其自组装纳米粒在制备抗肿瘤药物中的应用。
[0024]
本发明的有益效果:
[0025]
目前,cpi-613以单药的形式或联合其他化疗方案进行抗肿瘤治疗,多数临床结果不尽人意,未能达到预期的治疗效果。本发明设计的药物制剂通过提高cpi-613在肿瘤细胞线粒体中的蓄积增强了cpi-613的抗肿瘤效果。在细胞水平上表现为对多种肿瘤细胞系的更强的生长抑制以及显著降低线粒体膜电位从而诱导肿瘤细胞凋亡;在体内实验中,5mg/kg的剂量下能显著抑制异位种植荷瘤小鼠(bx pc-3)的肿瘤生长,且5mg/kg和20mg/kg的剂量均能显著延长小鼠的生存期。这提示我们,线粒体靶向修饰的、有氧化应激响应的cpi-613可以达到提高其抗肿瘤效果的目的,为临床上进一步提高其抗肿瘤疗效提供了方向,具有借鉴意义。
附图说明
[0026]
图1为实施例1合成的中间产物ts的质谱图。
[0027]
图2为实施例1合成的最终产物tsc的质谱图。
[0028]
图3为实施例1合成的最终产物tsc的核磁氢谱图。
[0029]
图4为实施例3的化学稳定性图。
[0030]
图5为实施例4的ros响应体外药物释放图。
[0031]
图6为实施例4的cpio的高效液相色谱图。
[0032]
图7为实施例4的cpio的质谱图。
[0033]
图8为实施例5的tsc纳米粒细胞毒性图。
[0034]
图9为实施例6的不同细胞系用jc-1与cpi-613和tsc nps孵育6h后的荧光照片
[0035]
图10为实施例6的绿色荧光和红色荧光的比值量化图。
[0036]
图11为实施例7的肿瘤生长趋势图。其中,a为低剂量组小鼠肿瘤体积变化图,b为高剂量组小鼠肿瘤体积变化图。
[0037]
图12为实施例7的小鼠体重变化图。
[0038]
图13为实施例7的小鼠生存期图。
具体实施方式
[0039]
下面通过实施例进一步说明本发明,但并不代表本发明局限于此实施例中,任何
本领域的技术人员能思之的变化均应落在本发明范围内。
[0040]
实施例1
[0041]
cpi-613线粒体靶向小分子前药(tsc)的合成方法,包括如下步骤:
[0042]
取5-羧戊基三苯基溴化磷(0.4570g,1mmol)、edci(0.3834g,2mmol)、hobt(0.1351g,1mmol)溶于15ml二氯甲烷(dcm)中,搅拌至其充分溶解。冰浴搅拌活化0.5h后加入含硫二甘醇(1.4534g,11.8mmol)的dcm溶液,室温下反应24h,用饱和氯化钠水溶液洗涤2遍,萃取,用无水硫酸钠在搅拌状态下干燥3h,过滤,通过减压旋蒸除去有机溶剂。通过制备液相(乙腈:水=70:30)分离纯化,再经减压蒸馏除去溶剂,得到黄色油状中间产物,记为ts(0.4931g,产率为87.9%)。经hplc分析测得其纯度在98%以上。取适量化合物ts溶于乙腈,利用ms确定样品分子量。质谱结果如图1,esi-ms(m/z):[m]
=481.39,[m h]
=482.38。
[0043]
取cpi-613(0.1943g,0.5mmol)、edci(0.1917g,1mmol)、hobt(0.0075g,0.5mmol)溶于10ml二氯甲烷(dcm)中,搅拌至其充分溶解。冰浴搅拌活化0.5h后加入含ts(0.3366g,0.6mmol)的dcm溶液室温下反应24h,用饱和氯化钠水溶液洗涤2遍,萃取,用无水硫酸钠在搅拌状态下干燥3h,过滤,通过减压旋蒸除去有机溶剂。通过制备液相(乙腈:水=80:20)分离纯化,再经减压蒸馏除去溶剂,得到淡黄色油状cpi-613线粒体靶向小分子前药(tsc)(0.1613g,产率为37.9%)。经hplc分析测得其纯度在98%以上。质谱结果如图2,核磁氢谱结果如图3。esi-ms(m/z):[m]
=851.66,[m h]
=852.65。tsc的核磁氢谱图分析如下:1h-nmr(400mhz,dmso-d6),δ7.421-7.410(m,25h),4.226-4.141(t,4h),2.811-2.765(t,4h),2.811-2.765(t,4h),2.508-2.438(m,4h),2.309-2.263(t,4h),2.091-2.968(m,1h),1.822-1.628(m,6h),1.401-1.269(m,8h)。
[0044]
实施例2
[0045]
cpi-613线粒体靶向小分子前药自组装纳米粒的制备工艺参数筛选及制备方法,包括如下步骤:
[0046]
本实施例以tpgs为修饰剂,采用纳米沉淀法制备纳米颗粒。取一定量的tsc与tpgs溶于有机溶剂,逐滴滴加到高速搅拌状态下的水中。室温下敞口搅拌直至有机溶剂挥尽,用水定容,即得目标纳米制剂。
[0047]
考察有机溶剂的种类及比例对纳米粒的影响:选用无水乙醇、乙腈、丙酮作考察的有机溶剂,固定tpgs添加质量为cpi-613线粒体靶向小分子药物质量的20%,考察有机溶剂与水的比例分别为1:15、1:9、1:7、1:4和1:2(v/v)时tsc浓度为2mg/ml纳米粒的粒径及pdi,如表1所示。
[0048]
表1不同溶剂比对纳米粒的影响
[0049][0050][0051]
由实验结果可知,有机溶剂的比例越高,粒径越小越均匀,其中乙醇作为有机相的纳米粒更均匀;乙醇比例逐渐增大,粒径及pdi的变化均不明显,说明再增加有机相的比例对减小粒径和pdi的帮助不大,且有机溶剂比例越大,挥发溶剂的时间越长;除乙醇:水=1:4和1:2外,制剂常温放置1h后粒径有明显的增大,因此,后续选择乙醇:水=1:4的比例进行处方筛选。
[0052]
考察修饰剂的添加比例对纳米粒的影响:tpgs具有两亲性,是常用的纳米制剂稳定剂,且研究发现tpgs还是p-gp抑制剂,可进一步增加药物在肿瘤细胞中的蓄积。本实施例采用分子量为2000的tpgs作为tsc自组装纳米粒的peg修饰剂。采用乙醇:水=1:4(v/v)的比例,加入质量为药物质量的0%、10%、20%和30%的tpgs,制备tsc浓度为2mg/ml的自组装纳米粒,采用马尔文粒径测定仪测定其粒径及pdi,结果如表2。
[0053]
表2不同tpgs添加比例对纳米粒的影响
[0054][0055]
由实验结果可知,tpgs添加的比例越高,平均粒径越小,pdi分布越均匀。在tpgs添加比例由20%增加到30%时,粒径变化不明显,为尽可能的少添加tpgs,后续的实验选择20%的tpgs添加量。
[0056]
考察tsc药物浓度对纳米粒的影响:为了降低后续动物实验中的给药体积,本实施例考虑将制剂制备成浓度更高的纳米粒,因此考察了乙醇:水=1:4(v/v),tpgs添加量为tsc质量的20%条件下,不同tsc药物浓度对纳米粒的粒径、pdi及实际药物浓度的影响。制备0.05mg/ml的tsc乙腈溶液作为外标。取100μl的制剂溶液并用乙腈稀释至0.05mg/ml,hplc进样分析,计算实际与理论的药物含量比例,结果如表3。
[0057]
表3不同药物浓度对纳米粒的影响
[0058][0059][0060]
由实验结果可知,纳米粒中实际药物浓度总小于其理论浓度,可推测在制备制剂时存在一定的药物损失。随着药物浓度从1mg/ml增加到6mg/ml,粒径和pdi逐渐减小,实际药物浓度逐渐升高,当药物浓度增加到8mg/ml时,粒径和pdi均有轻微的增大,实际药物含量有所降低。因此,我们最终选择制备6mg/ml的纳米制剂进行后续的实验。
[0061]
最终制备乙醇:水=1:4,tpgs添加量为tsc质量的20%,药物浓度为6mg/ml的自组装纳米制剂,记为tsc@nps。
[0062]
实施例3
[0063]
cpi-613线粒体靶向小分子前药自组装纳米粒稳定性评价
[0064]
将实施例2中制得的tsc@nps放置于4℃、25℃、37℃条件下储存,分别在2h,4h,6h取样,用马尔文粒径测定仪测定粒径及粒径分布。其物理稳定性如表1,化学稳定性如图4。由表4可知,tsc@nps只能在短时间内保存在4℃环境中,应现配现用。而由图4可知,tsc@nps在4℃、25℃、37℃条件下均没有tsc含量的明显下降,说明tsc@nps的化学稳定性较好。
[0065]
表4tsc@nps在不同温度储存条件下的物理稳定性
[0066]
[0067]
考察tsc@nps在血清中的稳定性。在10ml含10%(v/v)胎牛血清的pbs中加入1ml的tsc@nps,在37℃摇床中振荡,每2h取一次样品,每次取200μl,并再补进200μl空白释放介质。采用沉淀蛋白法提取药物(tsc)。在含制剂的样品溶液(含tsc@nps的胎牛血清的pbs溶液)中加入1.8ml的乙腈,涡旋1min使溶液充分混匀,静置,在3000rpm/min条件下低温(4℃)离心5min,取上清液,过滤后进样用hplc分析。以0h的样品中的tsc药物含量为标准,计算药物含量的变化情况,如表5。结果表明,tsc@nps的粒径及粒径分布较为均匀,药物含量随时间的增长有轻微下降。
[0068]
表5tsc@nps粒径、粒径分布、药物含量在含血清的pbs溶液中的变化情况
[0069][0070]
实施例4
[0071]
tsc@nps ros响应的体外药物释放
[0072]
在ph 7.4的pbs中添加不同浓度的过氧化氢(0mm,1mm,5mm,10mm)作为释放介质。在30ml的释放介质中加入100μl的tsc@nps,分别在0.5、1、2、4、6、8、10、12、24h取样,并向体系内补进相同体积的空白介质,继续孵育。将样品用乙腈稀释至0.02mg/ml,采用hplc检测释放出化合物的量以及tsc减少的量,并制备同等浓度的游离tsc溶液和cpi-613溶液作对照。实验过程中发现tsc的含量随过氧化氢的增加逐步降低,说明过氧化氢确实促进了tsc的分解,但并没有检测到对应的cpi-613的生成,反而在新的峰位上产生了新峰,并且新峰的最终峰面积与加入的过氧化氢的量成正比。在这里我们将新峰命名为cpio,以便区分。分别以tsc累积减少量和cpio累积生成量绘制成释放曲线,如图5,a为tsc累积减少量图,b为cpio累积增加量图,其中cpio的定量以后续cpi-613与过量过氧化氢氧化所得产物为标准。
[0073]
由图5可知,过氧化氢的添加量越多,tsc分解的越快越完全。而cpio的产生十分迅速,并且在不添加过氧化氢时几乎无产生,推测新峰是被过氧化氢氧化得到的产物,那么cpio有可能是cpi-613经过氧化氢氧化而得到的。为验证此推测,将cpi-613溶于乙腈,得到0.02mg/ml的乙腈溶液,加入10mm及过量的过氧化氢,充分反应后,用hplc分析,发现所得图像与释放实验过程中10mm组完全释放后所得的一致,如图6,其中a为制剂(tsc@nps)释放实验中10mm过氧化氢浓度下的液相色谱图,b为cpi-613经10mm过氧化氢处理的液相色谱图,c为cpi-613经过量过氧化氢处理的液相色谱图。说明cpio确实是cpi-613经过氧化氢氧化得来的产物。
[0074]
由以上结果表明,释放过程中产生的新峰是cpi-613经过氧化氢氧化所得的产物,且10mm的过氧化氢并不能使cpi-613的氧化反应进行完全。将得到的新的化合物用ms确定
其分子量以分析其结构,如图7。由图可知,[m-h]-分别为403.30728、419.13641、435.13120、451.12643。说明cpio中包含几种cpi-613的氧化产物。由于其结构相似,极性相近,难以在以上色谱条件中分离显示出来,所以在hplc中表现为一个单峰。由于hplc中并没有出现其他的杂物峰,可以以tsc的累积减少量绘制的释放曲线为标准,即随着过氧化氢浓度的增加,药物释放量也增加。至于发挥抗肿瘤效果的具体是cpi-613还是cpi-613的某种氧化产物并不影响实验的进行,不在本说明的研究范围内。
[0075]
实施例5
[0076]
cpi-613线粒体靶向小分子前药纳米粒(tsc@nps)的细胞毒性测试
[0077]
采用mtt法,考察药物对不同细胞系的毒性作用,计算半数生长抑制浓度(ic
50
)以验证cpi-613线粒体靶向小分子前药纳米粒的增毒效果。选用的细胞系为:人原位胰腺癌细胞bx pc-3;小鼠前列腺癌细胞rm-1;小鼠胰腺癌细胞pan-02;小鼠胚胎成纤维细胞nih-3t3。当培养皿中细胞长至约90%融合度时,消化细胞并收集细胞沉淀,再加入适量新鲜的培养液重新分散细胞,吸取10μl细胞悬液滴在计数板上,在倒置显微镜下计数。将细胞按照其响应的数目接种至96孔板中(rm-1:1.0
×
104cells/ml;pan-02:2
×
104cells/ml;bx pc-3:3
×
104cells/ml;nih-3t3:3
×
104cells/ml),贴壁在培养箱中培养12h。弃去旧培养液,将游离的cpi-613、cpi-613与tpp的物理混合物、tsc、tsc@nps按照倍比稀释法用对应的培养液稀释成不同的梯度浓度,向每孔中加入含药培养液(n=6),再将细胞置于培养箱中继续培养48h。随后在避光的条件下,向每孔中加入20μl mtt溶液,再放回培养箱孵育4h。弃去含药培养液,用滤纸将残留溶液吸附干净,然后向每孔中加入200μl dmso,在避光条件下振摇孔板10min,用酶标仪在570nm波长下测定各孔的吸光度。
[0078]
细胞毒结果如图8所示:a为bx pc-3细胞系的存活率曲线图,b为rm-1细胞系的存活率曲线图,c为pan-02细胞系的存活率曲线图,d为nih-3t3细胞系的存活率曲线图。细胞半数生长抑制浓度(ic
50
)如表6。
[0079]
表6cpi-613、cpi-613与tpp物理混合物、tsc、tsc@nps对不同细胞系的细胞半数生长抑制浓度表
[0080][0081]
当ic
50
值越小,说明抑制细胞生长所需药物浓度越小,即相同剂量下对细胞的毒性越大。由结果可知,tsc@nps在不同程度上降低了bx pc-3,rm-1,pan-02细胞的ic
50
,但对于nih-3t3细胞来说并没有明显的增毒效果,这说明tsc@nps对不同的细胞系有不同的敏感度,并且对于正常的小鼠成纤维细胞来说,tsc@nps的毒性与cpi-613相似。
[0082]
实施例6
[0083]
cpi-613线粒体靶向小分子前药纳米粒(tsc@nps)对不同细胞系线粒体膜电位的
影响jc-1是一种检测线粒体膜电位的荧光探针。当线粒体膜电位较高时,jc-1聚集在线粒体基质中,形成聚合物,产生红色荧光;当线粒体膜电位较低时,jc-1不能聚集在线粒体基质中,成为单体,产生绿色荧光。所以可以用红色荧光与绿色荧光的比例来考察线粒体膜电位的变化。所以以线粒体膜电位变化为指标,考察cpi-613及tsc@nps对不同细胞系线粒体膜电位的影响。
[0084]
当bx pc-3、rm-1、pan-02和nih-3t3细胞长至约90%融合度时,将细胞消化下来后,用新鲜培养基重新分散,计数,并将细胞稀释至1
×
105cells/ml,取1ml接种至铺有细胞爬片的12孔板中,在细胞培养箱中贴壁培养12h。用新鲜培养基分别将cpi-613和tsc@nps稀释至50μm。弃去旧培养基,替换为新鲜培养基和含药培养基继续孵育6h。然后每孔加入1ml jc-1染色工作液,充分混匀后在孵育20min。孵育结束后吸取上清液,并用jc-1染色缓冲液洗涤两次。将缓冲液除尽,取出细胞爬片,将其倒扣在滴有10μl抗荧光猝灭封片剂的载玻片上,放在激光共聚焦显微镜下观察,设定绿光波长为488nm,红光波长为561nm,结果如图9,a、b、c、d分别代表bx pc-3、rm-1、pan-02和nih-3t3细胞系,以绿色荧光与红色荧光的比值作为指标,用来表征线粒体膜电位的变化,其荧光量化图如图10。比值越大,说明线粒体膜电位降低的越多,诱导细胞凋亡的能力越强。
[0085]
结果表明,tsc@nps在不同程度上都更加有力地降低了细胞线粒体膜电位,说明tsc@nps相比于cpi-613增强了线粒体靶向性,进一步增加了对肿瘤细胞凋亡的诱导。
[0086]
实施例7
[0087]
cpi-613线粒体靶向小分子前药纳米粒(tsc@nps)的体内药效学研究
[0088]
将生长状态良好的bx pc-3细胞经胰酶消化后,计数,离心收集细胞,用冷pbs重新分散稀释细胞后置于冰盒中保存待用。将pbs分散均匀的肿瘤细胞悬液接种于balb/c-nu裸鼠腋下的皮下组织(5
×
106个细胞,200μl),以建立bx pc-3荷瘤小鼠模型。待荷瘤小鼠肿瘤体积长到100mm3时,将42只小鼠随机分为7组,分别记为cpi-613-h组,cpi-613-l组,tsc-h组,tsc-l组,tsc@nps-h组,tsc@nps-l组和对照组(control组),h、l的剂量分别对应的是20mg/kg和5mg/kg(按照cpi-613计算)。各组小鼠每周静脉注射给药一次,对照组给予h组相同体积的生理盐水。第一次给药后,记录小鼠肿瘤的体积及体重,来评价tsc@nps的药效。给药后,每两天用游标卡尺测量并记录各组小鼠体重及肿瘤大小,以此评价药物的急性毒性和药效。肿瘤生长大小结果如图11,小鼠体重变化如图12,小鼠生存期如图13。
[0089]
由结果可知,低剂量组中,tsc@nps具有最显著的抑制肿瘤生长的效果,而游离cpi-613对肿瘤的生长几乎无抑制作用;高剂量组中,cpi-613和tsc@nps抑瘤效果显著,且程度相似,没有明显的差别。通过记录小鼠的体重,发现小鼠体重没有明显降低,说明给药组别没有明显的急性毒性。低剂量组中,tsc@nps组的小鼠中位生存期分别是对照组的1.25倍,cpi-613组的1.3倍;高剂量组中,tsc@nps组的小鼠中位生存期分别是对照组的1.64倍,cpi-613组的1.28倍。说明相比于对照组及cpi-613-l组来说,tsc@nps在高低剂量中都明显提高了小鼠的生存期。
[0090]
以上所述的实施例对本发明的技术方案和有益效果进行了详细说明,应理解的是以上所述仅为本发明的具体实施例,并不用于限制本发明,凡在本发明的原则范围内所做的任何修改、补充和等同替换等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。