1.本发明涉及磁制冷材料技术领域。更具体地,涉及一种钆基硼酸盐晶体及其制备方法和应用。
背景技术:
2.磁热效应是所有磁性材料的固有性质。磁制冷材料主要依靠等温磁化和绝热去磁过程来实现对周围环境的降温:外加磁场为零时,材料内磁矩方向杂乱无序,磁熵较大;等温条件下施加磁场,磁矩取向趋于一致,磁熵减小,磁场对材料做功使体系绝热温度上升,向环境放热;其后在绝热条件下撤去外加磁场,磁矩恢复杂乱状态,磁熵增大,体系绝热温度下降,向外界环境吸热,达到制冷的目的。磁制冷技术是基于材料的磁热效应(mce)以获得极低温度的技术,较传统制冷技术,其有高效率、低能耗、绿色环保的优势,被誉为绿色制冷技术,因此也受到世界各国的重视。
3.磁制冷材料的选择要求磁性分子具有大的自旋基态、小的磁各向异性、高的磁密度、合适的磁交换以及低能量的激发自旋态。gd
3
离子具有半充满的4f电子壳层,基态自旋大,磁各向异性可忽略,钆基化合物可作为良好的低温磁制冷材料。因此,本发明旨在提供一种在磁制冷材料领域具有较大应用前景的新型钆基硼酸顺磁盐晶体材料。
技术实现要素:
4.本发明的第一个目的在于提供一种钆基硼酸盐晶体。
5.本发明的第二个目的在于提供一种钆基硼酸盐晶体的制备方法。
6.本发明的第三个目的在于提供一种钆基硼酸盐晶体在用作磁制冷材料中的应用。
7.为达到上述目的,本发明采用下述技术方案:
8.第一方面,本发明提供一种钆基硼酸盐晶体,所述钆基硼酸盐晶体的化学式为ba5gd3(bo3)6f。
9.进一步,所述钆基硼酸盐晶体属单斜晶系,空间群为p21/c,其晶胞参数为:/c,其晶胞参数为:α=γ=90
°
,β=99.116(7)
°
,z=1。
10.其中,稀土离子gd
3
具有高自旋基态和较小磁各向异性,但是磁性阳离子gd
3
的体积较大,常见的几种配位环境中的配体较多,本发明选择体积和相对分子质量小的配体,在构建化合物基本框架时,可以给体积较大的磁性阳离子提供尽可能多的填充空间,同时保证磁性阳离子之间存在一定的间距以较少磁相互作用,尽可能提高稀土/配体的质量比值来提高晶体磁密度。此外,小分子量的bo
33-作为配体,还可以降低晶体在实际应用过程中的加工难度并提高晶体的制冷稳定性。
11.第二方面,本发明提供一种上述钆基硼酸盐晶体的制备方法,该方法包括以下步骤:
12.将含ba化合物、含gd化合物、含b化合物和含f化合物混匀,在有氧环境下,匀速升温至650℃-700℃,恒温预烧,一次冷却后研磨;在真空环境下,再次匀速升温至800℃-810
℃,恒温反应,二次冷却后研磨得到钆基硼酸盐晶体。
13.进一步,在上述方法中,所述含ba化合物、含gd化合物、含b化合物和含f化合物中,ba、gd、b和f的摩尔比为4-6:2-4:5-7:1;优选为5:3:6:1。其中,原料配比在本发明范围内可进一步减少目标产物的杂相比例。
14.所述含ba化合物为含ba的碳酸盐或含ba的氟化物。
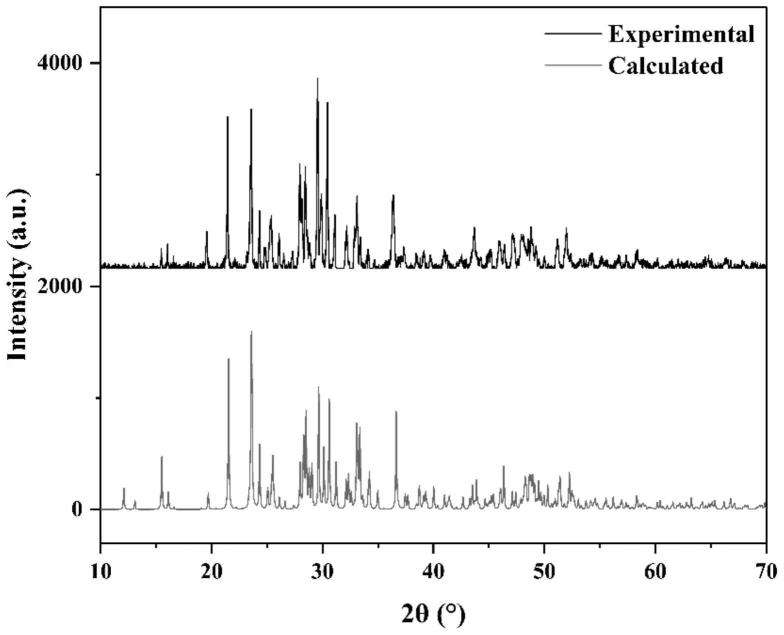
15.所述含gd化合物为含gd的氧化物。
16.所述含b化合物为h3bo3或b2o3。
17.所述含f化合物为baf2。
18.所述匀速升温或所述再次匀速升温的升温速率为30℃/h-40℃/h。
19.所述恒温预烧的时间为1d-3d。其中,恒温煅烧目的在于去除反应物中的h2o以及co2,并进行初步固相反应,所述有氧环境优选为空气气氛。
20.所述一次冷却是在40℃/h-50℃/h的降温速率下进行的。
21.所述二次冷却是在30℃/h-40℃/h的降温速率下进行的。
22.第五方面,本发明提供一种上述钆基硼酸盐晶体在用作磁制冷材料中的应用。
23.另需注意的是,如果没有特别说明,本发明所记载的任何范围包括端值以及端值之间的任何数值以及以端值或者端值之间的任意数值所构成的任意子范围。本发明中制备方法如无特殊说明则均为常规方法,所用的原料如无特别说明均可从公开的商业途径获得或根据现有技术制得。
24.本发明的有益效果
25.1)本发明提供的钆基硼酸盐晶体属于顺磁盐材料,具有可忽略的磁滞效应,用作磁制冷材料具有较高的制冷效率。
26.2)本发明提供的钆基硼酸盐晶体,在3k,δμ0h=9t条件下表现出的最大磁熵变值为29.86j kg-1
k-1
,在作为磁制冷剂方面具有巨大潜力。
27.3)本发明提供的钆基硼酸盐晶体的制备方法方便快捷,易于操作,有较大的应用前景。
附图说明
28.图1示出实施例1制备的ba5gd3(bo3)6f晶体的xrd图。
29.图2示出实施例1制备的ba5gd3(bo3)6f晶体的结构示意图。
30.图3示出实施例1制备的ba5gd3(bo3)6f晶体的红外光谱图。
31.图4示出实施例1制备的ba5gd3(bo3)6f晶体的热重曲线图。
32.图5示出实施例1制备的ba5gd3(bo3)6f晶体的变温磁化率曲线以及居里外斯拟合曲线图。
33.图6示出实施例1制备的ba5gd3(bo3)6f晶体的变温变场磁化强度图。
34.图7示出实施例1制备的ba5gd3(bo3)6f晶体的arrott曲线图。
35.图8示出实施例1制备的ba5gd3(bo3)6f晶体的磁熵变化图。
具体实施方式
36.下面通过实施例对本发明进行具体描述,有必要在此指出的是本实施例只用于对
本发明进行进一步说明,不能理解为对本发明保护范围的限制,该领域的技术熟练人员可以根据以上发明的内容做出一些非本质的改进和调整。在不冲突的情况下,本发明中的实施例及实施例中的特征可以相互组合。
37.实施例1
38.采用高温固相法制备粉末状ba5gd3(bo3)6f晶体,包括如下步骤:
39.称取baco3:2.22g(11.25mmol),baf2:0.22g(1.25mmol),gd2o3:1.36g(3.75mmol),b2o3:0.52g(7.5mmol)混匀,装入φ20mm
×
20mm的开口铂金坩埚中,将其压实,放入马弗炉中,在空气环境下以30℃/h的加热速率升温至700℃,恒温预烧2d,再以50℃/h的速率冷却至室温,研磨后得到初步预烧产物。后将预烧产物倒入石英玻璃管中抽真空密封,放入马弗炉中,以30℃/h的加热速率升温至810℃,恒温反应2d后,以40℃/h的速率冷却至室温,研磨,制得ba5gd3(bo3)6f晶体的粉末样品。
40.将本例制备的ba5gd3(bo3)6f晶体样品进行如下测试:
41.结构表征:
42.采用xrd对本例得到的ba5gd3(bo3)6f晶体进行表征,结果如图1所示,图中显示,ba5gd3(bo3)6f属单斜晶系,空间群为p21/c,其晶胞参数为:/c,其晶胞参数为:α=γ=90
°
,β=99.116(7)
°
,z=1。
43.本例制得的ba5gd3(bo3)6f结构示意图见图2所示。
44.图3是本例得到的ba5gd3(bo3)6f晶体红外光谱表征结果,图中显示,bo
33-的不对称伸缩振动峰在1265,1213和1165cm-1
,bo
33-的对称伸缩振动峰在916cm-1
,bo
33-的弯曲振动峰位于740和586cm-1
。红外光谱表明ba5gd3(bo3)6f晶体中b的配位方式是bo3,与实际结构相吻合。
45.热稳定性测试:
46.该ba5gd3(bo3)6f晶体的热重分析结果如图4所示,表明此晶体材料在室温到其熔点1235℃的温度范围内稳定性良好,不存在相变以及质量损失。
47.磁性测试:
48.采用quantum design ppms-9综合物性系统在2k-300k范围内,磁场0t-9t条件下进行以下磁热效应研究:
49.在温度范围为2k-300k,磁场范围为0-9t的条件下测得ba5gd3(bo3)6f晶体的变温磁化率以及变温磁化率倒数曲线如图5所示。根据居里-外斯定理对变温磁化率倒数曲线进行线性拟合,可得该化合物为顺磁盐材料,c=7.56emu k mol-1
,θ=0.98k,正的外斯常数说明了ba5gd3(bo3)6f晶体的极其弱的铁磁耦合作用,适用于用作磁制冷材料。
50.在温度范围为2k-10k,磁场范围为0-9t的条件下测得的ba5gd3(bo3)6f晶体的变温变场磁化强度图,如图6所示。曲线显示随着磁场强度的增强,ba5gd3(bo3)6f晶体的磁化强度逐渐增加,并在温度2k、磁场9t时达到饱和值6.72nμ
β
和理论值7nμ
β
十分接近。
51.ba5gd3(bo3)6f晶体的相变类型可以根据banerjee准则:用变温变场的磁化强度数据来估算,所得结果如图7的arrott曲线,曲线上每点的斜率均为正,说明该晶体材料的磁相变属于二级磁相变。
52.ba5gd3(bo3)6f晶体的磁熵变化可根据maxwell公式:用变温变场的磁化强度数据
来估算,所得结果如图8的磁熵曲线,可知测试范围内晶体材料在3k,δμ0h=9t时表现出最大磁熵变值29.86j kg-1
k-1
。
53.实施例2
54.采用高温固相法制备粉末状ba5gd3(bo3)6f晶体,包括如下步骤:
55.称取baco3:2.22g(11.25mmol),baf2:0.22g(1.25mmol),gd2o3:1.36g(3.75mmol),h3bo3:0.93g(15mmol)混匀,装入φ20mm
×
20mm的开口铂金坩埚中,将其压实,放入马弗炉中,在空气环境下以30℃/h的加热速率升温至700℃,恒温预烧2d,再以50℃/h的速率冷却至室温,研磨后得到初步预烧产物。后将预烧产物倒入石英玻璃管中抽真空密封,放入马弗炉中,以30℃/h的加热速率升温至810℃,恒温反应2d后,以40℃/h的速率冷却至室温,研磨,制得ba5gd3(bo3)6f晶体的粉末样品。
56.采用xrd对本例得到的ba5gd3(bo3)6f晶体进行表征,结果与实施例1基本一致。
57.实施例3
58.采用高温固相法制备粉末状ba5gd3(bo3)6f晶体,包括如下步骤:
59.称取baco3:2.22g(11.25mmol),baf2:0.22g(1.25mmol),gd2o3:1.36g(3.75mmol),h3bo3:0.46g(7.5mmol)和b2o3:0.26g(3.75mmol)混匀,装入φ20mm
×
20mm的开口铂金坩埚中,将其压实,放入马弗炉中,在空气环境下以30℃/h的加热速率升温至700℃,恒温预烧2d,再以50℃/h的速率冷却至室温,研磨后得到初步预烧产物。后将预烧产物倒入石英玻璃管中抽真空密封,放入马弗炉中,以30℃/h的加热速率升温至810℃,恒温反应2d后,以40℃/h的速率冷却至室温,研磨,制得ba5gd3(bo3)6f晶体的粉末样品。
60.采用xrd对本例得到的ba5gd3(bo3)6f晶体进行表征,结果与实施例1基本一致。
61.实施例4
62.采用高温固相法制备粉末状ba5gd3(bo3)6f晶体,包括如下步骤:
63.称取baco3:2.22g(11.25mmol),baf2:0.22g(1.25mmol),gd2o3:1.36g(3.75mmol),h3bo3:0.46g(7.5mmol)和b2o3:0.26g(3.75mmol)混匀,装入φ20mm
×
20mm的开口铂金坩埚中,将其压实,放入马弗炉中,在空气环境下以30℃/h的加热速率升温至700℃,恒温预烧1d,再以50℃/h的速率冷却至室温,研磨后得到初步预烧产物。后将预烧产物倒入石英玻璃管中抽真空密封,放入马弗炉中,以30℃/h的加热速率升温至810℃,恒温反应2d后,以40℃/h的速率冷却至室温,研磨,制得ba5gd3(bo3)6f晶体的粉末样品。
64.采用xrd对本例得到的ba5gd3(bo3)6f进行表征,结果与实施例1基本一致。
65.对比例1
66.采用高温固相法制备晶体,包括如下步骤:
67.称取baco3:2.96g(15mmol),gd2o3:1.45g(4mmol),gdf3:0.21g(1mmol)和b2o3:0.63g(9mmol)混匀,装入φ20mm
×
20mm的开口铂金坩埚中,将其压实,放入马弗炉中,在空气环境下以30℃/h的加热速率升温至700℃,恒温预烧1d,再以50℃/h的速率冷却至室温,研磨后得到初步预烧产物。后将预烧产物倒入石英玻璃管中抽真空密封,放入马弗炉中,以30℃/h的加热速率升温至810℃,恒温反应2d后,以40℃/h的速率冷却至室温,研磨,制得粉末样品。
68.采用xrd对本例得到的粉末样品进行表征,结果显示本例粉末样品的xrd与实施例1不符,即未能合成目标产物ba5gd3(bo3)6f晶体。
69.对比例2
70.采用高温固相法制备晶体,包括如下步骤:
71.称取baco3:2.22g(11.25mmol),bacl2:0.26g(1.25mmol),gd2o3:1.36g(3.75mmol),b2o3:0.52g(7.5mmol)混匀,装入φ20mm
×
20mm的开口铂金坩埚中,将其压实,放入马弗炉中,在空气环境下以30℃/h的加热速率升温至700℃,恒温预烧2d,再以50℃/h的速率冷却至室温,研磨后得到初步预烧产物。后将预烧产物倒入石英玻璃管中抽真空密封,放入马弗炉中,以30℃/h的加热速率升温至810℃,恒温反应2d后,以40℃/h的速率冷却至室温,研磨,制得粉末样品。
72.采用xrd对本例得到的粉末样品进行表征,结果显示本例粉末样品的xrd谱图与实施例1不符,即未能合成目标产物ba5gd3(bo3)6f晶体。
73.对比例4
74.采用高温固相法制备晶体,包括如下步骤:
75.称取baco3:2.22g(11.25mmol),baf2:0.22g(1.25mmol),gd2o3:1.36g(3.75mmol),b2o3:0.52g(7.5mmol)混匀,装入φ20mm
×
20mm的开口铂金坩埚中,将其压实,放入马弗炉中,在空气环境下以30℃/h的加热速率升温至700℃,恒温预烧2d,再以50℃/h的速率冷却至室温,研磨后得到初步预烧产物。空气中将预烧产物倒入石英玻璃管中,放入马弗炉,以30℃/h的加热速率升温至810℃,恒温反应2d后,以40℃/h的速率冷却至室温,研磨,制得粉末样品。
76.采用xrd对本例得到的粉末样品进行表征,结果显示本例粉末样品的xrd谱图与实施例1不符,即未能合成目标产物ba5gd3(bo3)6f晶体。
77.对比例5
78.采用水溶液法制备晶体,包括如下步骤:
79.称取baco3:2.22g(11.25mmol),baf2:0.22g(1.25mmol),gd2o3:1.36g(3.75mmol),b2o3:0.52g(7.5mmol)混匀放置于100ml玻璃烧杯中,将其溶解于体积浓度为40%的浓硝酸中,将其放置于200℃的烘箱中至烘干。将得到的白色粉末放置于φ20mm
×
20mm的开口铂金坩埚中,将其压实,放入马弗炉中,在空气环境下以30℃/h的加热速率升温至700℃,恒温预烧2d,再以50℃/h的速率冷却至室温,研磨后得到初步预烧产物。后将预烧产物倒入石英玻璃管中抽真空密封,放入马弗炉中,以30℃/h的加热速率升温至810℃,恒温反应2d后,以40℃/h的速率冷却至室温,研磨,制得粉末样品。
80.采用xrd对本例得到的粉末样品进行表征,结果显示本例粉末样品的xrd谱图与实施例1不符,即未能合成目标产物ba5gd3(bo3)6f晶体。
81.显然,本发明的上述实施例仅仅是为清楚地说明本发明所做的举例,而并非是对本发明的实施方式的限定,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引申出的显而易见的变化或变动仍处于本发明的保护范围之列。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。