1.本发明属于电极催化剂技术领域,具体涉及一种共轭微孔聚合物催化剂及其制备方法和应用。
背景技术:
2.近年来,具有超高能量密度的环保型锌空气电池(zabs)被认为是新一代可再生能源存储和转换设备的最佳选择之一。尽管zabs具有优越的技术特点,但其应用上仍面临一些重大挑战,其中最关键的是能量转换过程中阴极发生的氧还原反应(orr)的迟滞动力学,它决定了zabs的整体效率。目前最先进的催化剂是贵金属pt基催化剂,除了成本较高外,这类催化剂还存在资源稀缺、稳定性低以及对甲醇的耐受性差等问题,阻碍了zabs的广泛应用。
3.开发具有足够高的活性和长期稳定性的低成本的非铂orr催化剂是多年来研究的重点。经过几十年的不断发展,各种非贵金属催化剂被开发出来,包括过渡金属氧化物/磷化物/氮化物/氮氧化合物/硫化物、过渡金属合金、无金属催化剂,以及同时掺杂过渡金属和杂原子的碳质材料。这些材料的共同特点是具有极大的凹凸不平和多孔结构,目的是使活性位点最大限度地暴露。然而,如何对催化剂的结构、孔隙度、电导率和掺杂构型进行精细修饰,避免金属基颗粒严重的自聚集,一直是研究的重点。
4.为了解决这些问题,需要研究新的催化剂,以实现金属颗粒在多孔碳基体中的均匀分散和精细包裹,确保活性位点的最大利用。
技术实现要素:
5.本发明要解决的技术问题是:提供一种共轭微孔聚合物催化剂,在碱性和中性介质中均表现出良好的orr性能,具有良好的长期运行稳定性和甲醇免疫性能,优于20wt%pt/c催化剂;本发明还提供其制备方法和在电化学中的应用,该催化剂作为电池的阴极催化剂使用,电池比容量和能量密度高,循环可持续性长,优于20wt%pt/c催化剂的电池。
6.本发明所述的共轭微孔聚合物催化剂的制备方法,包括以下步骤:(1)4-羟基邻苯二甲腈的制备:将碳酸钾、亚硝酸钠和二甲基亚砜混合,再加入4-硝基邻苯二甲腈进行反应,反应完毕后,将产物倒入稀盐酸溶液中,抽滤,将得到的沉淀依次进行洗涤、干燥,得到4-羟基邻苯二甲腈;(2)六-(3,4)-氰基-苯氧基环三磷腈的制备:将步骤(1)得到的4-羟基邻苯二甲腈、三聚氯化磷腈、氢化钠和四氢呋喃混合进行反应,反应完毕后,将产物倒入蒸馏水中,过滤,将得到的沉淀依次进行洗涤、干燥,得到六-(3,4)-氰基-苯氧基环三磷腈;(3)fep-cmp的制备:将步骤(2)得到的六-(3,4)-氰基-苯氧基环三磷腈和无水氯化铁在惰性气体氛围
下搅拌混合,升温至90~110℃反应15~20min,再加入喹啉,继续升温至110~130℃反应30~40min,随后升温至150~170℃继续回流反应18~20h,然后冷却过滤,将得到的沉淀依次进行洗涤、索氏提取、干燥,得到fep-cmp;(4)fep-900的制备:将步骤(3)得到的fep-cmp在惰性气体氛围下,升温至900
±
5℃碳化2~2.5h,得到最终产物共轭微孔聚合物催化剂,记为fep-900。
7.步骤(1)中,4-硝基邻苯二甲腈、碳酸钾、亚硝酸钠的摩尔比为1:(1.1~1.2):(1.1~1.2); 4-硝基邻苯二甲腈与二甲基亚砜(dmso)的质量体积比为1g:(8~9)ml。
8.步骤(1)中,反应温度为24~30℃,反应时间为24~30h。
9.步骤(1)中,稀盐酸溶液的浓度优选为0.1~0.2 mol/l。
10.步骤(1)中,洗涤时,优选采用蒸馏水进行洗涤;干燥时,优选真空干燥,干燥温度80~90℃,干燥时间24~36h。
11.步骤(2)中,三聚氯化磷腈、4-羟基邻苯二甲腈、氢化钠的摩尔比为1:(6~6.5):(6~6.5);三聚氯化磷腈与四氢呋喃(thf)的质量体积比为1g:(43~45)ml。
12.步骤(2)中,反应温度为24~30℃,反应时间为4~6h。
13.步骤(2)中,洗涤时,依次采用水、异丙醇、丙酮进行洗涤;干燥时,优选真空干燥,干燥温度60~80℃,干燥时间24~36h。
14.步骤(3)和步骤(4)中,惰性气体优选为氮气或氩气。
15.步骤(3)中,六-(3,4)-氰基-苯氧基环三磷腈、无水氯化铁的摩尔比为1:(2~2.2);六-(3,4)-氰基-苯氧基环三磷腈与喹啉的质量体积比为1g:(7~8)ml。
16.步骤(3)中,洗涤时,依次采用乙醇、丙酮、甲醇、四氢呋喃、二氯甲烷进行洗涤;索氏提取时,采用四氢呋喃作为萃取剂,索氏提取时间为24~36h;干燥时,优选真空干燥,干燥温度100~110℃,干燥时间24~36h。
17.本发明所述的共轭微孔聚合物催化剂,由上述制备方法制备得到。
18.本发明所述的共轭微孔聚合物催化剂的应用,用于电化学中,优选作为电池的阴极催化剂。
19.与现有技术相比,本发明的有益效果如下:(1)本发明以六-(3,4-二氰苯氧基)环三磷腈为原料,在三氯化铁存在下进行自聚合,形成具有金属的多孔酞菁结构,并进行高温碳化,得到了fe、n、p共掺杂的碳基体,并形成了fe-n-c与fep纳米颗粒(fep-900),制备工艺简单,成本低;(2)本发明制备的共轭微孔聚合物催化剂fep-900为过渡金属(fe)和杂原子(n、p)共掺杂的新型碳基催化剂,具有良好的orr性能,在碱性和中性介质中均具有良好的长期运行稳定性和甲醇免疫能力;以fep-900为阴极催化剂的国产锌空气电池,具有比容量高、能量密度大、循环寿命长等特点,优于20wt%pt/c组成的电池。
附图说明
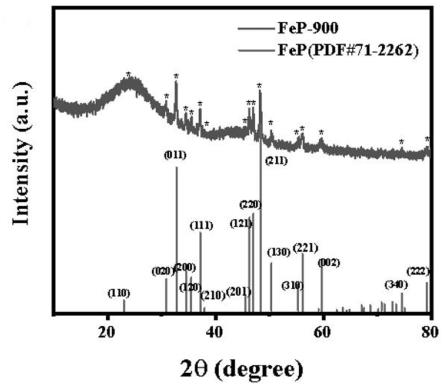
20.图1为本发明实施例1制备的fep-900的xrd表征;图2为本发明实施例1制备的fep-900的拉曼光谱;图3为本发明实施例1制备的fep-900的77k低温氮气吸收测量;
图4为本发明实施例1制备的fep-900的nldft孔径分布曲线;图5为本发明实施例1制备的fep-900的xps测量谱;图6为本发明实施例1制备的fep-900的高分辨率c的1s光谱;图7为本发明实施例1制备的fep-900的fe的2p谱;图8为本发明实施例1制备的fep-900的p的2p谱;图9为本发明实施例1制备的fep-900的n的1s谱;图10为本发明实施例1制备的fep-900的sem和tem图;图10中:a、比例尺为2μm的sem;b、比例尺为200nm的tem;c、比例尺为100nm的tem;d、比例尺为5 nm的hr-tem;图11为本发明实施例1制备的fep-900的mapping图;图11中:a、1
µ
m标尺下的透射电镜图;b、c元素在fep-900中的分布情况;c、n元素在fep-900中的分布情况;d、o元素在fep-900中的分布情况;e、p元素在fep-900中的分布情况;f、fe元素在fep-900中的分布情况;图12为在o2饱和溶液中,扫描速率为5mv
·s−1、1600rpm时,实施例1与对比例1-3制备的催化剂的lsv曲线;图13为本发明实施例1制备的fep-900在o2饱和溶液中,扫描速率为5mv
·s−1,不同转速下的lsv曲线(从上往下的转速依次增加);图14为本发明实施例1制备的fep-900对应的拟合线;图15为实施例1与对比例1-3制备的催化剂在不同电位下的过氧化物收率和电子转移数;图16为本发明实施例1制备的fep-900的耐久性评价;图17为本发明实施例1制备的fep-900与对比例1的20wt%pt/c的甲醇耐受性试验比较;其中,图12-17为催化剂在浓度0.1m的koh溶液中的电化学性能表征;图18为本发明实施例1制备的fep-900在扫描速率为50mv
·s−1时的cv;图19为在o2饱和溶液中,扫描速率为5mv
·s−1、1600rpm时,实施例1与对比例1-3制备的催化剂的lsv曲线;图20为本发明实施例1制备的fep-900在400~2500rpm不同转速下的lsv曲线(从上往下的转速依次增加);图21为实施例1与对比例1-3制备的催化剂在不同电位下的过氧化物收率和电子转移数;图22为本发明实施例1制备的fep-900的耐久性评价;图23为本发明实施例1制备的fep-900与对比例1的20wt%pt/c的甲醇耐受性试验比较;其中,图19-23为催化剂在浓度0.1m的pbs溶液(磷酸盐平衡生理盐水)中的电化学性能表征;图24为本发明制备的催化剂组装后的锌空气电池示意图;图25为本发明实施例1制备的fep-900和对比例1的20wt%pt/c作为空气阴极锌空气电池的极化曲线和功率密度曲线;
图26为使用本发明实施例1制备的fep-900和对比例1的20wt%pt/c组装的锌空气电池在10ma
·
cm-2
电流密度下的比容量;图27为本发明实施例1制备的fep-900和对比例1的20wt%pt/c组装的锌空气电池在10ma
·
cm-2
电流密度下的长期稳定性试验;图28为本发明实施例1制备的fep-900和对比例1的20wt%pt/c组装锌空气电池的充放电循环性能。
具体实施方式
21.下面结合实施例对本发明作进一步说明。
22.实施例1采用本发明的制备方法制备fep-900催化剂:(1)4-羟基邻苯二甲腈的制备:在三颈烧瓶中加入碳酸钾(2.63g,19.063mmol)、亚硝酸钠(1.32g,19.063mmol)和dmso(24ml),混合溶解后,再加入4-硝基邻苯二甲腈(3g,17.33mmol),在30℃下连续搅拌反应24h,反应完毕后,将产物倒入400ml(0.1mol/l)稀盐酸溶液中,采用抽滤法收集沉淀的灰色固体,用大量蒸馏水冲洗,然后置于80℃真空干燥箱中干燥24h,得到4-羟基邻苯二甲腈;(2)六-(3,4)-氰基-苯氧基环三磷腈的制备:三颈烧瓶中分别加入三聚氯化磷腈(2g,5.75mmol)、nah(0.83g,34.5mmol)、thf(86ml)和4-羟基邻苯二甲腈(4.97g,34.5mmol)混合,在30℃下连续搅拌反应4h,反应完毕后,将产物依次用水、异丙醇、丙酮进行过滤洗涤,然后置于60℃真空干燥箱中干燥24h,得到六-(3,4)-氰基-苯氧基环三磷腈;(3)fep-cmp的制备:将六-(3,4)-氰基-苯氧基环三磷腈(0.99g,1mmol)和无水氯化铁(0.34g,2mmol)在氩气氛围下搅拌混合,升温至100℃反应15min,加入喹啉(7ml),继续升温至120℃反应30min,随后升温至160℃继续回流反应18h,然后冷却过滤,将得到的沉淀依次用乙醇、丙酮、甲醇、四氢呋喃、二氯甲烷进行过滤洗涤,然后用四氢呋喃作为萃取剂,进行索氏提取24h,将得到的产品置于100℃真空干燥箱中干燥24h,得到fep-cmp;(4)fep-900的制备:将fep-cmp在氩气氛围下,升温至900
±
5℃碳化2h,得到最终产物共轭微孔聚合物催化剂,记为fep-900。
23.本实施例制备的fep-900的物理表征见图1-9。其中图1为fep-900的xrd表征;图2为fep-900的拉曼光谱;图3为fep-900的77k低温氮气吸收测量;图4为fep-900的nldft孔径分布曲线;图5为fep-900的xps测量谱;图6为fep-900的高分辨率c的1s光谱;图7为fep-900的fe的2p谱;图8为fep-900的p的2p谱;图9为fep-900的n的1s谱。
24.如图1所示,可以观察到fep(jspds71-2262)的显著衍射峰位于22.9、30.8、32.7、34.5、35.4、37.1、37.9、45.5、46.2、46.9、48.3、50.3、55.3、56.1、59.6、74.5和79.1,分别为(110)、(020)、(011)、(200)、(120)、(111)、(201)、(121)、(220)、(211)、(130)、(310)、(221)、(002)、(340)和(222)晶体面。
25.图2为fep-900样品的拉曼光谱,可以检测到明显的d波段和g波段,分别以1347cm-1
和1600cm-1
为中心根据峰值强度计算的fep-900的d波段和g波段比值(id/ig)分别为0.846。
26.图3给出了合成fep-900样品的低温n2吸附-脱附等温线,fep-900表现为典型的iv型等温线,在低氮气压力(p/p0《0.01)时吸附量急剧增加,吸附和解吸曲线分支上出现明显的滞后环,表明丰富的微孔和中孔同时存在;同时,在高压范围(p/p0》0.9)再次发生快速吸附增长,表明大孔同时存在,基于非局域密度泛函理论(nldft)的孔径分布等温线,进一步表征了fep-900的层次性孔隙结构(图4),主峰以1.13nm为中心,宽峰为1.48~3.96nm。计算得出fep-900的bet比表面积为959.2m2·
g-1
,累计总孔体积为0.722m2·
g-1
,有利于增加反应物与活性位点的接触。
27.通过图5的x射线光电子能谱(xps)得到fep-900样品的整体表面组成和元素电子态,主要由c、fe、o、p和n元素组成。
28.图6中fep-900的c的1s光谱由c-o(290.09ev)、c-n(286.33ev)、c-p(284.76ev)和c=c/c-c(284.33ev)峰组成。可以看出,n和p元素都成功地进入了多孔碳基质中,有效地改变了碳结构,形成了有利于orr的活性位点。
29.图7中高分辨率fe的2p谱可以解卷积成8个峰,分别分布在707.3ev、711.31ev、712.54ev、715.39ev、718.15ev、722.49ev、725.69ev和728ev。在715.39ev和725.69ev处的两个峰分别属于fe
3
的2p
3/2
和2p
1/2
,在722.49ev和712.54ev处的两个峰分别属于fe
2
的2p
3/2
和2p
1/2
,在718.15ev和728ev的两个峰被分配到卫星峰,在711.31ev和707.3ev处的两个峰分别与fe-n和fe-p有关。
30.图8中p的2p区域分别在129.48(p-fe)、133.30(p-c)和134.14ev(p-o)出现三个峰。
31.图9中n的1s的高分辨率xps谱图在402.17ev、400.91ev、400.46ev、398.80ev和398.10ev处可以检测到明显的峰,分别属于氧化n、石墨n、吡啶n、fe-n和吡啶n键。在这些n种中,吡啶-n(17.31%)、石墨-n(29.58%)和fe-n(17.39%)是orr的常用活性位点。n、p共掺杂以及fep和fe-n原子位同时存在有利于电催化。
32.本实施例制备的fep-900的sem和tem表征见图10,其中a为比例尺为2μm的sem,b为比例尺为200nm的tem,c为比例尺为100nm的tem,d为比例尺为5 nm的hr-tem。
33.从图10可以看出,fep-900具有互穿大孔的块体,从透射结果也可以发现新形成的fep纳米颗粒被均匀分配到碳基体上。图d中fep-900的hr-tem可以检测到明显的d间距为0.199nm的晶格条纹,对应于fep的(210)面。
34.本实施例制备的fep-900的mapping表征见图11,其中a为1
µ
m标尺下的透射电镜图,b为c元素在fep-900中的分布情况,c为n元素在fep-900中的分布情况,d为o元素在fep-900中的分布情况,e为p元素在fep-900中的分布情况,f为fe元素在fep-900中的分布情况。
35.从图11可以看出,fep-900元素映射进一步证实了n在多孔碳基体上的均匀分布,而p和fe元素则高度集中在fep纳米颗粒出现的位置。同时,从fe元素映射中也可以检测到fe信号,该信号属于分散在碳基体上的孤立铁原子,这一结果进一步揭示了碳骨架中同时引入了原子fe-nx和fep。
36.实施例2采用本发明的制备方法制备fep-900催化剂:(1)4-羟基邻苯二甲腈的制备:
在三颈烧瓶中加入碳酸钾(2.75g,19.93mmol)、亚硝酸钠(1.37g,19.93mmol)和dmso(27ml),混合溶解后,再加入4-硝基邻苯二甲腈(3g,17.33mmol),在26℃下连续搅拌反应28h,反应完毕后,将产物倒入400ml(0.15mol/l)稀盐酸溶液中,采用抽滤法收集沉淀的灰色固体,用大量蒸馏水冲洗,然后置于90℃真空干燥箱中干燥36h,得到4-羟基邻苯二甲腈;(2)六-(3,4)-氰基-苯氧基环三磷腈的制备:三颈烧瓶中分别加入三聚氯化磷腈(2g,5.75mmol)、nah(0.90g,37.375mmol)、thf(90ml)和4-羟基邻苯二甲腈(5.39g,37.375mmol)混合,在24℃下连续搅拌反应6h,反应完毕后,将产物依次用水、异丙醇、丙酮进行过滤洗涤,然后置于80℃真空干燥箱中干燥30h,得到六-(3,4)-氰基-苯氧基环三磷腈;(3)fep-cmp的制备:将六-(3,4)-氰基-苯氧基环三磷腈(0.99g,1mmol)和无水氯化铁(0.36g,2.1mmol)在氮气氛围下搅拌混合,升温至110℃反应18min,加入喹啉(7.5ml),继续升温至130℃反应35min,随后升温至170℃继续回流反应19h,然后冷却过滤,将得到的沉淀依次用乙醇、丙酮、甲醇、四氢呋喃、二氯甲烷进行过滤洗涤,然后用四氢呋喃作为萃取剂,进行索氏提取30h,将得到的产品置于105℃真空干燥箱中干燥36h,得到fep-cmp;(4)fep-900的制备:将fep-cmp在氩气氛围下,升温至900
±
5℃碳化2.5h,得到最终产物共轭微孔聚合物催化剂,记为fep-900。
37.实施例3采用本发明的制备方法制备fep-900催化剂:(1)4-羟基邻苯二甲腈的制备:在三颈烧瓶中加入碳酸钾(2.87g,20.79mmol)、亚硝酸钠(1.43g,20.79mmol)和dmso(26ml),混合溶解后,再加入4-硝基邻苯二甲腈(3g,17.33mmol),在24℃下连续搅拌反应30h,反应完毕后,将产物倒入400ml(0.2mol/l)稀盐酸溶液中,采用抽滤法收集沉淀的灰色固体,用大量蒸馏水冲洗,然后置于85℃真空干燥箱中干燥30h,得到4-羟基邻苯二甲腈;(2)六-(3,4)-氰基-苯氧基环三磷腈的制备:三颈烧瓶中分别加入三聚氯化磷腈(2g,5.75mmol))、nah(0.86g,35.65mmol)、thf(88ml)和4-羟基邻苯二甲腈(5.14g,35.65mmol)混合,在26℃下连续搅拌反应5h,反应完毕后,将产物依次用水、异丙醇、丙酮进行过滤洗涤,然后置于70℃真空干燥箱中干燥36h,得到六-(3,4)-氰基-苯氧基环三磷腈;(3)fep-cmp的制备:将六-(3,4)-氰基-苯氧基环三磷腈(0.99g,1mmol)和无水氯化铁(0.38g,2.2mmol)在氩气氛围下搅拌混合,升温至90℃反应20min,加入喹啉(8ml),继续升温至110℃反应40min,随后升温至150℃继续回流反应20h,然后冷却过滤,将得到的沉淀依次用乙醇、丙酮、甲醇、四氢呋喃、二氯甲烷进行过滤洗涤,然后用四氢呋喃作为萃取剂,进行索氏提取36h,将得到的产品置于110℃真空干燥箱中干燥30h,得到fep-cmp;(4)fep-900的制备:将fep-cmp在氩气氛围下,升温至900
±
5℃碳化2h,得到最终产物共轭微孔聚合物
催化剂,记为fep-900。
38.对比例1市售20wt%pt/c催化剂。
39.对比例2本对比例与实施例1的不同之处仅在于,步骤(4)中碳化温度为800
±
5℃,将制备得到的共轭微孔聚合物催化剂记为fep-800。
40.对比例3本对比例与实施例1的不同之处仅在于,步骤(4)中碳化温度为1000
±
5℃,将制备得到的共轭微孔聚合物催化剂记为fep-1000。
41.以下以实施例1为例,对fep-900催化剂与对比例1-3催化剂的电化学性能进行研究。
42.所有电化学测试均采用传统的三电极系统进行,以测量室温下orr和oer的性能。对电极采用pt板,参比电极采用kcl饱和ag/agcl或sce(饱和甘汞电极)。考虑到ag/agcl或sce的电位对应的可逆氢电极(rhe),根据nernst方程(erhe=e
ag/agcl
0.059
×
ph 0.197v;erhe=e
hg/hgo 0.059
×
ph 0.098v),将测量的电位转换为rhe。
43.工作电极由直径为5.0mm的玻璃碳盘(rde)或由直径为3mm的玻璃碳盘(pt-ring)和内径为5mm、外径为7mm的外环(pt-ring)组成的旋转圆盘电极(rrde)。以75
µ
l去离子水、375
µ
l乙醇、25
µ
l5wt%nafion溶液和5mg催化剂或20wt%工业pt/c悬浮液超声振荡0.5h形成均相催化剂油墨,抛光后的玻璃碳基片作为工作电极,涂上8
µ
l催化剂油墨,自然干燥0.5h,得到均匀的催化层。
44.在chi-760电化学工作站采用循环伏安法(cv)和旋转圆盘电极(rde)研究了该催化剂对orr的催化活性。所有测试均在碱性(浓度0.1m的koh溶液)、中性(浓度0.1m的pbs溶液)条件下进行。循环伏安法(cv)首先在ar-和o
2-饱和氢氧化钾溶液(浓度0.1m)50mv
·
s-1
中进行。在o2饱和溶液中,用5mv
·
s-1
的线性扫描伏安法(lsv)测量orr极化曲线(400~2500rpm)。
45.催化剂在0.1m koh中的电化学性能表征结果如下:在o2饱和溶液中,扫描速率为5mv
·s−1、1600rpm时,实施例1与对比例1-3制备的催化剂的lsv曲线如图12所示,fep-900的催化活性最好,其起始电位eonset为1.03v(电流密度为0.1ma
·
cm-2
),半波电位e
1/2
为0.823v,与商业20wt%pt/c催化剂相当(eonset=1.01v,e1/2=0.821v),远远超过了其他对照催化剂。fep-900的极限电流密度(5.51ma
·
cm-2
)也高于fep-800(2.52ma
·
cm-2
)和fep-1000(2.74ma
·
cm-2
),接近pt/c(5.64ma
·
cm-2
)。这些结果表明fep-900在碱性介质中对orr具有较高的电催化活性。
46.实施例1制备的fep-900在o2饱和溶液中,扫描速率为5mv
·s−1,不同转速下的lsv曲线(从上往下的转速依次增加)如图13所示,转速的增加并没有改变初始电位,反而显著增加了电流密度,说明fep-900在碱性介质中是扩散控制orr过程。
47.实施例1制备的fep-900对应的拟合线如图14所示,fep-900在0.2~0.6v的测量电位呈良好的线性关系,n的计算平均值为3.97,表明一个一步4e为主的转移过程。
48.实施例1与对比例1-3制备的催化剂在不同电位下的过氧化物收率和电子转移数如图15所示,在测量电位下h2o2产率较低(小于4.86%),进一步验证了催化剂对o2还原的高
选择性。而在0.6v下的rrde测量得到的电子转移数为3.97,进一步证实了四电子氧还原过程。
49.实施例1制备的fep-900的耐久性评价如图16所示,在测量电压下运行20000s后,fep-900仍然保持其初始电流值的86.34%,高于20wt%pt/c的56.97%。
50.实施例1制备的fep-900与对比例1的20wt%pt/c的甲醇耐受性试验比较如图17所示,fep-900样品在注入甲醇400s时pt/c的剧烈波动相比之下,变化要小得多。这一结果表明fep-900对甲醇具有较好的免疫能力。
51.催化剂在浓度0.1m的pbs溶液中的电化学性能表征结果如下:实施例1制备的fep-900在扫描速率为50mv
·s−1时的cv如图18所示,fep-900在饱和ar溶液中呈近似矩形的cv曲线,而在饱和o2电解质中则出现明显的还原峰。
52.在o2饱和溶液中,扫描速率为5mv
·s−1、1600rpm时,实施例1与对比例1-3制备的催化剂的lsv曲线如图19所示,fep-900的e1/2达到了0.703v,超过了pt/c和fep-800与fep-1000。结果表明fep-900在中性介质中对orr仍然具有较高的电催化活性。
53.实施例1制备的fep-900在400~2500rpm不同转速下的lsv曲线(从上往下的转速依次增加)如图20所示,转速的增加在中性条件中也并没有改变初始电位,而是增加了电流密度,说明fep-900在中性介质中也是扩散控制orr过程。
54.实施例1与对比例1-3制备的催化剂在不同电位下的过氧化物收率和电子转移数如图21所示,在0.2~0.7v的测量电位下,双电子副产物的产率小于2.1%,平均转移电子数约为3.96,符合四电子氧还原过程。
55.实施例1制备的fep-900的耐久性评价如图22所示,fep-900在中性电解质中的运行耐久性也远好于商业pt/c,在20000s的连续循环后仍能保持其初始值的86.2%的电流,高于pt/c的70.5%,说明fep-900的耐久性很好。
56.实施例1制备的fep-900与对比例1的20wt%pt/c的甲醇耐受性试验比较如图23所示,fep-900在注入浓度3.0m的甲醇溶液后的电流衰减也远小于商业20wt%pt/c,证实了fep-900在中性条件下也具有良好的甲醇耐受能力。
57.将实施例1制备的fep-900用于制备锌空气电池,以锌板为阳极,负载催化剂的碳纸为空气阴极,浓度6m的koh溶液为电解液,将5mg催化剂分散到375
µ
l乙醇和125
µ
l浓度5wt%的nafion溶液(全氟磺酸型聚合物溶液)中制备催化剂油墨,然后用超声波分散0.5h,将含1.5mg催化剂的油墨负载在碳纸上,制成锌空气电池。采用对比例1的工业20wt%pt/c作为参考空气电极,通过双电极系统的电化学工作站(chi-760e)得到放电过程曲线,在多通道电池测试系统(land ct2001a)上,在电流密度为10ma
·
cm-2
的情况下,放电10min,充电10min,对电池的放电-充电性能进行测试。
58.实施例1制备的fep-900组装后的锌空气电池示意图如图24所示,锌板为阳极,fep-900负载碳纸(1cm
×
1cm)为空气阴极,电解液为浓度6.0m的koh溶液。
59.实施例1制备的fep-900和对比例1的20wt%pt/c作为空气阴极锌空气电池的极化曲线和功率密度曲线如图25所示,配备fep-900的电池的最大放电功率密度为208mw
·
cm
−2,优于20wt%pt/c(184mw
·
cm
−2)。
60.实施例1制备的fep-900和对比例1的20wt%pt/c组装的锌空气电池在10ma
·
cm-2
电流密度下的比容量如图26所示,fep-900驱动的电池在电流密度为10ma
·
cm
−2时的比容量约
为801mah
·
gzn
−1,高于pt/c催化的比容量(744mah
·
gzn
−1),接近zn的理论比容量(820mah
·
gzn-1
)。
61.实施例1制备的fep-900和对比例1的20wt%pt/c组装的锌空气电池在10ma
·
cm-2
电流密度下的长期稳定性试验如图27所示,恒流放电曲线显示,fep-900基电池可连续工作97h,优于20wt%pt/c基电池(78h),进一步证实了其优异的orr稳定性。
62.实施例1制备的fep-900和对比例1的20wt%pt/c组装锌空气电池的充放电循环性能如图28所示,可以清楚地看到,与pt/c相比,fep-900的充放电电压范围要小得多,初始放电和充电电压分别为1.0844v和2.0046v,20wt%pt/c初始放电和充电电压分别为1.2291v和2.3791v。经过1000次长周期后,fep-900的放电和充电电压分别变为1.0298v和1.9859v,20wt%pt/c的放电和充电电压分别变为1.0125v和2.0186v。上述结果验证了fep-900具有良好的活性和长期运行稳定性,适合在zabs中实际应用。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
