gsk840的合成方法
技术领域
1.本发明涉及有机合成技术领域,特别是涉及一种gsk840的合成方法。
背景技术:
2.受体相互作用蛋白激酶3(ripk3)是受体相互作用的丝氨酸/苏氨酸蛋白激酶家族成员,可通过ripk1-ripk3-混合谱系激酶结构域样蛋白途径或钙/钙调素依赖性蛋白激酶ⅱ介导坏死性凋亡途径,调节炎症反应和氧化应激,进而在心脏、脑、肝脏、肾脏等损伤和病毒感染中发挥重要作用。
3.目前有多种化合物可抑制ripk3,具有潜在的临床应用前景。其中较为典型的为gsk840,其结构如下所示:
[0004][0005]
gsk840作为一种ripk3抑制剂,能够高亲和力地结合ripk3激酶结构域并抑制激酶活性,ic
50
值为0.3nm。但是目前关于其合成路线研究较少。
技术实现要素:
[0006]
基于此,本发明提供一种gsk840的合成方法。
[0007]
本发明的第一方面,提供一种gsk840的合成方法,包括如下步骤:
[0008]
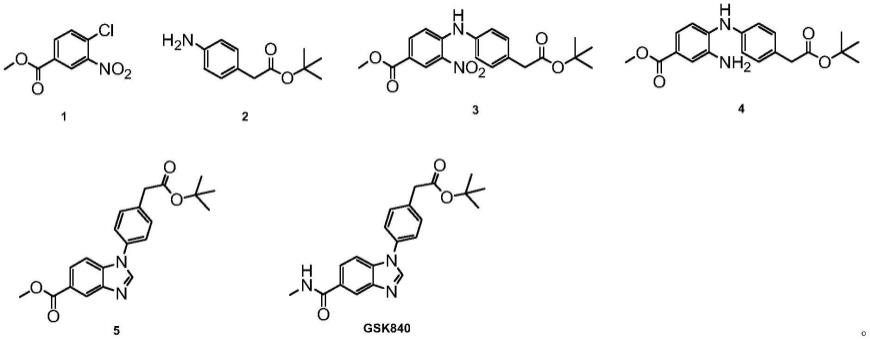
s100:将化合物1与化合物2进行取代反应,制备化合物3;
[0009]
s200:将化合物3进行还原反应,制备化合物4;
[0010]
s300:将化合物4进行关环反应,制备化合物5;
[0011]
s400:将化合物5进行胺酯交换反应,制备gsk840;
[0012]
其中,化合物1、化合物2、化合物3、化合物4、化合物5和gsk840的结构如下所示:
[0013][0014]
在其中一个实施例中,步骤s100中,取代反应的条件具有如下特征中的至少一个:
[0015]
(1)化合物1与化合物2的摩尔比为1:(1~2);
[0016]
(2)溶剂为n,n-二甲基甲酰胺、二甲基亚砜和四氢呋喃中的一种或多种;
[0017]
(3)反应温度为50℃~150℃;
[0018]
(4)反应时间为2h~12h。
[0019]
在其中一个实施例中,步骤s200中,还原反应的条件具有如下特征中的至少一个:
[0020]
(1)采用的还原剂为铁单质、锌单质和钯碳中的一种或多种;
[0021]
(2)采用的还原剂与化合物3的摩尔比为(2~7):1;
[0022]
(3)溶剂为乙醇、水、甲醇和醋酸中的一种或多种;
[0023]
(4)反应温度为0℃~100℃;
[0024]
(5)反应时间为1h~4h。
[0025]
在其中一个实施例中,步骤s300中,关环反应的条件具有如下特征中的至少一个:
[0026]
(1)采用的关环试剂为原甲酸三甲酯和甲酸中的一种或两种;
[0027]
(2)采用的关环试剂与化合物4的摩尔比为(0.01~0.1):1;
[0028]
(3)反应温度为20℃~100℃;
[0029]
(4)反应时间为2h~12h。
[0030]
在其中一个实施例中,步骤s400中,胺酯交换反应的条件具有如下特征中的至少一个:
[0031]
(1)采用的胺酯交换试剂为甲胺甲醇溶液、甲胺乙醇溶液和甲胺水溶液中的一种或多种;
[0032]
(2)采用的胺酯交换试剂与化合物5的摩尔比为(5~20):1;
[0033]
(3)反应温度为20℃~100℃;
[0034]
(4)反应时间为2h~12h。
[0035]
本发明的第二方面,提供gsk840的合成中间体3,其具有如下所示结构特征:
[0036][0037]
本发明的第三方面,提供第二方面所述的gsk840的合成中间体3的制备方法,通过
步骤s100制备得到。
[0038]
本发明的第四方面,提供gsk840的合成中间体4,其具有如下所示结构特征:
[0039][0040]
本发明的第五方面,提供第四方面所述的gsk840的合成中间体4的制备方法,通过步骤s100~步骤s200制备得到。
[0041]
本发明的第六方面,提供gsk840的合成中间体5,其具有如下所示结构特征:
[0042][0043]
本发明的第七方面,提供第六方面所述的gsk840的合成中间体5的制备方法,通过步骤s100~步骤s300制备得到。
[0044]
上述gsk840的合成方法,通过选择合适的原料和反应步骤,实现了gsk840的合成。同时该合成方法起始原料简单、便宜易得、反应步骤短、操作简便、后处理简单、收率高,具有潜在的工业化应用前景。
附图说明
[0045]
图1为实施例一中制备得到的化合物3的lc-ms谱图;
[0046]
图2为实施例二中制备得到的化合物4的lc-ms谱图;
[0047]
图3为实施例三中制备得到的化合物5的lc-ms谱图;
[0048]
图4为实施例四中制备得到的化合物gsk840的lc-ms谱图;
[0049]
图5为实施例四中制备得到的化合物gsk840的hplc谱图;
[0050]
图6为实施例四中制备得到的化合物gsk840的1h nmr谱图;
[0051]
图7为对比例1中产物的lc-ms谱图(无化合物5生成)。
具体实施方式
[0052]
以下结合具体实施例对本发明的gsk840的合成方法作进一步详细的说明。本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本发明公开内容理解更加透彻全面。
[0053]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。
[0054]
本文中,“一种或多种”指所列项目的任一种、任两种或任两种以上。
[0055]
本发明中,“第一方面”、“第二方面”、“第三方面”、“第四方面”、“第五方面”、“第六方面”、“第七方面”等仅用于描述目的,不能理解为指示或暗示相对重要性或数量,也不能理解为隐含指明所指示的技术特征的重要性或数量。
[0056]
本发明中,以开放式描述的技术特征中,包括所列举特征组成的封闭式技术方案,也包括包含所列举特征的开放式技术方案。
[0057]
本发明中,涉及到数值区间,如无特别说明,上述数值区间内视为连续,且包括该范围的最小值及最大值,以及这种最小值与最大值之间的每一个值。进一步地,当范围是指整数时,包括该范围的最小值与最大值之间的每一个整数。此外,当提供多个范围描述特征或特性时,可以合并该范围。换言之,除非另有指明,否则本文中所公开之所有范围应理解为包括其中所归入的任何及所有的子范围。
[0058]
本发明中涉及的百分比含量,如无特别说明,对于固液混合和固相-固相混合均指质量百分比,对于液相-液相混合指体积百分比。
[0059]
本发明中涉及的百分比浓度,如无特别说明,均指终浓度。所述终浓度,指添加成分在添加该成分后的体系中的占比。
[0060]
本发明中的温度参数,如无特别限定,既允许为恒温处理,也允许在一定温度区间内进行处理。所述的恒温处理允许温度在仪器控制的精度范围内进行波动。
[0061]
本发明中的室温一般指4℃~30℃,较佳地指20
±
5℃。
[0062]
本技术提供一种gsk840的合成方法,包括如下步骤:
[0063]
s100:将化合物1与化合物2进行取代反应,制备化合物3;
[0064]
s200:将化合物3进行还原反应,制备化合物4;
[0065]
s300:将化合物4进行关环反应,制备化合物5;
[0066]
s400:将化合物5进行胺酯交换反应,制备gsk840;
[0067]
其中,化合物1、化合物2、化合物3、化合物4、化合物5和gsk840的结构如下所示:
[0068][0069]
具体地,步骤s100中,化合物1和2在溶剂存在和加热条件下进行取代反应,制备化合物3:
[0070]
在其中一个示例中,化合物1与化合物2的摩尔比为1:(1~2)。具体地,化合物1与化合物2的摩尔比包括但不限于:1:1、1:1.3、1:1.5、1:1.7、1:2。
[0071]
在其中一个示例中,溶剂为n,n-二甲基甲酰胺、二甲基亚砜和四氢呋喃中的一种或多种。进一步地,溶剂为二甲基亚砜。
[0072]
在其中一个示例中,化合物1与溶剂的质量体积比为1g:(5~30)ml。具体地,化合
物1与溶剂的质量体积比包括但不限于(g:ml):1:5、1:8、1:10、1:12、1:15、1:20、1:25、1:30。
[0073]
在其中一个示例中,反应温度为50℃~150℃。具体地,反应温度包括但不限于:50℃、60℃、70℃、80℃、90℃、100℃、120℃、150℃。
[0074]
在其中一个示例中,反应时间为2h~12h。具体地,反应时间包括但不限于:2h、5h、8h、10h、12h。
[0075]
具体地,步骤s200中,在溶剂存在和还原剂存在条件下,化合物3上的硝基还原为氨基,制备化合物4:
[0076]
其中一个示例中,采用的还原剂为铁单质、锌单质和钯碳中的一种或多种。
[0077]
其中一个示例中,采用的还原剂与化合物3的摩尔比为(2~7):1。具体地,采用的还原剂与化合物3的摩尔比包括但不限于:2:1、3:1、4:1、5:1、6:1、7:1。
[0078]
其中一个示例中,溶剂为乙醇、水、甲醇和醋酸中的一种或多种。进一步地,溶剂为乙醇和水的组合。
[0079]
其中一个示例中,化合物3与溶剂的质量体积比为1g:(5~20)ml。具体地,化合物1与溶剂的质量体积比包括但不限于(g:ml):1:5、1:7、1:8、1:9、1:10、1:11、1:14、1:17、1:20。
[0080]
其中一个示例中,反应温度为0℃~100℃。具体地,反应温度包括但不限于:0℃、20℃、30℃、40℃、45℃、50℃、55℃、60℃、70℃、80℃、90℃、100℃。
[0081]
其中一个示例中,反应时间为1h~4h。具体地,反应时间包括但不限于:1h、2h、3h、4h。
[0082]
其中一个示例中,还原反应的条件还包括加入氯化铵。进一步地,化合物3与氯化铵的摩尔比选自1:(5~40)。具体地,化合物3与氯化铵的摩尔比包括但不限于:1:5、1:8、1:10、1:12、1:15、1:20、1:30、1:40。
[0083]
具体地,步骤s300中,在关环试剂存在的条件下,将化合物4进行关环反应,制备化合物5:
[0084]
其中一个示例中,采用的关环试剂为原甲酸三甲酯和甲酸中的一种或两种。进一步地,采用的关环试剂为原甲酸三甲酯。
[0085]
其中一个示例中,采用的关环试剂与化合物4的摩尔比为(0.01~0.1):1。具体地,采用的关环试剂与化合物4的摩尔比包括但不限于:0.01:1、0.02:1、0.03:1、0.04:1、0.05:1、0.07:1、0.1:1。
[0086]
其中一个示例中,反应温度为20℃~100℃。具体地,反应温度包括但不限于:20℃、30℃、40℃、50℃、60℃、70℃、80℃、100℃。
[0087]
其中一个示例中,反应时间为2h~12h。具体地,反应时间包括但不限于:2h、5h、8h、9h、10h、11h、12h。
[0088]
具体地,步骤s400中,在胺酯交换试剂存在的条件下,将化合物5进行胺酯交换反应,制备gsk840:
[0089]
其中一个示例中,采用的胺酯交换试剂为甲胺甲醇溶液、甲胺乙醇溶液和甲胺水溶液中的一种或多种。不作限制地,甲胺甲醇溶液的浓度为1m~3m,甲胺乙醇溶液的浓度为1m~3m,甲胺水溶液的体积浓度为30%~50%。进一步地,采用的胺酯交换试剂为甲胺乙醇
溶液。
[0090]
其中一个示例中,采用的胺酯交换试剂与化合物5的摩尔比为(5~20):1。具体地,采用的胺酯交换试剂与化合物5的摩尔比包括但不限于:5:1、6:1、7:1、8:1、9:1、10:1、12:1、15:1、20:1。
[0091]
其中一个示例中,反应温度为20℃~100℃。具体地,反应温度包括但不限于:20℃、50℃、60℃、70℃、80℃、90℃、100℃。
[0092]
其中一个示例中,反应时间为2h~12h。具体地,反应时间包括但不限于:2h、5h、8h、9h、10h、11h、12h。
[0093]
另外,本技术还提供gsk840的合成中间体3,其具有如下所示结构特征:
[0094][0095]
上述的gsk840的合成中间体3的制备方法,通过前述步骤s100制备得到。
[0096]
本技术还提供gsk840的合成中间体4,其具有如下所示结构特征:
[0097][0098]
上述的gsk840的合成中间体4的制备方法,通过前述步骤s100~步骤s200制备得到。
[0099]
本技术还提供gsk840的合成中间体5,其具有如下所示结构特征:
[0100][0101]
上述的gsk840的合成中间体5的制备方法,通过前述步骤s100~步骤s300制备得到。
[0102]
以下为具体实施例。如无特别说明实施例采用的原料均为市售产品。
[0103]
实施例一 化合物3的合成
[0104][0105]
往化合物1(3.00g,13.9mmol,1.00eq)的二甲基亚砜(30ml)的溶液中加入化合物2(4.33g,20.9mmol,1.50eq)。该混合溶液70℃下搅拌反应10个小时。lcms监测反应显示有化
合物3生成。将反应液倒入40ml水中,乙酸乙酯萃取(30ml
×
2),合并有机相,有机相相继用水(25ml
×
2)、饱和氯化钠水溶液(30ml
×
1)洗涤,无水硫酸钠干燥,过滤,浓缩得到粗品。粗品经柱层析(100-200目硅胶,石油醚/乙酸乙酯=100:1~90:1)得到黄色固体化合物3(2.80g,7.25mmol,52.1%yield)。
[0106]
lc-ms:(m na)
:409.1。如图1所示。
[0107]
实施例二 化合物4的合成
[0108][0109]
往化合物3(2.80g,7.25mmol,1.00eq)的乙醇(25.0ml)溶液中加入氯化铵(3.88g,72.4mmol,10.0eq)的水(5.0ml)溶液。然后在氮气保护下,铁粉(1.62g,29.0mmol,4.00eq)分批加入到上述混合物中。该混合物在50℃下搅拌反应2个小时。lcms监测反应结束后,反应液过滤,滤液浓缩得到黄色固体化合物4(2.50g,7.01mmol,96.8%yield),直接用于下一步无需纯化。
[0110]
lc-ms:(m h)
:357.1。如图2所示。
[0111]
实施例三 化合物5的合成
[0112][0113]
化合物4(2.50g,7.01mmol,1.00eq)与原甲酸三甲酯(25ml,0.23mmol,0.03eq)的混合物在50℃搅拌反应10个小时。lcms监测反应结束后,反应液浓缩得到黄色固体化合物5(2.50g,6.82mmol,97.2%yield),直接用于下一步无需纯化。
[0114]
lc-ms:(m h)
:367.2。如图3所示。
[0115]
实施例四 gsk840的合成
[0116][0117]
往100ml封管中加入化合物5(3.00g,8.19mmol,1.00eq)与2m甲胺乙醇溶液(30.0ml,7.33eq)。此混合物在80℃搅拌反应10个小时。lcms监测反应结束后,反应液浓缩
得到粗品。粗品经制备hplc(流动相为[水(甲酸):乙腈]=22%-52%,22min)得到2.4g白色固体gsk840。hnmr显示内含有~0.96%甲酸,此白色固体用30ml二氯甲烷溶解后使用水(30ml
×
2)洗涤,合并有机相,浓缩得到白色固体gsk840(2.20g,6.02mmol,91.6%yield,99.6%purity)。
[0118]
lc-ms:(m 1)
:366.0。如图4所示。
[0119]
hplc:220nm,rt=2.240min。如图5所示。
[0120]1h nmr:(400mhz,dmso-d6)δ8.65(s,1h),8.50-8.49(m,1h),8.29(s,1h),7.87-7.85(d,j=8.8hz,1h),7.66-7.64(d,j=8.0hz,3h),7.52-7.50(d,j=8.0hz,2h),3.70(s,2h),2.82-2.81(d,j=4.0hz,3h),1.44(s,9h)。如图6所示。
[0121]
对比例1
[0122]
本对比例按照实施例一~二制备得到化合物4后,按照如下步骤制备化合物5:
[0123]
化合物4(200mg,561.14μmol,1eq)与甲酸(2ml)的混合物在50℃搅拌反应10个小时。lcms监测显示化合物4完全反应完,但是无化合物5生成。
[0124]
产物的lc-ms如图7所示,可见,此条件并不适用于化合物5的合成。
[0125]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0126]
以上所述实施例仅表达了本发明的几种实施方式,便于具体和详细地理解本发明的技术方案,但并不能因此而理解为对发明专利保护范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。应当理解,本领域技术人员在本发明提供的技术方案的基础上,通过合乎逻辑的分析、推理或者有限的试验得到的技术方案,均在本发明所附权利要求的保护范围内。因此,本发明专利的保护范围应以所附权利要求的内容为准,说明书及附图可以用于解释权利要求的内容。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。