一种n-取代吲哚类化合物及其合成方法
技术领域
1.本发明属于有机合成领域,具体涉及一种n-取代吲哚类化合物及其合成方法。
背景技术:
2.n-取代吲哚类化合物是一种杂环生物碱,广泛存在于各种天然化合物,生物活性化合物和药理活性化合物中。由于其独特的化学结构,大多数吲哚环类化合物都显示出重要的生物或化学性质,可用于合成染料、医药中间体以及具有生物活性的天然化合物,是一类非常重要的杂环类精细化工中间体。传统合成n-取代吲哚类化合物主要依赖于fischer合成法,[a)b p,eilbracht p.org.lett.,2008,10,3433],该方法存在反应条件严苛、化学反应效率低的缺点。氧化反应合成法以邻氨基苯乙醇为原料,可在催化氧化作用下制备n-取代吲哚类化合物,[a)tsuji y,kotachi s,huh k t,et al.chemin form,1990,580],该反应需要价格比较昂贵的反应物,并且产物的分离过程比较复杂,在具体应用过程中的实用性较差。此外,使用金属催化是近年来发展的一种高效合成n-取代吲哚类化合物的策略[a)cera g,piscitelli s,chiarucci m.angew.chem.,int.ed.2012,51,9891],然而该方法必须使用昂贵的过渡金属催化剂。因此,现有技术中合成n-取代吲哚类化合物的方法出存在反应条件严苛、化学反应效率低的缺点,以及原料来源复杂昂贵与采用过渡金属做催化剂成本较高的问题。
技术实现要素:
[0003]
针对现有技术中存在的问题,本发明的目的在于提供一种n-取代吲哚类化合物及其合成方法,以n-取代芳香胺类化合物和碳酸亚乙烯酯为反应原料,在有机溶剂中加热反应后经分离纯化得到n-取代吲哚类化合物,原料简便易得、反应操作简单、反应条件温和、生产成本低。
[0004]
本发明是通过以下技术方案来实现:
[0005]
一种n-取代吲哚类化合物的合成方法,其特征在于,包括以下步骤,
[0006]
向有机溶剂中加入n-取代芳香胺类化合物和碳酸亚乙烯酯、钇催化剂和配体,在惰性气体环境下加热反应后分离纯化得到的n-取代吲哚类化合物。
[0007]
优选的,所述n-取代吲哚类化合物的反应式为:
[0008][0009]
其中,r1选自氢、烷基、烷氧基、芳香基、酯基、卤素、硝基、氰基、三氟甲基或杂环;r2为烷基、支链烷基、芳香基或杂环。
[0010]
优选的,所述n-取代芳香胺类化合物、碳酸亚乙烯酯、钇催化剂和配体的物质的量之比为1:1-5:0.1-3:0.1-2。
[0011]
优选的,所述n-取代芳香胺类化合物在溶剂中的浓度为0.1-0.5摩尔/升。
[0012]
优选的,所述溶剂包括二甲苯、二氯甲烷、氯苯和甲苯中的一种或多种。
[0013]
优选的,所述钇催化剂为乙酸钇、三氟甲烷磺酸钇和氢氧化钇中的一种。
[0014]
优选的,所述配体为2,2
′‑
联吡啶、4,4
′
,4
″‑
三叔丁基-2,2
′
:6
′
,2
″‑
三联吡啶和4
′‑
溴-2,2
′
:6
′
,2
″‑
三联吡啶中的一种。
[0015]
优选的,所述n-取代芳香胺类化合物包括n-甲基-(3,4,5-三甲氧基)苯胺、n-甲基-4-苯基苯胺、n-环己基苯胺、n-异丁基苯胺、n-甲基-4-(甲氧基)苯胺、n-甲基-4-氯苯胺、n-甲基-4-硝基苯胺、n-甲基-苯并[d][1,3]二氧杂环戊烯-5-胺、n-甲基-4-氟苯胺、n-异丙基苯胺、n-甲基-4-(三氟甲氧基)苯胺、n-甲基-3-(叔丁基)苯胺、n-甲基-3-(卞氧基)苯胺、n-甲基-4-(三氟甲基硫代)苯胺、n,n,n'-三甲基-1,4-苯二胺、n-甲基-3-(氯乙基)苯胺和n-环丁基苯胺中的一种。
[0016]
优选的,所述惰性气体采用氮气,所述加热温度为65℃-160℃,搅拌时间为4h-24h。
[0017]
一种n-取代吲哚类化合物,由上述的合成方法制得,所述n-取代吲哚类化合物是结构式为:
[0018][0019]
其中,r1选自氢、烷基、烷氧基、芳香基、酯基、卤素、硝基、氰基、三氟甲基或杂环;r2为烷基、支链烷基、芳香基或杂环。
[0020]
与现有技术相比本发明具有以下有益的技术效果:
[0021]
本发明开创性的使用简便易得的n-取代芳香胺类化合物和碳酸亚乙烯酯为原料,在路易斯酸催化下发生环化反应,一步生成n-取代吲哚衍生物。相比于传统合成吲哚衍生物的方法,该方法使用廉价的路易斯酸催化剂,加入的吡啶配体可使催化剂的活性增强,并且原料碳酸亚乙烯酯可作为内部氧化剂,在不需外加氧化剂和碱的条件下消去co
32-,通过c-h/n-h氧化环化反应得到n-取代n-取代吲哚类化合物。该方法操作简单,无需多种添加剂,且目标化合物种类丰富。除此之外,本发明的方法底物的普适性较好,在优化的反应条件下,目标产物易于分离,在材料及医药领域具有潜在的应用价值。
附图说明
[0022]
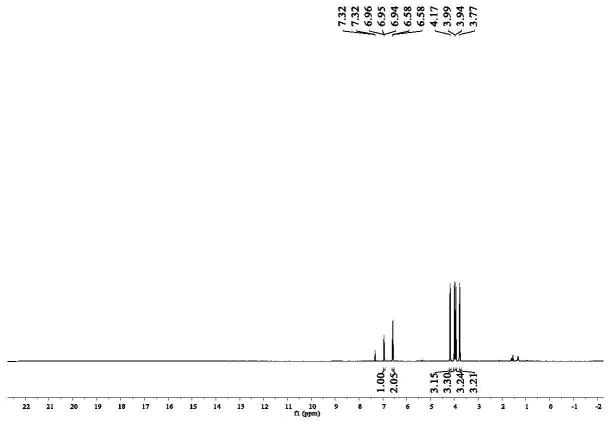
图1为实施例1所制备的产物的1h nmr谱图;
[0023]
图2为实施例1所制备的产物的
13
c nmr谱图;
[0024]
图3为实施例2所制备的产物的1h nmr谱图;
[0025]
图4为实施例2所制备的产物的
13
c nmr谱图;
[0026]
图5为实施例3所制备的产物的1h nmr谱图;
[0027]
图6为实施例3所制备的产物的
13
c nmr谱图;
[0028]
图7为实施例4所制备的产物的1h nmr谱图;
[0029]
图8为实施例4所制备的产物的
13
c nmr谱图;
[0030]
图9为实施例5所制备的产物的1h nmr谱图;
[0031]
图10为实施例5所制备的产物的
13
c nmr谱图;
[0032]
图11为实施例6所制备的产物的1h nmr谱图;
[0033]
图12为实施例6所制备的产物的
13
c nmr谱图;
[0034]
图13为实施例7所制备的产物的1h nmr谱图;
[0035]
图14为实施例7所制备的产物的
13
c nmr谱图;
[0036]
图15为实施例8所制备的产物的1h nmr谱图;
[0037]
图16为实施例8所制备的产物的
13
c nmr谱图;
[0038]
图17为实施例9所制备的产物的1h nmr谱图;
[0039]
图18为实施例9所制备的产物的
13
c nmr谱图;
[0040]
图19为实施例10所制备的产物的1h nmr谱图;
[0041]
图20为实施例10所制备的产物的
13
c nmr谱图;
[0042]
图21为实施例11所制备的产物的1h nmr谱图;
[0043]
图22为实施例11所制备的产物的
13
c nmr谱图;
[0044]
图23为实施例12所制备的产物的1h nmr谱图;
[0045]
图24为实施例12所制备的产物的
13
c nmr谱图;
[0046]
图25为实施例13所制备的产物的1h nmr谱图;
[0047]
图26为实施例13所制备的产物的
13
c nmr谱图;
[0048]
图27为实施例14所制备的产物的1h nmr谱图;
[0049]
图28为实施例14所制备的产物的
13
c nmr谱图;
[0050]
图29为实施例15所制备的产物的1h nmr谱图;
[0051]
图30为实施例15所制备的产物的
13
c nmr谱图。
[0052]
图31为实施例16所制备的产物的
13
c nmr谱图;
[0053]
图32为实施例16所制备的产物的1h nmr谱图;
[0054]
图33为实施例17所制备的产物的
13
c nmr谱图;
[0055]
图34为实施例17所制备的产物的1h nmr谱图;
[0056]
图35为实施例18所制备的产物的
13
c nmr谱图;
[0057]
图36为实施例18所制备的产物的1h nmr谱图;
具体实施方式
[0058]
下面结合具体的实施例对本发明做进一步的详细说明,所述是对本发明的解释而不是限定。
[0059]
一种n-取代吲哚类化合物的合成方法,向溶剂中加入如式1所示的n-取代芳香胺类化合物和如式2所示的碳酸亚乙烯酯和钇催化剂及配体,其中,n-取代芳香胺类化合物和碳酸亚乙烯酯、钇催化剂和配体的物质的量之比为1:1-5:0.1-3:0.1-2,向溶剂中加入n-取代芳香胺类化合物、碳酸亚乙烯酯、配体和催化剂后,n-取代芳香胺类化合物在溶剂中的浓度为0.1-0.5摩尔/升,然后在惰性气体保护下,60℃-160℃加热搅拌4h-24h后分离提纯即得到如式3所示的n-取代吲哚类化合物。
[0060][0061]
其中,r1选自氢、烷基、烷氧基、芳香基、酯基、卤素、硝基、氰基、三氟甲基或杂环;r2为烷基、支链烷基、芳香基、或杂环。
[0062]
所述的溶剂为二甲苯、二氯甲烷、氯苯和甲苯中的一种或多种。
[0063]
以下详细说明均是实施例的说明,旨在对本发明提供进一步的详细说明。除非另有指明,本发明所采用的所有技术术语与本技术所属领域的一般技术人员的通常理解的含义相同。本发明所使用的术语仅是为了描述具体实施方式,而并非意图限制根据本发明的实例性实施方式。
[0064]
实施例1
[0065]
4,5,6-trimethoxy-1-methyl-1h-indole的制备
[0066]
将0.2mmol的n-甲基-(3,4,5-三甲氧基)苯胺、0.3mmol的碳酸亚乙烯酯、0.01mmol的乙酸钇和0.02mmol的4,4
′
,4
″‑
三叔丁基-2,2
′
:6
′
,2
″‑
三联吡啶溶于装有1.2ml甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于90℃下加热搅拌12h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg白色固体化合物,产率为65%,所得产品结构式如下:
[0067][0068]
如图1和图2所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ6.95(s,1h),6.62
–
6.53(m,2h),4.17(s,3h),3.99(s,3h),3.94(s,3h),3.77(s,3h).
13
cnmr(100mhz,cdcl3):δ150.9,145.8,135.4,133.7,126.8,115.2,98.5,87.7,61.5,60.7,56.4,33.1.
[0069]
其中,tlc检测反应为薄层色谱法,将适宜的固定相涂布于玻璃板、塑料或铝基片上,成一均匀薄层。待点样、展开后,根据比移值(rf)与适宜的对照物按同法所得的色谱图的比移值(rf)作对比,用以进行药品的鉴别、杂质检查或含量测定的方法。薄层色谱法是快速分离和定性分析少量物质的一种很重要的实验技术,也用于跟踪反应进程。
[0070]
实施例2
[0071]
1-methyl-5-phenyl-1h-indole的制备
[0072]
将0.2mmol的n-甲基-4-苯基苯胺、0.3mmol的碳酸亚乙烯酯、0.01mmol的乙酸钇和0.02mmol的4
′‑
溴-2,2
′
:6
′
,2
″‑
三联吡啶溶于装有1.2ml甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于90℃下加热搅拌12h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,粗产物经硅胶柱层析分离,得到24.5mg白色固体化合物,产率为65%,所得产品结构式如下:
[0073][0074]
如图3和图4所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.93(s,1h),7.76
–
7.70(m,2h),7.59
–
7.49(m,3h),7.46(d,j=8.5hz,1h),7.39(t,j=7.4hz,1h),7.16(d,j=3.0hz,1h),6.62(d,j=3.0hz,1h),3.89(s,3h).
13
cnmr(100mhz,cdcl3):δ142.7,136.3,132.9,129.5,129.0,128.7,127.4,126.3,121.4,119.5,109.5,101.4,33.0.
[0075]
实施例3
[0076]
1-cyclohexyl-1h-indole的制备
[0077]
将0.2mmol的n-环己基苯胺、0.3mmol的碳酸亚乙烯酯、0.01mmol的三氟甲烷磺酸钇和0.02mmol的2,2
′‑
联吡啶溶于装有1.2ml二甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于90℃下加热搅拌12h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg无色油状化合物,产率为56%,所得产品结构式如下:
[0078][0079]
如图5和图6所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.71(d,j=7.9hz,1h),7.46(d,j=8.3hz,1h),7.31
–
7.22(m,2h),7.18(t,j=8.0hz,
[0080]
1h),6.62
–
6.53(m,1h),4.30(t,j=11.9hz,1h),2.28
–
2.14(m,2h),2.09
–
1.95(m,2h),1.91
–
1.71(m,3h),1.64
–
1.50(m,2h),1.40
–
1.28(m,1h).
13
c nmr(100mhz,cdcl3):δ135.5,128.5,124.1,121.1,121.0,119.2,109.5,101.0,55.1,33.6,26.0,25.7.
[0081]
实施例4
[0082]
1-isobutyl-1h-indole的制备
[0083]
将0.2mmol的n-异丁基苯胺、0.3mmol的碳酸亚乙烯酯、0.01mmol的三氟甲烷磺酸钇和0.02mmol的2,2
′‑
联吡啶溶于装有1.2ml甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于90℃下加热搅拌12h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg无色油状化合物,产率为64%,所得产品结构式如下:
[0084][0085]
如图7和图8所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.67(d,j=8.6hz,1h),7.37(d,j=7.6hz,1h),7.23(t,j=7.1hz,1h),7.17
–
7.09(m,2h),6.53(d,j=3.5hz,1h),
3.95(d,j=6.8hz,2h),2.30
–
2.18(m,1h),0.99
–
0.94(m,6h).
13
c nmr(100mhz,cdcl3):δ136.2,128.5,128.4,121.3,120.9,119.1,109.6,100.7,54.2,29.5,20.4.
[0086]
实施例5
[0087]
5-methoxy-1-methyl-1h-indole的制备
[0088]
将0.2mmol的n-甲基-4-(甲氧基)苯胺、0.3mmol的碳酸亚乙烯酯、0.01mmol的三氟甲烷磺酸钇和0.02mmol的2,2
′‑
联吡啶溶于装有1.2ml甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于90℃下加热搅拌12h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg白色固体化合物,产率为68%,所得产品结构式如下:
[0089][0090]
如图9和图10所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.24(d,j=8.9hz,1h),7.13(s,1h),7.05(d,j=2.4hz,1h),6.92(d,j=8.9hz,1h),6.43(d,j=2.3hz,1h),3.88(s,3h),3.79(s,3h).
13
c nmr(100mhz,cdcl3):δ154.1,132.2,129.3,128.8,111.9,109.9,102.7,100.4,56.0,32.9.
[0091]
实施例6
[0092]
5-chloro-1-methyl-1h-indole的制备
[0093]
将0.2mmol的n-甲基-4-氯苯胺、0.3mmol的碳酸亚乙烯酯、0.01mmol的三氟甲烷磺酸钇和0.02mmol的吡啶配体溶于装有1.2ml甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于90℃下加热搅拌12h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg无色油状化合物,产率为55%,所得产品结构式如下:
[0094][0095]
如图11和图12所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.63(s,1h),7.27(d,j=8.8hz,1h),7.21(d,j=8.7hz,1h),7.11(d,j=2.9hz,1h),6.47(d,j=3.0hz,1h),3.82(s,3h).
13
c nmr(100mhz,cdcl3):δ135.2,130.1,129.5,125.1,121.8,120.2,110.2,100.6,33.0.
[0096]
实施例7
[0097]
6-bromo-1-methyl-1h-indole的制备
[0098]
将0.2mmol的n-甲基-3-溴苯胺、0.08mmol的碳酸亚乙烯酯、0.08mmol的三氟甲烷磺酸钇催化剂和0.04mmol的4,4
′
,4
″‑
三叔丁基-2,2
′
:6
′
,2
″‑
三联吡啶溶于装有2.0ml二甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于90℃下加热搅拌12h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后
加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg无色油状化合物,产率为57%,所得产品结构式如下:
[0099][0100]
如图13和图14所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.58
–
7.51(m,2h),7.28(d,j=8.4hz,1h),7.07(d,j=3.2hz,1h),6.53(d,j=3.1hz,1h),3.80(s,3h).
13
c nmr(100mhz,cdcl3):δ137.6,129.4,127.4,122.6,122.1,115.2,112.3,101.3,32.8.
[0101]
实施例8
[0102]
1-methyl-5-nitro-1h-indole的制备
[0103]
将0.2mmol的n-甲基-4-硝基苯胺、0.3mmol的碳酸亚乙烯酯、0.01mmol的三氟甲烷磺酸钇和0.02mmol的2,2
′‑
联吡啶溶于装有1ml甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于110℃下加热搅拌12h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg黄色固体化合物,产率为51%,所得产品结构式如下:
[0104][0105]
如图15和图16所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ8.63(s,1h),8.17(d,j=9.1hz,1h),7.38(d,j=9.0hz,1h),7.25(d,j=3.3hz,1h),6.72(d,j=3.2hz,1h),3.91(s,3h).
13
c nmr(100mhz,cdcl3):δ141.7,139.4,132.0,127.7,118.1,117.2,109.0,103.9,33.3.
[0106]
实施例9
[0107]
5-methyl-5h-[1,3]dioxolo[4,5-f]indole的制备
[0108]
将0.2mmol的n-甲基-苯并[d][1,3]二氧杂环戊烯-5-胺、0.3mmol的碳酸亚乙烯酯、0.01mmol的三氟甲烷磺酸钇和0.02mmol的吡啶配体溶于装有1ml甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于90℃下加热搅拌12h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg无色油状化合物,产率为64%,所得产品结构式如下:
[0109][0110]
如图17和图18所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.06(s,1h),6.97(d,j=3.1hz,1h),6.83(s,1h),6.41(d,j=3.1hz,1h),5.98(s,2h),3.76(s,3h).
13
c nmr(100mhz,cdcl3):δ144.8,142.8,132.0,127.3,122.3,101.1,100.5,99.3,90.2,33.1.
[0111]
实施例10
[0112]
5-fluoro-1-methyl-1h-indole的制备
[0113]
将0.2mmol的n-甲基-4-氟苯胺、0.3mmol的碳酸亚乙烯酯、0.01mmol的三氟甲烷磺酸钇和0.02mmol的4
′‑
溴-2,2
′
:6
′
,2
″‑
三联吡啶溶于装有1.0ml甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于160℃下加热搅拌4h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg无色油状化合物,产率为65%,所得产品结构式如下:
[0114][0115]
如图19和图20所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.32
–
7.24(m,2h),7.13(d,j=3.0hz,1h),7.02(t,j=9.1hz,1h),6.49(d,j=3.1hz,1h),3.84(s,3h).
13
c nmr(100mhz,cdcl3):δ157.9(d,j=232.5hz),133.4,130.3,128.7(d,j=10.3hz),109.8(d,j=33.4hz),109.7,105.5(d,j=23.1hz),100.9(d,j=4.6hz),33.0.
[0116]
实施例11
[0117]
1-isopropyl-1h-indole的制备
[0118]
将0.2mmol的n-异丙基苯胺、0.3mmol的碳酸亚乙烯酯、0.04mmol的三氟甲烷磺酸钇和0.02mmol的4,4
′
,4
″‑
三叔丁基-2,2
′
:6
′
,2
″‑
三联吡啶溶于装有1.6ml甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于65℃下加热搅拌24h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg无色油状化合物,产率为60%,所得产品结构式如下:
[0119][0120]
如图21和图22所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.68(d,j=7.9hz,1h),7.43(d,j=8.3hz,1h),7.29
–
7.22(m,2h),7.15(t,j=6.9hz,1h),6.57(d,j=3.2hz,1h),4.80
–
4.68(m,1h),1.62
–
1.57(m,6h).
13
c nmr(100mhz,cdcl3):δ135.6,128.7,123.5,121.2,121.0,119.2,109.4,101.2,47.0,22.7.
[0121]
实施例12
[0122]
1-methyl-5-(trifluoromethoxy)-1h-indole的制备
[0123]
将0.2mmol的n-甲基-4-(三氟甲氧基)苯胺、0.3mmol的碳酸亚乙烯酯、0.01mmol的三氟甲烷磺酸钇和0.02mmol的2,2
′‑
联吡啶溶于装有0.8ml氯苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌8h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg无色油状化合物,产率为54%,所得产品结构式如下:
[0124][0125]
如图23和图24所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.53(s,1h),7.33(d,j=8.8hz,1h),7.18
–
7.12(m,2h),6.55(d,j=3.1hz,1h),3.85(s,3h).
13
c nmr(100mhz,cdcl3):δ142.9,135.1,130.5,128.5,120.9(q,j=253.5hz),115.5,113.2,109.7,101.4,33.0.
[0126]
实施例13
[0127]
6-(tert-butyl)-1-methyl-1h-indole的制备
[0128]
将0.2mmol的n-甲基-3-(叔丁基)苯胺、0.3mmol的碳酸亚乙烯酯、0.01mmol的三氟甲烷磺酸钇和0.02mmol的4
′‑
溴-2,2
′
:6
′
,2
″‑
三联吡啶溶于装有1.2ml甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于90℃下加热搅拌12h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg无色油状化合物,产率为61%,所得产品结构式如下:
[0129][0130]
如图25和图26所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.64(d,j=8.4hz,1h),7.38(s,1h),7.29(d,j=8.4hz,1h),7.08(d,j=3.1hz,1h),6.51(d,j=3.1hz,1h),3.86(s,3h),1.50(s,9h).
13
c nmr(100mhz,cdcl3):δ145.0,136.8,128.6,126.2,120.3,117.7,105.3,100.5,34.9,32.7,31.9.
[0131]
实施例14
[0132]
6-(benzyloxy)-1-methyl-1h-indole的制备
[0133]
将0.2mmol的n-甲基-3-(卞氧基)苯胺、0.3mmol的碳酸亚乙烯酯、0.06mmol的氢氧化钇和0.02mmol的4,4
′
,4
″‑
三叔丁基-2,2
′
:6
′
,2
″‑
三联吡啶溶于装有1.8ml氯苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于90℃下加热搅拌12h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg无色油状化合物,产率为65%,所得产品结构式如下:
[0134][0135]
如图27和图28所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.59
–
7.51(m,3h),7.48
–
7.37(m,3h),7.00(d,j=3.1hz,1h),6.96
–
6.90(m,2h),6.48(d,j=3.1hz,1h),5.20(s,2h),3.77(s,3h).
13
c nmr(100mhz,cdcl3):δ155.5,137.6,137.4,128.5,127.9,127.8,127.5,123.1,121.4,109.9,100.9,94.5,70.8,32.8.
[0136]
实施例15
[0137]
1-methyl-5-((trifluoromethyl)thio)-1h-indole的制备
[0138]
将0.2mmol的n-甲基-4-(三氟甲基硫代)苯胺、0.3mmol的碳酸亚乙烯酯、0.02mmol的三氟甲烷磺酸钇和0.04mmol的吡啶配体溶于装有1.8ml二甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于80℃下加热搅拌14h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg无色油状化合物,产率为52%,所得产品结构式如下:
[0139][0140]
如图29和图30所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ8.00(s,1h),7.52(d,j=8.5hz,1h),7.38(d,j=8.5hz,1h),7.15(d,j=4.4hz,1h),6.58(d,j=4.3hz,1h),3.84(s,3h).
13
c nmr(100mhz,cdcl3):δ137.7,130.4,130.3,130.0(q,j=306.0hz),129.3,129.3,113.6,110.0,101.7,32.9.
[0141]
实施例16
[0142]
n,n,1-trimethyl-1h-indol-5-amine的制备
[0143]
将0.2mmol的n,n,n'-三甲基-1,4-苯二胺、0.4mmol的碳酸亚乙烯酯、0.01mmol的三氟甲烷磺酸钇和0.02mmol的4,4
′
,4
″‑
三叔丁基-2,2
′
:6
′
,2
″‑
三联吡啶溶于装有1ml甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于90℃下加热搅拌12h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg无色油状化合物,产率为61%,所得产品结构式如下:
[0144][0145]
如图31和图32所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.26(d,j=8.8hz,1h),7.09(d,j=2.2hz,1h),7.03
–
6.95(m,2h),6.41(d,j=2.3hz,1h),3.79(s,3h),2.97(s,6h).
13
c nmr(100mhz,cdcl3):δ145.9,131.6,129.2,128.9,112.8,109.5,105.3,100.2,43.0,32.8.
[0146]
实施例17
[0147]
5-(2-chloroethyl)-1-methyl-1h-indole的制备
[0148]
将0.2mmol的n-甲基-3-(氯乙基)苯胺、1.0mmol的碳酸亚乙烯酯、0.6mmol的三氟甲烷磺酸钇和0.4mmol的2,2
′‑
联吡啶溶于装有2ml甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于160℃下加热搅拌1h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg无色油状化合物,产率为64%,所得产品结构式如下:
[0149][0150]
如图33和图34所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.52(s,1h),7.32(d,j=8.4hz,1h),7.14(d,j=8.3hz,1h),7.09(d,j=3.1hz,1h),6.50(d,j=3.1hz,1h),3.84
–
3.77(m,5h),3.22(t,j=7.6hz,2h).
13
c nmr(100mhz,cdcl3):δ135.9,129.2,129.0,128.8,122.5,120.8,109.2,100.7,45.7,39.5,32.8.
[0151]
实施例18
[0152]
1-cyclobutyl-1h-indole(n-huandingji)的制备
[0153]
将0.2mmol的n-环丁基苯胺、0.3mmol的碳酸亚乙烯酯、0.02mmol的三氟甲烷磺酸钇和0.01mmol的4,4
′
,4
″‑
三叔丁基-2,2
′
:6
′
,2
″‑
三联吡啶溶于装有2ml甲苯的耐压管内(配有磁力搅拌子),用气球充氮气至少3次,直至空气全部排空,封闭耐压管,于65℃下加热搅拌24h,tlc检测反应进程,待反应完全后将反应物冷却至室温,用ea稀释后加水萃取,合并有机相用无水硫酸钠干燥,过滤掉后留滤液,并减压旋蒸除去溶剂,粗产物经硅胶柱层析分离,得到24.5mg无色油状化合物,产率为58%,所得产品结构式如下:
[0154][0155]
如图35和图36所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ7.66(d,j=7.9hz,1h),7.40(d,j=8.2hz,1h),7.32
–
7.29(m,1h),7.23(t,j=7.7hz,1h),7.14(t,j=7.5hz,1h),6.55(d,j=2.9hz,1h),4.98
–
4.84(m,1h),2.69
–
2.42(m,4h),2.04
–
1.92(m,2h).
13
c nmr(100mhz,cdcl3):δ135.8,128.8,124.5,121.3,120.9,119.4,109.8,101.2,50.3,30.5,15.2。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
