1.本发明属于有机合成技术领域,具体涉及一种非共平面苯并咪唑二胺及其制备方法和应用。
背景技术:
2.二胺类化合物是一种重要的化工原料或中间体,可以作为单体和二酐反应合成性能优异的聚酰亚胺、聚酰胺或聚脲。通过在二胺类化合物中的n原子引入侧链基团,侧链基团的空间位阻和电子效应可以有效地减少聚合物分子间的电荷作用,使得到的薄膜无色透明。但是由于分子链的柔性过高,刚性降低,导致分子链不牢固,热膨胀系数过高。
3.热膨胀系数是透明聚酰亚胺(cpi)薄膜的重要性质,是衡量cpi薄膜热稳定性好坏的一个重要指标。在现有技术中,通过将刚性的酰胺键引入cpi的分子结构可以明显降低cpi薄膜的热膨胀系数;同时酰胺键通过在聚合物链中形成强的分子内或分子间氢键使得cpi薄膜具有较高的玻璃转化温度。例如文献“hasegawa m,watanabe y,tsukuda s,et al.solution-processable colorless polyimides with ultralow coefficients of thermal expansion for optoelectronic applications[j].polymer international,2016,65(9):1063~1073”中公开了引入酰胺键后能够明显降低聚酰亚胺薄膜的热膨胀系数,但是所得到的薄膜依然存在耐热性能较差的缺陷。
技术实现要素:
[0004]
本发明的目的在于提供一种非共平面苯并咪唑二胺及其制备方法和应用,本发明提供的非共平面苯并咪唑二胺能够在降低薄膜热膨胀系数的基础上提高耐热性能。
[0005]
为了实现上述目的,本发明提供如下技术方案:
[0006]
本发明提供了一种非共平面苯并咪唑二胺,具有式ⅰ所示结构:
[0007][0008]
所述x为-ch3、-cf3、-c2h5、、
[0009]
所述y为-ch3、-cf3、-f、-cl、-br、br、
[0010]
优选的,所述非共平面苯并咪唑二胺具有式ⅱ或式ⅲ所示结构:
[0011][0012]
本发明还提供了上述技术方案所述非共平面苯并咪唑二胺的制备方法,包括以下步骤:
[0013]
将具有式ⅳ所示结构的化合物、对硝基苯甲酰氯、缚酸剂和第一极性有机溶剂第一混合,进行缩合酰化反应,得到具有式
ⅴ
所示结构的化合物;
[0014][0015]
式ⅳ和式
ⅴ
中的x和y的种类和式ⅰ中的相同;
[0016]
将所述具有式
ⅴ
所示结构的化合物、加氢催化剂、还原剂和第二极性溶剂第二混合,进行还原反应,得到所述具有式ⅰ所示结构的化合物。
[0017]
优选的,所述第一极性溶剂包括二氯乙烷、二氯甲烷、苯、甲苯、氯仿、四氯化碳和四氢吠喃中的一种或几种;
[0018]
所述缚酸剂包括三乙胺、二异丙基乙胺、吡啶、碳酸钠、碳酸氢钠和碳酸钾中的一
种或几种。
[0019]
优选的,所述具有式ⅳ所示结构的化合物、对硝基苯甲酰氯和缚酸剂的摩尔比为1:(2~6):(1.5~6);
[0020]
所述具有式ⅳ所示结构的化合物和第一极性有机溶剂的用量比为1g:10~50ml。
[0021]
优选的,所述缩合酰化的温度为-20℃~40℃,时间为12~24h。
[0022]
优选的,所述加氢催化剂包括钯碳、铂炭、活性镍和铑碳中的一种或几种;
[0023]
所述第二极性有机溶剂包括四氢吠喃、乙醇、甲醇、异丙醇、二甲基甲酰胺、二甲基乙酰胺、n-甲基吡咯烷酮、1,4-二氧六环、乙酸乙醋、苯、甲苯和二甲苯中的一种或几种;
[0024]
所述还原剂包括锡、氯化亚锡、水合肼、甲酸、和甲脒亚磺酸中的一种或几种。
[0025]
优选的,所述具有式
ⅴ
所示结构的化合物、还原剂和加氢催化剂的摩尔比为1:(5~20):(0.5~1.5);
[0026]
所述具有式
ⅴ
所示结构的化合物和第二极性溶剂的用量比为1g:7~15ml。
[0027]
优选的,所述还原反应的温度为40~100℃,时间为8~24h,压力为0.5~3.0mpa。
[0028]
本发明还提供了上述技术方案所述的非共平面苯并咪唑二胺或上述技术方案所述制备方法制备得到的非共平面苯并咪唑二胺在合成聚酰亚胺、聚酰胺或聚脲中的应用。
[0029]
本发明提供了一种非共平面苯并咪唑二胺,具有式ⅰ所示结构。本发明提供的化合物是一种苯并咪唑和苯环上都带有侧基的新型苯并咪唑二胺,双侧基的引入促使苯并咪唑环和苯环扭转,形成非共平面结构,可以降低二胺分子本身的共轭效应;进一步的,通过在聚酰亚胺结构式中同时引入苯并咪唑结构和双酰胺结构,能够使本发明提供的苯并咪唑二胺作为单体制备得到的透明聚酰亚胺薄膜,在具有较低的热膨胀系数的基础上,具有优异的耐热性能。
附图说明
[0030]
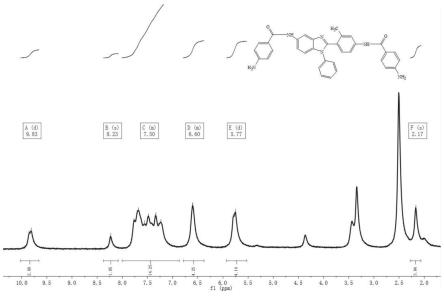
图1为实施例1得到的n-(2-(2-甲基-4-(4-硝基苯甲酰胺)苯基)-1-苯基-1h-苯并咪唑-5-基)-4-硝基苯甲酰胺的核磁氢谱图;
[0031]
图2为实施例1得到的4-氨基-n-(4-(5-(4-氨基苯甲酰胺)-1-苯基-1h-苯并咪唑-2-基)-3-甲基苯基)苯甲酰胺的核磁氢谱图;
[0032]
图3为实施例2得到的n-(1-甲基-2-(2-甲基-4-(4-硝基苯甲酰胺)苯基)-1h-苯并咪唑-5-基)-4-硝基苯甲酰胺的核磁氢谱图;
[0033]
图4为实施例2得到的4-氨基-n-(4-(5-(4-氨基苯甲酰胺)-1-甲基-1h-苯并咪唑-2基)-3-甲基苯基)苯甲酰胺的核磁氢谱图;
[0034]
图5为应用例得到的自支撑的透明聚酰亚胺薄膜的热稳定性测试结果;
[0035]
图6为应用例得到的自支撑的透明聚酰亚胺薄膜的玻璃转化温度测试结果;
[0036]
图7为应用例得到的自支撑的透明聚酰亚胺薄膜热膨胀系数测试结果。
具体实施方式
[0037]
本发明提供了一种非共平面苯并咪唑二胺,具有式ⅰ所示结构:
[0038][0039]
所述x为-ch3、-cf3、-c2h5、、
[0040]
所述y为-ch3、-cf3、-f、-cl、-br、br、
[0041]
在本发明的具体实施例中,所述非共平面苯并咪唑二胺优选具有式ⅱ或式ⅲ所示结构:
[0042][0043]
本发明还提供了上述技术方案所述非共平面苯并咪唑二胺的制备方法,包括以下步骤:
[0044]
将具有式ⅳ所示结构的化合物、对硝基苯甲酰氯、缚酸剂和第一极性有机溶剂第一混合,进行缩合酰化反应,得到具有式
ⅴ
所示结构的化合物;
[0045][0046]
式ⅳ和式
ⅴ
中的x和y的种类和式ⅰ中的相同;
[0047]
将所述具有式
ⅴ
所示结构的化合物、加氢催化剂、还原剂和第二极性溶剂第二混合,进行还原反应,得到所述具有式ⅰ所示结构的化合物。
[0048]
本发明实施例中,合成非共平面苯并咪唑二胺的合成路线如式ⅵ所示:
[0049]
所述式ⅵ中,x和y的种类和式ⅰ中的一致,在此不再赘述。
[0050]
在本发明中,若无特殊说明,所有制备原料均为本领域技术人员熟知的市售产品
[0051]
本发明将具有式ⅳ所示结构的化合物、对硝基苯甲酰氯、缚酸剂和第一极性有机溶剂第一混合,进行缩合酰化反应,得到具有式
ⅴ
所示结构的化合物;
[0052][0053]
式ⅳ和式
ⅴ
中的x和y的种类和式ⅰ中的相同。
[0054]
在本发明中,所述缚酸剂优选包括三乙胺、二异丙基乙胺、吡啶、碳酸钠、碳酸氢钠和碳酸钾中的一种或几种;当所述缚酸剂为上述选择中的两种以上时,本发明对具体物质的比例没有特殊的限定,按照任意比例混合均可。
[0055]
在本发明中,所述第一极性溶剂优选包括二氯乙烷、二氯甲烷、苯、甲苯、氯仿、四氯化碳和四氢吠喃中的一种或几种;当所述第一极性有机溶剂为上述选择中的两种以上时,本发明对具体物质的比例没有特殊的限定,按照任意比例混合均可。
[0056]
在本发明中,所述具有式ⅳ所示结构的化合物、对硝基苯甲酰氯和缚酸剂的摩尔比优选为1:(2~6):(1.5~6),进一步优选为1:(2.5~5.5):(2.0~5.5),进一步优选为1:(3.0~5.0):(2.5~5.0)。在本发明中,所述具有式ⅳ所示结构的化合物和第一极性有机溶剂的用量比优选为1g:10~50ml,进一步优选为1g:20~40ml,更优选为1g:25~30ml。
[0057]
在本发明中,所述第一混合的过程优选为:将具有式ⅳ所示结构的化合物、缚酸剂和部分第一极性有机溶剂预混,采用冰水浴将所得混合物的温度降至5~10℃,得到预混液;
[0058]
将对硝基苯甲酰氯溶于剩余第一极性有机溶剂,将得到的对硝基苯甲酰氯溶液滴加至所述预混液中。
[0059]
在本发明中,所述预混液中具有式ⅳ所示结构的化合物的质量浓度优选为4~15%,进一步优选为5~12%,更优选为8~10%。在本发明中,所述对硝基苯甲酰氯溶液中对硝基苯甲酰氯的质量浓度优选为8~20%,进一步优选为10~15%。
[0060]
本发明对所述预混和再混合的过程没有特殊的限定,采用本领域技术人员熟知的过程进行即可。
[0061]
在本发明中,所述滴加的滴加速度优选为0.5~1.5ml/min,时间优选为0.7~1.2ml/min。本发明优选在滴加的过程中控制预混液的温度为0~20℃。本发明对所述控制温度的方法没有特殊的要求,按照本领域技术人员熟知的方法进行即可。
[0062]
在本发明中,所述缩合酰化反应的温度优选为-20℃-40℃,进一步优选为0℃~20℃;时间优选为12~24h,进一步优选为16~20h。在本发明的具体实施例中,所述缩合酰化反应在室温下进行。
[0063]
在本发明的具体实施例中,优选采用tlc监测反应结束。
[0064]
所述缩合酰化反应完成后,本发明还优选包括对反应得到的产物进行后处理;所述后处理优选为:
[0065]
将所述缩合酰化反应得到的产物进行第一减压抽滤,得到粗产物;
[0066]
将所述粗产物和甲醇混合后第二减压抽滤,重复3~4次;
[0067]
将所述第二抽滤得到的固体产物干燥。
[0068]
本发明对所述第一减压抽滤的过程没有特殊的限定,采用本领域技术人员熟知的过程进行即可。
[0069]
在本发明中,所述混合在搅拌的条件下进行,所述搅拌的时间优选为2~3h。本发明对所述搅拌的转速没有特殊的限定,采用本领域技术人员熟知的进行即可。
[0070]
本发明对所述干燥的过程没有特殊的限定,采用本领域技术人员熟知的过程进行即可。
[0071]
在本发明的实施例中,所述具有式
ⅴ
所示结构的化合物的产率具体为84%。
[0072]
得到所述具有式
ⅴ
所示结构的化合物后,本发明将所述具有式
ⅴ
所示结构的化合物、加氢催化剂、还原剂和第二极性溶剂第二混合,进行还原反应,得到所述具有式ⅰ所示结构的化合物。
[0073]
在本发明中,所述加氢催化剂优选包括钯碳、铂炭、活性镍和铑碳中的一种或几种;当所述加氢催化剂为上述选择中的两种以上时,本发明对具体物质的比例没有特殊的限定,按照任意比例混合均可。
[0074]
在本发明中,所述第二极性有机溶剂优选包括四氢吠喃、乙醇、甲醇、异丙醇、二甲基甲酰胺、二甲基乙酰胺、n-甲基吡咯烷酮、1,4-二氧六环、乙酸乙醋、苯、甲苯和二甲苯中的一种或几种;当所述第二极性有机溶剂为上述选择中的两种以上时,本发明对具体物质的比例没有特殊的限定,按照任意比例混合均可。
[0075]
在本发明中,所述还原剂优选包括锡、氯化亚锡、水合肼、甲酸和甲脒亚磺酸中的一种或几种。在发明中,所述还原剂优选以还原剂水溶液的形式进行添加;本发明对所述还原剂水溶液的浓度没有特殊的限定,采用本领域技术人员熟知的即可。在本发明的具体实
施例中,所述水合肼水溶液中水合肼的质量浓度优选为80%。
[0076]
在本发明中,所述具有式
ⅴ
所示结构的化合物、还原剂和加氢催化剂的摩尔比优选1:(5~20):(0.5~1.5),进一步优选为1:(10~15):(1.0~1.2)。
[0077]
在本发明中,所述具有式
ⅴ
所示结构的化合物和第二极性溶剂的用量比优选为1g:7~15ml,进一步优选为1g:10~12ml。
[0078]
在本发明中,所述第二混合的过程优选为:将所述具有式
ⅴ
所示结构的化合物、加氢催化剂和第二极性有机溶剂混合,然后滴加还原剂溶液。在本发明中,所述还原剂溶液的滴加速度优选为0.3~0.7ml/min,时间优选为0.4~0.6ml/min。
[0079]
在本发明中,所述还原反应的温度优选为40~100℃,进一步优选为50~90℃,更优选为60~80℃;时间优选为8~24h;压力优选为0.5~3.0mpa。在本发明中,所述还原反应优选在充有惰性气体的容器中进行。
[0080]
在本发明的具体实施例中,优选采用tlc监测反应结束。
[0081]
所述还原反应完成后,本发明还优选包括对得到的还原产物进行后处理;所述后处理优选为:
[0082]
将所述还原反应得到的产物进行过滤,分别得到回收的加氢催化剂和滤液;
[0083]
将所述滤液和水混合进行冷却结晶,经减压抽滤后干燥,得到所述具有式ⅰ所示结构的化合物。
[0084]
在本发明的具体实施例中,所述过滤采用的滤头的孔径优选为0.22μm。在本发明中,所述过滤的次数优选为2~3次。在本发明中,经过多次的过滤,能够充分的回收产物中的加氢催化剂。
[0085]
本发明对所述混合、减压抽滤和干燥的过程没有特殊的限定,采用本领域技术人员熟知的过程进行即可。
[0086]
本发明在苯并咪唑环和苯环上引入的侧基(即x和y基团)种类多变,能够克服二胺单体结构单一的劣势,将本发明的二胺单体应用于聚合材料的合成中,能够得到多种不同结构的聚合材料,具有广阔的应用前景。
[0087]
本发明提供的制备方法原料价廉易得、工艺安全性高、操作简便、生产成本较低、反应产率较高、适合工业化生产。
[0088]
本发明还提供了上述技术方案所述的非共平面苯并咪唑二胺或上述技术方案所述制备方法制备得到的非共平面苯并咪唑二胺在合成聚酰亚胺、聚酰胺或聚脲中的应用。
[0089]
在本发明中,所述聚酰亚胺的制备方法优选包括以下步骤:
[0090]
将非共平面苯并咪唑二胺、1,2,4,5-环己烷四甲酸二酐、异喹啉和间甲酚混合,经聚合反应得到所述聚酰亚胺。
[0091]
在本发明中,所述非共平面苯并咪唑二胺和1,2,4,5-环己烷四甲酸二酐的摩尔比优选为1:1~1.3。在本发明中,所述非共平面苯并咪唑二胺和异喹啉的用量比优选为1g:0.3~0.5ml。在本发明中,所述混合的混合液的固含量优选为15%~20%。
[0092]
在本发明中,所述混合的过程优选为:
[0093]
将非共平面苯并咪唑二胺和间甲酚一级混合,得到一级混合液;
[0094]
将所述一级混合液和1,2,4,5-环己烷四甲酸二酐二级混合,得到二级混合液;
[0095]
将所述二级混合液和异喹啉三级混合。
[0096]
在本发明中,所述一级混合优选在搅拌的条件下进行;所述搅拌的转速优选为400r/min;时间优选为6~8h。在本发明中,所述一级混合优选在氮气气氛下进行。
[0097]
在本发明中,所述二级混合优选在搅拌的条件下进行。本发明对所述搅拌的条件参数没有特殊的限定,采用本领域技术人员熟知的即可。在本发明中,所述二级混合的时间优选为1~3h。
[0098]
本发明对所述三级混合的过程没有特殊的限定,采用本领域技术人员熟知的过程进行即可。
[0099]
在本发明中,所述聚合反应的温度优选为180~220℃,时间优选为6~10h。在本发明中,所述聚合反应优选在搅拌的条件下进行;所述搅拌的转速优选为200~500r/min。
[0100]
所述聚合反应完成后,本发明还优选包括对得到的产物进行后处理;所述后处理优选包括依次进行的冷却、沉淀析出、过滤、洗涤和干燥。
[0101]
本发明对所述冷却的过程没有特殊的限定,采用本领域技术人员熟知的过程进行即可。
[0102]
在本发明中,所述沉淀析出的过程优选为:将冷却后的产物和乙醇混合,进行沉淀析出。本发明对所述乙醇的添加量没有特殊的限定,采用本领域技术人员熟知的即可
[0103]
本发明对所述过滤的过程没有特殊的限定,采用本领域技术人员熟知的过程进行即可。
[0104]
在本发明中,所述洗涤优选包括依次进行乙醇洗涤和水洗涤。本发明对所述乙醇洗涤和水洗涤的过程没有特殊的限定,采用本领域技术人员熟知的即可。
[0105]
本发明对所述干燥的过程没有特殊的限定,采用本领域技术人员熟知的即可。
[0106]
得到所述聚酰亚胺后,本发明还优选包括将所述聚酰亚胺溶解于n-甲基吡咯烷酮中进行密封保存。在本发明中,所述聚酰亚胺和n-甲基吡咯烷酮的质量比优选为1:25。
[0107]
为了进一步说明本发明,下面结合附图和实施例对本发明提供的一种非共平面苯并咪唑二胺及其制备方法和应用进行详细地描述,但不能将它们理解为对本发明保护范围的限定。
[0108]
实施例1
[0109]
在本实施例中,非共平面苯并咪唑二胺的结构(式ⅱ)如下所示:
[0110][0111]
中文名称为4-氨基-n-(4-(5-(4-氨基苯甲酰胺)-1-苯基-1h-苯并咪唑-2-基)-3-甲基苯基)苯甲酰胺;
[0112]
制备方法为:
[0113]
向反应瓶中加入10.0g 2-(4-氨基-2-甲基苯基)-1-苯基-1h-苯并咪唑-5-胺、9.65g三乙胺和70ml四氢呋喃后,采用冰水浴将反应体系降温至5℃,得到预混物;将17.71g对硝基苯甲酰氯溶解于130ml四氢呋喃中,然后以1ml/min的滴加速度滴加至预混物中,1h滴加完成,确保在滴加的过程中反应体系的温度不超过10℃;在室温反应16h,采用tlc确定
反应结束。反应结束后将反应瓶中的产物进行减压抽滤,得到23.1g粗产物;将22.73g粗产物加入到反应瓶中,并倒入120ml甲醇,搅拌6h后再次抽滤,重复3次后,将抽滤得到的产物进行干燥,得到16.35gn-(2-(2-甲基-4-(4-硝基苯甲酰胺)苯基)-1-苯基-1h-苯并咪唑-5-基)-4-硝基苯甲酰胺,产率为84%,结构核磁确认,核磁氢谱图如图1所示,结构如式ⅶ所示:
[0114][0115]
向反应瓶中加入10.0g n-(2-(2-甲基-4-(4-硝基苯甲酰胺)苯基)-1-苯基-1h-苯并咪唑-5-基)-4-硝基苯甲酰胺、1g钯碳和100ml乙醇,然后以0.5ml/min的滴加速度滴加6.54g质量浓度为80%的水合肼,2h滴加完成,在80℃加热36h进行还原反应,tlc确定反应结束。反应结束后用0.22μm的滤头过滤反应液中的钯碳,重复3次确保过滤完全。向过滤后的反应液中加入400ml的水进行冷却结晶,将得到的晶体经减压抽滤和干燥,得到7.39g4-氨基-n-(4-(5-(4-氨基苯甲酰胺)-1-苯基-1h-苯并咪唑-2-基)-3-甲基苯基)苯甲酰胺(式ⅱ所示结构),产率为82%,结构核磁确认,核磁氢谱图如图2所示。
[0116]
实施例2
[0117]
在本实施例中,非共平面苯并咪唑二胺的结构(式ⅲ)如下所示:
[0118][0119]
中文名称为4-氨基-n-(4-(5-(4-氨基苯甲酰胺)-1-甲基-1h-苯并咪唑-2基)-3-甲基苯基)苯甲酰胺;
[0120]
制备方法为:
[0121]
向反应瓶中加入10.0g 2-(4-氨基-2-甲基苯基)-1-甲基-1h-苯并[d]咪唑-5-胺、9.65g三乙胺和70ml四氢呋喃后,采用冰水浴将反应体系降温至5℃,得到预混物;将17.71g对硝基苯甲酰氯溶解于130ml四氢呋喃中,然后以1ml/min的滴加速度滴加至预混物中,1h滴加完成,确保在滴加的过程中反应体系的温度不超过10℃;在室温反应16h,采用tlc确定反应结束。反应结束后将反应瓶中的产物进行减压抽滤,得到23.1g粗产物;将23.1g粗产物加入到反应瓶中,并倒入120ml甲醇,搅拌6h后再次抽滤,重复3次后,将抽滤得到的产物进行干燥,得到18.34g n-(1-甲基-2-(2-甲基-4-(4-硝基苯甲酰胺)苯基)-1h-苯并咪唑-5-基)-4-硝基苯甲酰胺,产率为84%,结构核磁确认,核磁氢谱图如图3所示,结构如式
ⅷ
所示:
[0122][0123]
向反应瓶中加入10.0g n-(1-甲基-2-(2-甲基-4-(4-硝基苯甲酰胺)苯基)-1h-苯并咪唑-5-基)-4-硝基苯甲酰胺、1g钯碳和100ml乙醇,然后以0.5ml/min的滴加速度滴加7.28g质量浓度为80%的水合肼,2h滴加完成,在80℃加热36h进行还原反应,tlc确定反应结束。反应结束后用0.22μm的滤头过滤反应液中的钯碳,重复3次确保过滤完全。向过滤后的反应液中加入400ml的水进行冷却结晶,将得到的晶体经减压抽滤和干燥,得到7.39g4-氨基-n-(4-(5-(4-氨基苯甲酰胺)-1-甲基-1h-苯并咪唑-2基)-3-甲基苯基)苯甲酰胺(式ⅲ所示结构),产率为82%,结构核磁确认,核磁氢谱图如图4所示。
[0124]
应用例
[0125]
将10g实施例1得到的4-氨基-n-(4-(5-(4-氨基苯甲酰胺)-1-苯基-1h-苯并咪唑-2-基)-3-甲基苯基)苯甲酰胺和49g间甲酚混合,在氮气气氛下、以400r/min的搅拌速度搅拌6h,得到一级混合液;将4.056g 1,2,4,5-环己烷四甲酸二酐加入上述一级混合液中,搅拌60min,待固体粉末溶解后得到二级混合液;将0.2g异喹啉加入上述二级混合液中,在转速为300r/min、温度为200℃反应时间8h;反应完成后,将反应体系冷却后倒入大量的乙醇中,收集沉淀后,用乙醇和水充分洗涤后干燥,得到所述聚酰亚胺。
[0126]
将10g上述得到的聚酰亚胺溶于250gn-甲基吡咯烷酮中,待完全溶解后,进行密封保存,得到均一的聚酰亚胺溶解液。
[0127]
性能测试
[0128]
将应用例得到的均一的聚酰亚胺溶解液在干净平整的玻璃片上进行涂膜,涂膜器的厚度控制在20μm;
[0129]
将涂好的玻璃片转移至马弗炉中,按照80℃/2h,150℃/2h,250℃/2h的升温程序除去溶剂;待马弗炉降至室温后将玻璃板取出并放入50℃的热水中,得到的聚酰亚胺膜从玻璃板上剥离下来,得到自支撑的透明聚酰亚胺薄膜;
[0130]
测试例1
[0131]
(1)将得到的自支撑的透明聚酰亚胺薄膜进行热稳定性(t
d5%
)测试,测试条件为:采用tga550,在氮气气氛,加热速率为10℃/min条件下,温度区间为室温到800℃;测试结果如图5和表1所示;
[0132]
(2)对得到的自支撑的透明聚酰亚胺薄膜的玻璃转化温度进行测试,测试条件为:使用美国ta公司的q800型动态热机械分析仪在空气氛围下以10℃/min的升温速率进行升温,测试频率为1hz,取损耗角正切tanδ曲线的峰值作为聚酰亚胺薄膜的玻璃化转变温度,测试结果如图6和表1所示;
[0133]
(3)对得到的自支撑的透明聚酰亚胺薄膜的热膨胀系数(cte)进行测试;测试条件为:使用美国ta公司q400型热机械分析仪在氮气氛围下以5℃/min的升温速率进行加热,预加载静态力设置为0.05n;测试前,样品在玻璃化转变温度以下20℃进行退火处理;
[0134]
热膨胀系数cte按照以下公式计算:
[0135]
cte=δl/(l
·
δt);
[0136]
其中,l是薄膜样条的原始长度;δt是样品选取的温度取值范围(100-250℃);δl是在100-250℃温度范围内薄膜样品的尺寸变化量;
[0137]
测试结果如图7和表1所示;
[0138]
表1自支撑的透明聚酰亚胺薄膜测试结果
[0139] t
d5%
(℃)tg(℃)cte(ppm/k)测试结果44839434
[0140]
根据表1和图5、6、7可知,由本发明提供的非共平面苯并咪唑二胺为单体合成的聚酰亚胺有优异的热性能。
[0141]
尽管上述实施例对本发明做出了详尽的描述,但它仅仅是本发明一部分实施例,而不是全部实施例,还可以根据本实施例在不经创造性前提下获得其他实施例,这些实施例都属于本发明保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。