1.本技术涉及一种芳基乙烯内酯类化合物及其制备方法、应用,属于有机合成领域。
背景技术:
2.近年来,高性能聚合物因其突出的机械性能、优异的热稳定性能和极佳的耐溶剂性能等,被广泛用于航空航天、国防军事、精密机械、生物医疗等领域,对我们的人类生活和社会发展都具有十分重要的意义。传统的高性能聚合物中的共价交联虽然赋予了材料坚固的机械强度、极高的热稳定性以及耐溶剂腐蚀性等优异性能,但是降解和回收利用面临挑战。目前高性能聚合物的回收途径主要是利用高温分解和溶剂分解,这类方法存在回收条件苛刻、能源浪费和污染排放等缺陷。因此,这些问题限制了高性能高分子材料进一步的发展,开发聚合物降解回收的新方法成为了近年来的研究热点。
3.动态共价键通常被定义为动态可逆的化学键,当前的研究表明,利用动态键构筑可回收聚合物是解决交联聚合物回收问题的一种重要策略。然而,现有利用动态共价体系构筑的可降解和可回收的交联聚合物报道较少,且底物适用范围较为受限。例如有文献将环氧树脂经动态二硫键交联,成功开发了一类具有可再加工性、可修复性和可回收性的热固性复合材料。有文献利用动态共价二酮烯胺键构建一种基于伯胺类单体的聚合物材料,利用酸碱反应回收单体并再循环到新材料中。有文献利用硼氮配位提高硼酸酯稳定性,将其引入聚氨酯交联网络中,并通过交换反应实现材料的降解和循环回收。上述例子均针对一类官能团,未能利用同一体系实现对多种类型底物的动态共价交联。而且,动态共价聚合物由于其可逆性容易受外界刺激的影响,在固有稳定性方面存在不足,将限制材料的实际应用,例如二硫键易被还原和硼酸酯易水解等。多胺、多醇和多硫醇在天然和合成分子及材料中广泛存在,实现上述化合物的光控交联和回收具有重要意义。因此,发现一种能与多类多功能亲核试剂进行动态共价反应并通过光照实现其高效控制从而用于聚合物可控交联及降解回收的体系具有重要意义。
技术实现要素:
4.根据本技术的一个方面,提供了一种化合物i,所述化合物i选自具有式i所示结构式的化合物中的任一种,其可以用于制备交联剂化合物ii。
5.根据本技术的第一方面,提供了一种化合物i,所述化合物i选自具有式i所示结构式的化合物中的任一种:
[0006][0007]
在式i中,r1选自卤素、具有式ii-1所示结构式的基团中的任一种;
[0008]
r2选自卤素、具有式ii-2所示结构式的基团中的任一种;
[0009]
r1、r2不同时为卤素;
[0010]
r3选自c1~c6的烷基、取代的c1~c6的烷基i、c6~c
10
的芳基、取代的c6~c
10
的芳基i中的任一种;
[0011][0012]
在式ii-1和式ii-2中,r
5'
、r
5”均独立地选自c1~c6的烷基、取代的c1~c6的烷基ii、c6~c
10
的芳基、取代的c6~c
10
的芳基ii中的任一种;
[0013]r4'
、r
4”均独立地选自c6~c
10
的芳基、取代的c6~c
10
的芳基iii、c1~c6的烷基、取代的c1~c6的烷基iii、c2~c6的炔基、取代的c2~c6的炔基中的任一种;
[0014]
优选地,在化合物i中,r
5'
、r
5”为相同的基团。
[0015]
优选地,所述取代的c6~c
10
的芳基iii、所述取代的c1~c6的烷基iii中的取代基均独立地选自羟基、羧基中的任一种。
[0016]
可选地,所述取代的c2~c6的炔基中的取代基选自c1~c6的烷基;
[0017]
所述取代的c1~c6的烷基i、取代的c1~c6的烷基ii中的取代基均独立地选自卤素;
[0018]
所述取代的c6~c
10
的芳基i、取代的c6~c
10
的芳基ii中的取代基均独立地选自c1~c6的烷氧基、卤素、卤素取代的c1~c6的烷基、硝基中的任一种。
[0019]
优选地,所述r
4'
、r
4”均独立地选自乙炔基、苯酚基、苯甲酸基。
[0020]
根据本技术的第二方面,提供了一种上述化合物i的制备方法,所述制备方法包括:
[0021]
将含有化合物a、化合物b、碱源和催化剂i的物料i,反应i,即可得到所述化合物i;
[0022]
所述化合物a选自具有式iii所示结构式的化合物中的任一种:
[0023][0024]
在式iii中,r6、r7独立地选自卤素;
[0025]
所述化合物b选自具有式iv所示结构式的化合物中的任一种:
[0026][0027]
r5选自c1~c6的烷基、取代的c1~c6的烷基ii、c6~c
10
的芳基、取代的c6~c
10
的芳基ii中的任一种;
[0028]
r4选自c6~c
10
的芳基、取代的c6~c
10
的芳基iii、c1~c6的烷基、取代的c1~c6的烷基iii、c2~c6的炔基、取代的c2~c6的炔基中的任一种;
[0029]
优选地,所述取代的c6~c
10
的芳基iii、所述取代的c1~c6的烷基iii中的取代基均独立地选自羟基、羧基中的任一种;
[0030]
所述取代的c2~c6的炔基中的取代基选自c1~c6的烷基;
[0031]
所述取代的c1~c6的烷基i、取代的c1~c6的烷基ii中的取代基均独立地选自卤素;
[0032]
所述取代的c6~c
10
的芳基i、取代的c6~c
10
的芳基ii中的取代基均独立地选自c1~c6的烷氧基、卤素、卤素取代的c1~c6的烷基、硝基中的任一种。
[0033]
可选地,所述催化剂i选自双三苯基磷二氯化钯、四三苯基膦钯、1,1'-双二苯基膦二茂铁二氯化钯、醋酸钯中的至少一种;
[0034]
所述碱源选自氟化铯、碳酸钾、碳酸钠、磷酸钾中的至少一种。
[0035]
优选地,r6、r7均为溴。
[0036]
可选地,r5选自甲基,r4选自苯基。
[0037]
可选地,所述碱源和化合物a的摩尔比为(3~5):1;
[0038]
所述催化剂i和化合物a的摩尔比为(0.05~0.1):1。
[0039]
可选地,当r1和r2为相同基团时,化合物a与化合物b的摩尔比为1:1~2:1。
[0040]
可选地,当r1和r2为不同基团,且r1和r2都不为卤素时,所述制备方法包括:
[0041]
(1)将含有化合物a、化合物b、碱源和催化剂i的物料ii,反应ii,得到r1或r2为卤素的化合物i;
[0042]
(2)将含有所述步骤(1)中得到的r1或r2为卤素的化合物i、化合物b、碱源和催化剂i的物料iii,反应iii,即可得到所述化合物i;
[0043]
在所述步骤(1)中,所述化合物a和化合物b的摩尔比为1:1~1:2;
[0044]
在所述步骤(2)中,所述r1或r2为卤素的化合物i、化合物b的摩尔比为1:1~1:4。
[0045]
可选地,当r1和r2为不同基团,且r1和r2都不为卤素时,在制备化合物i的步骤(1)中,化合物b中的r4基团选自c6~c
10
的芳基、c1~c6的烷基;在制备化合物i的步骤(2)中,化合物b 中的r4基团选自取代的c6~c
10
的芳基、取代的c1~c6的烷基i、c2~c6的炔基、取代的c2~c6的炔基中的任一种。
[0046]
可选地,所述反应i的条件为:温度为20~90℃;时间为3~24h;
[0047]
所述反应ii的条件为:温度为20~30℃;时间为12~24h;
[0048]
所述反应iii的条件为:温度为80~90℃;时间为3-6h。
[0049]
可选地,在所述物料i、物料ii和物料iii中还包括相转移催化剂i和溶剂i;
[0050]
所述溶剂i包括水和有机溶剂i。
[0051]
可选地,所述有机溶剂i选自甲苯、1,4-二氧六环、四氢呋喃中的至少一种;
[0052]
所述相转移催化剂i选自苄基三乙基氯化铵、四丁基溴化铵、四丁基氟化铵、十八冠六中的至少一种。
[0053]
可选地,所述有机溶剂i和所述水的体积比为1:1~6:1;
[0054]
所述相转移催化剂i和化合物a的摩尔比为0.03~0.07:1。
[0055]
根据本技术的第三方面,提供了一种化合物ii,所述化合物ii具有式
ⅴ
所示结构式的化合物、具有式
ⅴ
'所示结构式的化合物中的任一种:
[0056]r9-r
8-r
10
ꢀꢀ
式
ⅴ
[0057][0058]
其中,r9、r
10
、r
11
、r
12
、r
13
独立地选自具有式vi所示结构式的取代基中的任一种;
[0059][0060]
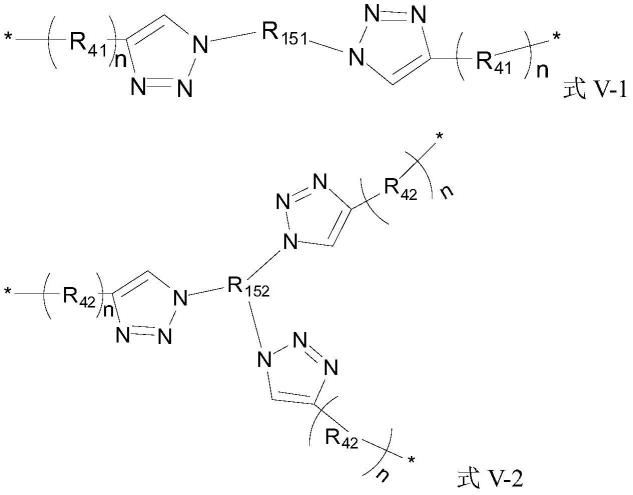
r8选自具有式v-1所示结构式的基团、具有式v-2所示结构式的基团、具有式v-3所示结构式的基团、具有式v-4所示结构式的基团、具有式v-5所示结构式的基团、具有式v-6所示结构式的基团、具有式v-7所示结构式的基团、具有式v-8所示结构式的基团中的任一种:
[0061][0062]
*-r
43-o-r
153-o-r
43-*式v-3
[0063][0064][0065]
其中,n选自0或1;r
151
、r
153
、r
156
、r
157
独立地选自c1~c6的亚烷基、*-r”'-r'-r
”‑
*、 *-r
0-o-r
00-*中的任一种;
[0066]
其中,r'选自c6~c
10
的亚芳基、取代的c6~c
10
的亚芳基中的任一种;
[0067]
r”、r”'、r0、r
00
独立地选自c1~c6的亚烷基、取代的c1~c6的亚烷基i中的任一种;
[0068]r152
、r
154
、r
155
、r
158
独立地选自c1~c6的次烷基、
中的任一种;
[0069]
ra选自c6~c
10
的次芳基、取代的c6~c
10
的次芳基中的任一种;
[0070]r41
、r
42
、r
43
、r
44
、r
45
、r
46
、r
47
、r
48
均独立地选自c6~c
10
的亚芳基中的任一种;
[0071]
rb、rc、rd、rf、rg、rh独立地选自c1~c6的亚烷基、取代的c1~c6的亚烷基ii中的任一种;
[0072]
re选自c1~c6的次烷基、取代的c1~c6的次烷基中的任一种;
[0073]
优选地,所述取代的c6~c
10
的亚芳基中的取代基选自c1~c6的烷基;
[0074]
所述取代的c1~c6的亚烷基i中的取代基选自卤素、c1~c6的烷氧基、硝基中的任一种;
[0075]
所述取代的c6~c
10
的次芳基中的取代基选自卤素、c1~c6的烷基、c1~c6的烷氧基、卤素取代的 c1~c6的烷基、硝基中的任一种;
[0076]
所述取代的c1~c6的亚烷基ii中的取代基选自卤素、c1~c6的烷氧基、硝基中的任一种;
[0077]
所述取代的c1~c6的次烷基中的取代基选自卤素、c1~c6的烷氧基、硝基中的任一种。
[0078]
可选地,r8选自下述基团中的任一种:
[0079][0080]
本技术中的化合物ii由于结构复杂,以下用简式表示一下本技术中的化合物ii,以更加清晰的阐述本技术。
[0081]
作为本技术一优选地实施方式,所述化合物ii的结构简式如下:
[0082][0083]
其中,基团表示:
[0084]
优选地,r8选自下述基团中的任一种:
[0085][0086]
可选地,上述化合物5的合成过程如下:
[0087][0088]
可选地,上述化合物9的合成原料如下:
[0089][0090]
可选地,上述化合物23的合成原料如下:
[0091][0092]
可选地,上述化合物11的合成原料如下:
[0093][0094]
可选地,上述化合物13的合成原料如下:
[0095][0096]
根据本技术的第四方面,提供了一种上述化合物ii的制备方法,所述制备方法包括:将含有化合物i、化合物c和催化剂ii的物料iv,反应iv,即可得到所述化合物ii;
[0097]
所述化合物i选自上述化合物i、根据上述方法制备得到的化合物i中的任一种;
[0098]
在所述化合物i中,r1、r2均不为卤素;
[0099]
且r
4'
、r
4”中至少有一个基团选自所述取代的c6~c
10
的芳基iii、所述取代的c1~c6的烷基iii、 c2~c6的炔基、取代的c2~c6的炔基中的任一种;
[0100]
所述取代的c6~c
10
的芳基iii、所述取代的c1~c6的烷基iii中的取代基均独立地选自羟基、羧基中的任一种;
[0101]
所述取代的c2~c6的炔基中的取代基选自c1~c6的烷基;
[0102]
所述化合物c选自具有式vii-1所示结构式的基团、具有式vii-2所示结构式的基团中的任一种:
[0103]r17-r
16-r
18
式vii-1
[0104][0105]
其中,r
16
选自r
151
、r
153
、r
156
、r
157
中的任一种;
[0106]r16'
选自r
152
、r
154
、r
155
、r
158
中的任一种;
[0107]r17
、r
18
、r
17'
、r
18'
、r
19'
独立地选自n
3-*、卤素、羟基、氨基中的任一种。
[0108]
可选地,所述催化剂ii由抗坏血酸和碘化亚铜发生反应制备得到。
[0109]
可选地,所述催化剂ii和所述化合物i的摩尔比为(0.05~0.1):1。
[0110]
可选地,所述化合物i和所述化合物c的摩尔比为1:0.5~3:2。
[0111]
可选地,所述反应iv的条件为:温度为20~25℃;时间为4-12h。
[0112]
可选地,在所述物料iv中还包括相转移催化剂ii和有机溶剂ii。
[0113]
可选地,所述有机溶剂ii选自四氢呋喃、1,4-二氧六环中的至少一种;
[0114]
所述相转移催化剂ii选自三[(1-苄基-1h-1,2,3-三唑-4-基)甲基]胺、n,n-二异丙基乙胺中的至少一种。
[0115]
可选地,所述相转移催化剂ii和所述化合物i的摩尔比为(0.2~0.3):1。
[0116]
可选地,本技术中的二噻吩乙烯内酯(化合物ii)的制备路线如下式所示:
[0117]
通过分步的suzuki偶联,再click反应或者亲核取代反应或者酯化酰胺化等反应,可集成式制备多功能二噻吩乙烯内酯类化合物。
[0118]
根据本技术的第五方面,提供了一种交联剂,所述交联剂选自上述化合物ii、根据上述方法制备得到的化合物ii中的任一种。
[0119]
根据本技术的第六方面,提供了一种化合物ii的调控方法i,所述调控方法i包括:
[0120]
将化合物ii进行紫外光照射处理,光环化后,得到调控产物a;和/或;
[0121]
将所述调控产物a进行可见光照射处理,开环后,得到所述化合物ii;
[0122]
其中,所述调控产物a选自具有式i所示结构式的化合物、具有式i'所示结构式的化合物中的任一种:
[0123]r9-r
8-r
10
式i
[0124][0125]
其中,r9、r
10
、r
11
、r
12
、r
13
独立地选自具有式ii所示结构式的取代基中的任一种;
[0126][0127]
r8选自具有式v-1所示结构式的基团、具有式v-2所示结构式的基团、具有式v-3所示结构式的基团、具有式v-4所示结构式的基团、具有式v-5所示结构式的基团、具有式v-6所示结构式的基团、具有式v-7所示结构式的基团、具有式v-8所示结构式的基团中的任一种:
[0128][0129]
*-r
43-o-r
153-o-r
43-*式v-3
[0130]
[0131][0132]
其中,n选自0或1;r
151
、r
153
、r
156
、r
157
独立地选自c1~c6的亚烷基、*-r”'-r'-r
”‑
*、 *-r
0-o-r
00-*中的任一种;
[0133]
其中,r'选自c6~c
10
的亚芳基、取代的c6~c
10
的亚芳基中的任一种;
[0134]
r”、r”'、r0、r
00
独立地选自c1~c6的亚烷基、取代的c1~c6的亚烷基i中的任一种;
[0135]r152
、r
154
、r
155
、r
158
独立地选自c1~c6的次烷基、中的任一种;
[0136]
ra选自c6~c
10
的次芳基、取代的c6~c
10
的次芳基中的任一种;
[0137]
rb、rc、rd、rf、rg、rh独立地选自c1~c6的亚烷基、取代的c1~c6的亚烷基ii中的任一
种;
[0138]
re选自c1~c6的次烷基、取代的c1~c6的次烷基中的任一种;
[0139]
所述化合物ii选自上述化合物ii、根据上述方法得到的化合物ii中的至少一种;
[0140]
优选地,所述取代的c6~c
10
的亚芳基中的取代基选自c1~c6的烷基;
[0141]
所述取代的c1~c6的亚烷基i中的取代基选自卤素、c1~c6的烷氧基、硝基中的任一种;
[0142]
所述取代的c6~c
10
的次芳基中的取代基选自卤素、c1~c6的烷基、c1~c6的烷氧基、卤素取代的 c1~c6的烷基、硝基中的任一种;
[0143]
所述取代的c1~c6的亚烷基ii中的取代基选自卤素、c1~c6的烷氧基、硝基中的任一种;
[0144]
所述取代的c1~c6的次烷基中的取代基选自卤素、c1~c6的烷氧基、硝基中的任一种;
[0145]
优选地,所述紫外光的波长为280nm~350nm;
[0146]
所述可见光的波长为450nm~700nm;
[0147]
优选地,所述紫外光照射的时间为:1-2h;
[0148]
所述可见光照射的时间为2-4h。
[0149]
可选地,通过紫外-可见光可以对化合物ii的光致变色性质进行控制,以化合物5为例,光致变色过程如下式所示:
[0150][0151]
当使用313nm的紫外光对其进行照射时,化合物5(me)由开环的o-5(me)(无色)形式光环化成 c-5(me)(深蓝色)形式如式3所示。
[0152]
根据本技术的第七方面,提供了一种化合物ii的调控方法ii,其特征在于,所述调控方法ii包括:
[0153]
(ii-1)将化合物ii进行紫外光照射处理i,光环化后,得到调控产物a;
[0154]
(
ⅱ‑
2)利用亲核试剂对所述调控产物a进行交联调控,形成动态共价键,得到聚合物b;
[0155]
所述化合物ii选自上述化合物ii、根据上述方法得到的化合物ii中的至少一种。
[0156]
可选地,对所述聚合物b进行可见光照射处理、紫外光照射处理、碱处理、酸处理、亲核试剂处理中的至少一种来实现对所述聚合物b的降解、回收或自修复。
[0157]
可选地,所述亲核试剂选自胺类化合物、醇类化合物、硫醇类化合物中的任一种;
[0158]
所述胺类化合物中至少含有两个氨基;
[0159]
所述醇类化合物中至少含有两个羟基;
[0160]
所述硫醇类化合物中至少含有两个巯基。
[0161]
可选地,所述亲核试剂选自明胶、季戊四醇四巯基乙酸酯中的任一种。
[0162]
可选地,所述亲核试剂为硫醇类化合物;
[0163]
在所述步骤(ii-2)中,所述硫醇类化合物中的巯基会与所述调控产物a中与or
3-*基团相连的碳活性位点在酸性条件下交联反应a,得到含有c-s键的聚合物b-1;
[0164]
所述c-s键为动态共价键。
[0165]
优选地,所述交联反应a的条件为:温度为20~25℃;时间为2-4h;
[0166]
优选地,所述硫醇类化合物和所述化合物ii的摩尔质量比为0.25mol:8~12mg。
[0167]
可选地,所述含有c-s键的聚合物b-1和正丙硫醇在酸性条件下混合反应b,即可实现所述含有 c-s键的聚合物b-1的降解;然后向分解后的混合物中加入所述硫醇类化合物,即可重新得到所述含有c-s键的聚合物b-1,实现所述含有c-s键的聚合物b-1的回收。
[0168]
优选地,所述混合反应b的条件为:温度为20~25℃;时间为1-3h。
[0169]
可选地,对所述含有c-s键的聚合物b-1进行可见光照射处理2-4h,开环后,即可得到聚合物 b-2。
[0170]
可选地,对所述聚合物b-2进行紫外光照射处理,光环化并分解c,即可实现所述聚合物b-2的降解。
[0171]
可选地,所述光环化并分解c的条件为:温度为20~25℃;时间为1-3h。
[0172]
可选地,通过紫外-可见光和外界刺激可调控该类分子(化合物ii)与多胺类、多醇类、多硫醇类等亲核试剂的交联以及所得聚合物的降解和回收。以下以四巯基乙酸季戊四醇酯(petma)和明胶为例,对本技术中的化合物ii的上述性质进行描述:
[0173]
紫外-可见光和化学刺激调控该类分子与petma的交联/降解如式-1所示:
[0174][0175]
以化合物5为例,将化合物o-5的氘代乙腈溶液置于313nm的紫外氙灯下照射,o-5光环化成 c-5达到光稳态后加入季戊四醇四巯基乙酸酯和甲基磺酸,形成聚合物25。向该聚合物溶液中加入正丙硫醇,聚合物25分解,得到化合物26和petma,向该溶液中再加入petma,则重新生成聚合物25。对聚合物25给予可见光(λ
irr
=650nm)的照射将其切换成聚合物27,加入正丙硫醇聚合物 27没有降解。再用313nm照射40分钟聚合物再次降解如附图5所示。具体过程可见实施案例3。从而通过光的调控既可以得到难以降解的稳定聚合物,也可以完成聚合物的降解与回收。
[0176]
可选地,所述亲核试剂为胺类化合物;
[0177]
在所述步骤(ii-2)中,所述胺类化合物中的氨基会与所述调控产物a中与or
3-*基团相连的碳活性位点在酸性条件下反应d,得到含有c-n键的聚合物b-3;
[0178]
所述c-n键为动态共价键。
[0179]
可选地,所述反应d的条件为:温度为20~25℃;时间为0.5~1h;
[0180]
所述胺类化合物和所述调控产物a的质量比为4~6:1。
[0181]
可选地,所述含有c-n键的聚合物b-3在酸性条件下分解e,即可实现所述含有c-n键的聚合物 b-3的降解。
[0182]
可选地,所述分解e的条件为:温度为20~25℃;时间为0.25~0.5h。
[0183]
可选地,对所述含有c-n键的聚合物b-3进行可见光照射处理2-4h,开环后,即可得到聚合物b-4。
[0184]
可选地,向所述含有c-n键的聚合物b-3分解后的混合物中加入碱源,即可得到所述含有c-n 键的聚合物b-3,实现所述含有c-n键的聚合物b-3的回收。
[0185]
可选地,对所述聚合物b-4进行紫外光照射处理1~2h,光环化后,即可得到所述含
有c-n键的聚合物b-3。
[0186]
可选地,将所述含有c-n键的聚合物b-3分成两部分,再将所述两部分靠近,经过0.5~1h,所述两部分可自修复成所述含有c-n键的聚合物b-3。
[0187]
紫外-可见光和酸碱刺激调控该类分子与明胶的交联/降解如式-2和式-3所示:
[0188][0189]
以化合物5(me)为例,将化合物o-5(me)的氘代乙腈溶液置于313nm的紫外氙灯下照射,o-5(me) 光环化成c-5(me)达到光稳态后加入甲基磺酸,c-5(me)原位分解为c-5(h)。再加入明胶水溶液,得到凝胶28。将28置于1m盐酸中,28分解为c-5(h)并完全溶解。将28置于650nm可见光照射下切换成凝胶29,此时再置于1m盐酸中,聚合物29不降解。用313nm对聚合物29进行照射,再次分解为c-5(h)如附图6所示。具体过程可见实施案例3。从而通过光的调控即可以得到难以降解的稳定聚合物,也可以完成交联聚合物的降解。
[0190][0191]
以化合物5(me)为例,将化合物o-5(me)的氘代乙腈溶液置于313nm的紫外氙灯下照射,o-5(me) 光环化成c-5(me)达到光稳态后加入甲基磺酸,c-5(me)原位分解成c-5(h)。
再加入明胶水溶液,得到凝胶28。将28置于1m盐酸中,28分解为c-5(h)。向该溶液中再加入1m氢氧化钠,凝胶28重新生成,再给予650nm可见光的照射,凝胶28切换成凝胶29如附图7所示。具体过程可见实施案例 3。从而通过光和酸碱的调控完成交联聚合物的可控降解和循环回收。
[0192]
紫外-可见光调控该类分子与明胶的自修复如式-4所示:
[0193][0194]
以化合物5(me)为例,将化合物o-5(me)的氘代乙腈溶液置于313nm的紫外氙灯下照射,o-5(me) 光环化成c-5(me)达到光稳态后加入甲基磺酸,c-5(me)原位分解成c-5(h)。再加入明胶水溶液,得到凝胶28。用光滑的刀片将凝胶28均匀的分成两片,再将两片凝胶靠近,两片凝胶完成自修复。将凝胶28置于650nm可见光照射下切换成凝胶29。同样地,将凝胶29均匀切成两片,未观察到凝胶完成自修复如附图8所示。具体过程可见实施案例3。从而通过光可以完成对聚合物自修复的精准控制。
[0195]
本技术中,c1~c6指所包含的碳原子数。对所述“取代烷基”、“取代芳基”的碳原子限定,是指烷基、芳基本身所含的碳原子数,而非取代后的碳原子数。如c1~c6的取代烷基,指碳原子数为1~10 的烷基上,至少一个氢原子被取代基取代。
[0196]
本技术中,“烷基”是由烷烃化合物分子上失去任意一个氢原子所形成的基团。所述烷烃化合物包括直链烷烃、支链烷烃、环烷烃、带有支链的环烷烃。
[0197]
本技术中,“芳基”是芳香族化合物分子上失去芳香环上一个氢原子所形成的基团;如甲苯失去苯环上甲基对位的氢原子所形成的对甲苯基。
[0198]
本技术中,“炔基”是由炔烃化合物分子上失去任意一个氢原子所形成的基团。所述炔烃化合物包括直链炔烃、支链炔烃、环炔烃、带有支链的环炔烃。
[0199]
本技术中,“亚烷基”是由烷烃化合物分子上失去任意两个氢原子所形成的基团。所述烷烃化合物包括直链烷烃、支链烷烃、环烷烃、带有支链的环烷烃。
[0200]
本技术中,“亚芳基”是由芳香族化合物分子的芳香环上失去任意两个氢原子所形成的基团。
[0201]
本技术中,“次烷基”是由烷烃化合物分子上失去任意三个氢原子所形成的基团。所述烷烃化合物包括直链烷烃、支链烷烃、环烷烃、带有支链的环烷烃。
[0202]
本技术中,“次芳基”是由芳香族化合物分子的芳香环上失去任意三个氢原子所形成的基团。
[0203]
可选地,“苯酚基”为苯酚的苯环上失去任意一个氢原子所形成的基团。
[0204]
可选地,“苯甲酸基”为苯甲酸的苯环上失去任意一个氢原子所形成的基团。
[0205]
本技术能产生的有益效果包括:
[0206]
本发明公布了一种化合物i,其可以用于制备基于二噻吩乙烯光致变色开关的多
功能交联剂(化合物ii),通过紫外-可见光来调控环链异构进而控制反应性,用于聚合物的可控交联、降解及循环回收。该类分子中的二噻吩乙烯部分对光具有刺激响应性,可通过紫外-可见光照射实现结构改变,从而导致反应性产生显著变化,可用于胺、醇和硫醇的动态共价键合和释放,实现c-n,c-o和c-s共价键的光控锁定和激活。通过交联剂中的多个含环链异构位点的二噻吩乙烯基元,实现了该类分子对多胺类、多醇类和多硫醇类亲核试剂的交联,获得聚合物网络。利用上述动态共价键的光控激活实现聚合物可控降解回收,而光照锁定动态键后聚合物具有优异稳定性,从而实现材料稳定性和可降解性、可回收性的双向切换。该发明在高性能聚合物材料的调控方面具有很好的应用前景。
附图说明
[0207]
图1为化合物3的核磁共振氢谱图;
[0208]
图2为化合物4的核磁共振氢谱图;
[0209]
图3为化合物5的核磁共振氢谱图;
[0210]
图4为化合物5的乙腈溶液313nm光照前(虚线)后(实线)吸收光谱,浓度为50μm;
[0211]
图5是紫外-可见光和亲核试剂调控c-s键聚合物的可逆交联与降解中样品颜色变化图;
[0212]
图6为紫外-可见光和酸调控c-n键聚合物的降解中样品颜色变化图;
[0213]
图7为紫外-可见光和酸碱调控c-n键聚合物的降解回收循环中样品颜色变化图;
[0214]
图8为紫外-可见光和调控c-n键聚合物的自修复中样品颜色变化图。
具体实施方式
[0215]
下面结合实施例详述本技术,但本技术并不局限于这些实施例。
[0216]
如无特别说明,本技术的实施例中的原料均通过商业途径购买。
[0217]
本技术实施例中使用的仪器型号为400mhz布鲁克biospin avance iii光谱仪;perkin-elmerlambda 365光谱仪。
[0218]
实施例1
[0219]
中间体3的合成:称取1mol粘溴酸的衍生物,1mol芳基硼酸,0.05molbnet3n
cl-和 0.05molpdcl2(pph3)2于双口圆底烧瓶中,安装回流装置和氮气保护装置,抽真空氮气置换三次,用注射器经橡胶塞加入甲苯和含4mol氟化铯的水溶液(v
水
:v
甲苯
=1:1,氟化铯的水溶液浓度为2m),室温反应24小时。反应液用二氯甲烷萃取三次,合并有机相用无水硫酸钠干燥,旋转蒸发仪除去溶剂后柱层析(sio2)分离,洗脱剂为石油醚:乙酸乙酯=50:1。可得化合物3,产率73%。
[0220]
本实施例中,r3为甲基。
[0221]
化合物3的核磁共振氢谱:1h nmr(cd3cn):δ=7.67(d,j=8.4hz,2h),7.56(s,1h),
7.47(t,j= 7.6hz,2h),7.39(t,j=7.2hz,1h),6.32(s,1h),3.56(s,3h),2.59(s,3h)。具体谱图见附图1。
[0222]
中间体4的合成:称取1mol化合物3,3mol(5-乙炔基-2-甲基噻吩-3-基)硼酸,0.05molbnet3n
cl-和0.05molpdcl2(pph3)2于双口圆底烧瓶中,安装回流装置和氮气保护装置,抽真空氮气置换三次,用注射器经橡胶塞加入甲苯和含4mol氟化铯的水溶液(2m)(v
水
:v
甲苯
=1:1),加热至90℃恒温4 小时。反应液冷却至室温后二氯甲烷萃取三次,合并有机相用无水硫酸钠干燥,旋转蒸发仪除去溶剂后柱层析(sio2)分离,洗脱剂为石油醚:乙酸乙酯=30:1。可得化合物4,产率66%。
[0223]
化合物4的核磁共振氢谱:1h nmr(cd3cn):δ=7.62
–
7.59(m,2h),7.47
–
7.43(tt,j=7.6,2.0 hz,2h),7.39
–
7.35(tt,j=7.6,1.2hz,1h),7.31(s,1h),7.24(s,1h),6.35(s,1h),3.72(s,1h),3.61(s, 3h),2.09(s,3h),2.07(s,3h)。具体谱图见附图2。
[0224]
目标产物5的合成:称取1mol上述合成的化合物4,0.5mol1,4-双(叠氮甲基)苯,0.1mol碘化亚铜,0.2mol三[(1-苄基-1h-1,2,3-三唑-4-基)甲基]胺作为相转移催化剂,0.3mol抗坏血酸钠于圆底烧瓶中,安装氮气保护装置,抽真空氮气置换三次,用注射器经橡胶塞加入5ml四氢呋喃,室温反应4 小时。用乙酸乙酯萃取三次,合并有机相用无水硫酸钠干燥,旋转蒸发仪除去溶剂后柱层析(sio2) 分离,洗脱剂为石油醚:乙酸乙酯=2:1。可得化合物5,产率85%。
[0225]
化合物5的核磁共振氢谱:1h nmr(cd3cn):δ=8.00(s,2h),7.57
–
7.55(dt,j=7.2,2.0hz,4h), 7.43
–
7.39(tt,j=8.0,1.6hz,4h),7.37
–
7.31(m,8h),7.26(s,2h),7.34(s,2h),5.57(s,4h),3.59(s, 6h),2.09(s,6h),2.05(s,6h)。具体谱图见附图3。
[0226]
通过上述方法可得多功能二噻吩乙烯内酯化合物,并通过核磁共振、质谱等手段表征化合物。
[0227]
实施例2
[0228]
将实施例1中合成的化合物5配制成50μm的乙腈标准溶液,测定其吸收光谱,再对其进行313 nm紫外光照射达到光稳态后(这里我们对其进行过核磁和光谱的跟踪,继续光照不变,即可认为达到光稳态),再测定其吸收光谱,结果如图4所示,化合物o-5(me)(无色)的吸收光谱在300nm左右,光环化成c-5(me)(深蓝色)的最大吸收光谱在577nm左右。
[0229]
实施例3
[0230]
对c-s键聚合物降解回收的调控:将o-5(me)(10mg)溶解在cd3cn(400μl)中,置于紫外光下(λ
irr
=313nm)照射80分钟,然后添加0.25mol季戊四醇四巯基乙酸酯和3.0mol甲基磺酸,反应达到热力学平衡后(这里是通过观察产物基本不变,基本认为达到热力学平衡)向该聚合物溶液中加20mol正丙硫醇,2小时后聚合物25完全分解得到化合物26和petma,向该溶液中再加10mol 的petma,则重新生成聚合物25。用可见光(λ
irr
=650nm)对聚合物25照射240分钟将其切换成聚合物27,加入20mol正丙硫醇聚合物27基本没有变化。再用紫外线(λ
irr
=313nm)照射40分钟聚合物再次完全降解,此过程中样品的颜色变化图如图5所示。
[0231][0232]
对c-n键聚合物降解的调控:将o-5(me)(10mg)溶解在cd3cn(400μl)中,置于紫外光下 (λ
irr
=313nm)照射80分钟,然后加入2mol甲基磺酸原位水解得到c-5(h)。20分钟后,加入溶于 600μl水中的50mg明胶,反应达到热力学平衡(这里是通过观察产物基本不变,基本认为达到热力学平衡)后得到凝胶28。取一小块凝胶28置于1m盐酸中,10分钟后完全降解为c-5(h)。用可见光(λ
irr
=650nm)对聚合物28照射240分钟将其切换成聚合物29,然后取一小块凝胶置于1m盐酸中,一星期后依然保持完整。再用紫外线(λ
irr
=313nm)照射40分钟聚合物再次完全降解为c-5(h),此过程中样品的颜色变化图如图6所示。
[0233][0234]
[0235]
对c-n键聚合物降解回收循环的调控:将o-5(me)(10mg)溶解在cd3cn(400μl)中,置于紫外光下(λ
irr
=313nm)照射80分钟,然后加入2mol甲基磺酸原位水解得到c-5(h)。20分钟后,加入溶于600μl水中的50mg明胶,反应达到热力学平衡(这里是通过观察产物基本不变,基本认为达到热力学平衡)后得到凝胶28。取一小块凝胶28置于1m盐酸中,10分钟后完全降解为c-5(h)。再加入1m氢氧化钠(2ml),凝胶28再次生成。用可见光(λ
irr
=650nm)对聚合物28照射240分钟可将其切换成凝胶29,此过程中样品的颜色变化图如图7所示。
[0236][0237][0238]
对c-n键聚合物自修复的调控:将o-5(me)(10mg)溶解在cd3cn(400μl)中,置于紫外光下(λ
irr
=313nm)照射80分钟,然后加入2mol甲基磺酸原位水解得到c-5(h)。20分钟后,加入溶于600μl水中的50mg明胶,反应达到热力学平衡后(这里是通过观察产物基本不变,基本认为达到热力学平衡)得到凝胶28。将凝胶28加热融化置于圆形磨具中,待其固化成型后取出,用刀片将其均匀切成两片,将两片凝胶靠近,一小时后完成自修复。用可见光(λ
irr
=650nm)对聚合物28照射240分钟可将其切换成凝胶29。同样地,用刀片将其均匀切成两片,将两片凝胶靠近,一星期后也未完成自修复。再对其两片凝胶进行313nm紫外光照射80分钟,1小时后再次完成自修复,此过程中样品的颜色变化图如图8所示。
[0239][0240][0241]
以上所述,仅是本技术的几个实施例,并非对本技术做任何形式的限制,虽然本技术以较佳实施例揭示如上,然而并非用以限制本技术,任何熟悉本专业的技术人员,在不脱
离本技术技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。