:
1.本发明属于医药技术领域,涉及多西他赛-油酸甘油三酯前药及其脂质制剂,具体涉及淋巴介导转运的多西他赛-油酸甘油三酯前药及其脂质制剂和在制备口服化疗药物中的应用。
背景技术:
:
2.现如今,全球癌症负担逐年增加,多西他赛(docetaxel,dtx)作为一线广谱抗癌化疗药物广泛应用于多种肿瘤的临床治疗。多西他赛临床采用静注给药,然而市售静注溶液剂采用吐温-80和乙醇助溶,带来辅料相关毒副作用,限制了临床应用。口服化疗具有患者顺应性高,方便给药,治疗成本低等优点,但是多西他赛的溶解度低,p-糖蛋白外排以及严重的首过效应使其口服生物利用度极低,目前还没有上市的多西他赛口服制剂。因此,开发低首过效应,高口服生物利用度的多西他赛口服制剂仍然是研究的热点。
3.口服药物淋巴转运可以避免首过效应,是提高口服吸收的一个有效方式。一种提高淋巴转运的策略是设计药物的类甘油三酯前药。长链甘油三酯1位和3位的脂肪酸被胰脂肪酶特异性水解,2位脂肪酸几乎不水解,将2位脂肪酸置换成药物,使前药模拟甘油三酯在肠道中的消化过程,以2-甘油单酯前药的形式进入肠上皮细胞进行再酯化,参与脂蛋白的组装,进而促进淋巴转运。已有很多研究者做了类甘油三酯前药的实例,然而这些实例所教导的结构在提高药物口服上几乎无效,一个重要的原因是可断裂连接链的缺失,导致母药不释放。在cn106715456a中,发明人引入了自消除连接链,促进母药睾酮的系统释放。然而,对于多西他赛,一个化疗药物,大量的系统暴露会带来严重的副反应,因此,该连接链对多西他赛口服递药系统的设计是无指导意义的。
4.现有技术中,医药工作者将多西他赛制备成甘油三酯前药并将其制备成纳米乳剂,通过模拟天然甘油三酯淋巴转运口服吸收的特点,改善多西他赛的口服吸收。然而,纳米乳剂具有储存不稳定性,制备繁琐,批间重现性差等缺点,并且在纳米乳剂的制备中可能加入了大量的助乳化剂,如脱氧胆酸钠等,多次给药可能会带来严重的胃肠道副反应,因此,设计安全简易的类甘油三酯前药口服剂型是十分必要的。
技术实现要素:
5.针对多西他赛口服吸收差的问题,本发明基于天然甘油三酯淋巴转运机制,首先提供了多西他赛-油酸甘油三酯前药,并将该前药制备成多西他赛-油酸甘油三酯前药的脂质制剂,制备工艺简单,重复性强,便于产业化,性质均一稳定,该前药脂质制剂能够促进难溶性药物的淋巴转运,提高其口服利用度,并在此基础上,引入还原敏感连接键,在促进抗肿瘤药物口服吸收的同时,使其能够在靶部位特异性释放,提高疗效,降低毒性。
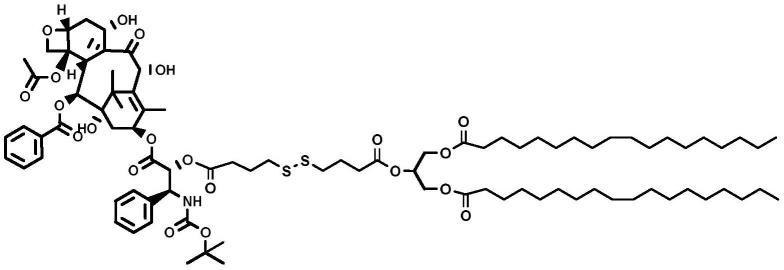
6.本发明的第一个目的是提供多西他赛-油酸甘油三酯前药或其几何异构体、药学上可接受的盐、水合物、溶剂化物:
[0007][0008]
本发明的第二个目的是提供上述化合物的制备方法。具体制备方法如下:
[0009]
(a)1,3-二油酸甘油酯的合成:油酸溶于二氯甲烷中,在1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci)和4-二甲氨基吡啶(dmap)催化作用下与1,3-二羟基丙酮发生成酯反应,再经硼氢化钠氢化反应得1,3-二油酸甘油酯。
[0010][0011]
(b)将二硫代二丁酸溶于适量乙酸酐中,室温下搅拌成二硫代二丁酸酐,与1,3-二油酸甘油酯在edci及dmap存在的条件下反应得到中间产物,与多西他赛在dmap及edci的催化条件下酯化反应得到目标前药。
[0012][0013]
本发明的第三个目的是提供一种简单安全的含多西他赛-油酸甘油三酯前药的药物组合物,所述的药物组合物为多西他赛-油酸甘油三酯前药的脂质制剂,包含多西他赛-油酸甘油三酯前药和辅料,所述辅料包括磷脂类乳化剂、短链醇或醚类助乳化剂、液态油类油相,按重量百分比计,液态油类油相占脂质制剂的30%-85%,磷脂类乳化剂占脂质制剂的10%-45%,短链醇或醚类助乳化剂占脂质制剂的5%-25%;多西他赛-油酸甘油三酯前药占辅料总重量的1-10%。
[0014]
进一步地,本发明所述的药物组合物按重量百分比计,液态油类油相占脂质制剂的55%-85%,磷脂类乳化剂占脂质制剂的10%-35%,短链醇或醚类助乳化剂占脂质制剂的5%-10%。
[0015]
优选地,本发明所述的药物组合物按重量百分比计,液态油类油相占脂质制剂的
55%-80%,磷脂类乳化剂占脂质制剂的15%-35%,短链醇或醚类助乳化剂占脂质制剂的5%-10%。
[0016]
进一步地,多西他赛-油酸甘油三酯前药占辅料总重量的2-4%。
[0017]
优选地,多西他赛-油酸甘油三酯前药占辅料总重量的3-4%。
[0018]
其中,所述的磷脂为蛋黄卵磷脂,大豆卵磷脂,合成磷脂类如1,2-二辛酰基-sn-甘油-3-磷酸胆碱,1,2-二癸酰基-sn-甘油-3-磷酸胆碱,优选为蛋黄卵磷脂;
[0019]
所述液态油选自长链甘油三酯、混合长链甘油酯、中链甘油三酯或其组合,包括:橄榄油、杏仁油、介花油、蓖麻油、椰子油、玉米油、棉籽油、鱼油、棕榈仁油、棕榈油、花生油、菜籽油、红花油、芝麻油、鲨鱼肝油、大豆油、向日葵油、氢化椰子油、氢化棉籽油、氢化棕榈油、氢化大豆油、部分氢化大豆油、氢化植物油、三辛酸甘油酯中、maisine、peceol的一种或几种,其中优选橄榄油。
[0020]
所述短链醇或醚类助乳化剂为二乙二醇单乙基醚(transcutol hp)、乙醇、丙二醇等,其中优选transcutol hp。
[0021]
更进一步地,短链醇或醚类助乳化剂、磷脂类乳化剂、液态油类油相的重量比为:1:2-4:5-20。
[0022]
本发明所述的多西他赛-油酸甘油三酯前药的脂质制剂通过如下方法制备:
[0023]
将多西他赛-油酸甘油三酯前药、短链醇或醚类助乳化剂、磷脂类乳化剂、液态油类油相混合,超声使辅料和前药混合均一,即得到前药的脂质制剂。
[0024]
或将磷脂类乳化剂和短链醇或醚类助乳化剂溶于液态油中,超声使其混合均匀,得到均一空白制剂。取多西他赛-油酸甘油三酯前药,加入上述空白制剂,超声使前药完全溶于其中,即得到均一的脂质口服制剂。
[0025]
当所述的磷脂类乳化剂为蛋黄卵磷脂、短链醇或醚类助乳化剂为二乙二醇单乙基醚、液态油类油相为橄榄油时,且液态油类油相占脂质制剂的55%-80%,磷脂类乳化剂占脂质制剂的15%-35%,短链醇或醚类助乳化剂占脂质制剂的5%-10%时,所得到的多西他赛-油酸甘油三酯前药的脂质制剂具有最佳的粒径和粒径分布,具有最好的稳定性,且能明显提高多西他赛的口服生物利用度。
[0026]
本发明首次将液态有(尤其是橄榄油)、磷脂以及二乙二醇单乙基醚进行均匀混合制备成空白脂质制剂,并将所合成的还原敏感多西他赛-油酸甘油三酯前药溶解到上述制剂中,脂质制剂的制备方法简单,无需高温加热,探头超声等工序,只需简单的搅拌或超声溶解,工艺简单,便于产业化,重复性强;得到的脂质制剂为均一的油溶液状态,相对纳米制剂的热力学不稳定状态,本发明提供的脂质制剂更稳定,便于储存;避免了脱氧胆酸钠的使用,本发明所提供的脂质制剂所使用的辅料均为fda批准的药用辅料,口服后不会造成胃肠道毒性;载药量大大提升,在制备以及患者的依从性上会得到大大改善;此外,该脂质制剂高度适用于所设计的多西他赛-油酸甘油三酯前药,显著改善了多西他赛的口服生物利用度。
[0027]
本发明的优势在于:
[0028]
1、本发明基于天然甘油三酯吸收的机制,设计了多西他赛-油酸甘油三酯前药,促进了多西他赛的淋巴转运,避免了首过效应,进而提高了其口服吸收。
[0029]
2、本发明以还原敏感的二硫键连接桥连接甘油三酯骨架和难溶性药物,在促进药
物口服吸收的同时,使药物能够在靶部位特异性释放,增效减毒。
[0030]
3、本发明所制备的脂质口服制剂,制备工艺简单,易于产业化,均一稳定,相较于现有技术中的纳米乳剂,避免了脱氧胆酸钠的使用,简化了制备工艺,无加热过程和探头超声。
附图说明
[0031]
图1为多西他赛-油酸甘油三酯前药的1h-nmr谱。
[0032]
图2为多西他赛-油酸甘油三酯前药的高分辨质谱图。
[0033]
图3为实例2制备的多西他赛-油酸甘油三酯前药纳米乳剂的粒径稳定性。
[0034]
图4为多西他赛-油酸甘油三酯前药在不同辅料中的溶解度。
[0035]
图5为多西他赛-油酸甘油三酯前药的基于中链甘油三酯的自微乳筛选伪三元相图。
[0036]
图6为多西他赛-油酸甘油三酯前药的基于中链的自微乳和橄榄油溶液在sd大鼠的药时曲线。
[0037]
图7多西他赛-油酸甘油三酯前药脂质制剂合用脱氧胆酸钠在sd大鼠的药时曲线。
[0038]
图8为合用/不合用脱氧胆酸钠(胆盐)的多西他赛-油酸甘油三酯前药脂质制剂在sd大鼠的药时曲线。
[0039]
图9为多西他赛-油酸甘油三酯前药脂质制剂在比格犬药动实验中的药时曲线。
[0040]
图10为多西他赛-油酸甘油三酯前药脂质制剂的胃肠道毒性。
[0041]
图11为多西他赛-油酸甘油三酯前药脂质制剂的体内口服抗肿瘤实验图。
[0042]
a:肿瘤生长曲线b:小鼠体重变化图c:肿瘤实物图d:荷瘤率。
具体实施方式
[0043]
本发明通过以下实施例对本发明作进一步阐述,但并不受限于此。
[0044]
实例1多西他赛-甘油三酯前药制备(oatg)
[0045]
结构如下
[0046][0047]
将11.28g(40mmol)油酸溶于二氯甲烷中,在edci和dmap催化作用下与1.75g(20mmol)1,3-二羟基丙酮发生成酯反应过夜,将反应液浓缩,重蒸水洗2次,用氯仿萃取水层,有机层经饱和食盐水洗1次,无水硫酸钠干燥有机层,旋蒸除溶剂。柱层析分离,分离条件正己烷-乙酸乙酯(30:1-15:1)。将2.9g(5mmol)上述产物溶解于混合溶剂(thf:苯:水10:2:1)100ml中,加入0.3g(7.8mmol)硼氢化钠,于5℃下反应30min,加入0.9ml冰乙酸停止反应,加入80ml氯仿与反应液混合,用重蒸水洗涤1次,4%碳酸氢钠溶液洗涤一次,饱和食盐
水洗涤一次,无水硫酸钠干燥,旋蒸除溶剂得1,3-二油酸甘油酯。将2.1g(8.8mmol)4,4
’‑
二硫代二丁酸溶于9ml乙酸酐中,于室温下反应2h,旋蒸溶剂浓缩并复溶于适量无水二氯甲烷中,并加入620mg(1mmol)1,3-二油酸甘油酯,100mg dmap。抽真空氮气保护于室温反应12小时。旋蒸除溶剂,柱层析分离得到中间产物(条件为正己烷:乙酸乙酯=15:1-8:1)。将中间产物680mg(0.83mmol)溶解于适量无水二氯甲烷中,加入175mg(0.9mmol)edci,45mg(0.36mmol)dmap,803mg(1mmol)多西他赛,抽真空氮气保护于室温反应48小时。旋蒸除溶剂,制备液相分离得到终产物(制备液相条件为纯乙腈)。
[0048]
采用核磁共振测定1h-nmr氢谱来确定实施例1中前药的结构,选用的溶剂为cdcl3,结果如图1。对应特征峰解析:多西他赛特征峰:δ8.12(d,j=7.3hz,2h),7.61(t,j=7.4hz,1h),7.51(t,j=7.8hz,2h),7.36(m,5h),6.25(s,1h);1,3-二油酸甘油酯骨架特征峰:2.06
–
1.98(m,8h),1.28(dd,j=19.6,7.7hz,40h),0.88(t,j=7.0hz,6h);
[0049]
采用高分辨质谱测定实例1中前药的分子量,结果如图2所示:
[0050]
esi-hrms:calcd.for c
90h135
nnao
21
s2[m na
]1652.8896 found 1652.8860。
[0051]
上述前药具体合成路线如下所示:
[0052][0053]
实例2 oatg纳米乳剂的稳定性
[0054]
将12mg多西他赛-油酸甘油三酯前药溶解于400mg橄榄油中,预热至60℃。称取120mg蛋黄卵磷脂,40mg脱氧胆酸钠,溶解于4ml去离子水中,预热至60℃。将油相缓慢滴入搅拌的水相中,持续搅拌3min形成初乳,将初乳冰浴探头超声10min,超声功率500w,得oatg纳米乳剂。并在第1,2,5,15,30天测定了乳剂的粒径,发现乳剂粒径随时间明显增大,最后甚至出现了分层的现象,暴露出了乳剂具有不稳定性的缺陷,结果如图3所示,oatg纳米乳剂的载药量仅为0.262%,可见,将多西他赛-油酸甘油三酯制备成纳米乳剂,不仅载药量低,而且稳定性差。
[0055]
实例3多西他赛-甘油三酯前药脂质制剂的处方优化
[0056]
类甘油三酯前药的口服吸收高度依赖于脂质消化。因此,oatg的最佳载体是基于脂质的药物递送系统(lbdds)。在lbdds的多种类型中,自微乳药物递送系统(smedds)独具
优势,包括高载药量和易于放大生产规模。
[0057]
3.1基于中链甘油三酯(mct)和长链甘油三酯(lct)的oatg自微乳的处方初步筛选和制备
[0058]
首先测定了oatg在不同油溶性辅料中的溶解度,如图4所示,发现oatg在三辛酸甘油酯,二乙二醇单乙基醚中的溶解度最高,遂将其作为油相,助乳化剂,大量文献报道吐温80能促进药物淋巴转运,因此采用吐温-80作为乳化剂。建立伪三元相图来寻找最适宜的三项比例。如图5所示,在图中多点围成的比例下制备出的制剂,可以通过模拟胃内搅动形成自微乳。经过进一步优化后,发现当三辛酸甘油酯/吐温80/transcutol hp=26.5/63/10.5(重量比)时,所形成的乳剂粒径较小且较为均一。
[0059]
根据溶解度和形成乳剂的粒径,pdi筛选之后,开发了一种基于中链甘油三酯的自微乳递药系统。根据前药在各辅料中溶解度,以多西他赛-油酸甘油三酯前药与上述辅料总重量比为1:20制备多西他赛-油酸甘油三酯脂质制剂。
[0060]
对按照如上组方制备的多西他赛-油酸甘油三酯脂质制剂进行体内药动实验,同时设置oatg的橄榄油溶液(长链甘油三酯lct)组进行比较。
[0061]
药动结果如图6所示,与基于mct(中链甘油三酯)的smedds相比,oatg橄榄油(lct)溶液组的auc(0-8h)显着增加(2.6倍),可见,虽然按照伪三元相图设计的组方在在药学上所形成的乳剂粒径较小且较为均一,但其在体内的药动学性质却低于长链甘油三酯的溶液组的药动学性质,即基于中链甘油三酯的自微乳并未表现较好的体内药动学性质。而基于lct的配方比基于mct的配方表现了更好的体内药动学性质,因此长链甘油三酯lct更适合oatg的自微乳的设计。
[0062]
结果表明:oatg的主要机制是模仿长链甘油三酸酯的口服吸收过程,该过程通过消化,吸收进入肠细胞,并再酯化组装成乳糜微粒,转运到淋巴系统然后循环。与lct共同给药,而不是mct,可以增强oamg(通过肠液消化oatg后获得的产物)在肠中的溶解度,从而改善口服吸收效率。
[0063]
因此,本发明选择长链甘油三酯lct作为油相,进行进一步的实验。
[0064]
3.2基于长链甘油三酯的oatg脂质制剂的处方筛选
[0065]
3.2.1油相的选择
[0066]
基于上述结果,为了保证脂质制剂的载药量,对oatg在几种lct基础的植物油中的溶解度测定,结果如表1。
[0067]
表1.oatg在几种植物油中的溶解度
[0068][0069]
根据溶解度结果,oatg脂质制剂的油相选择橄榄油。
[0070]
3.2.2助乳化剂的选择
[0071]
磷脂可以帮助改善类甘油三酯前药的口服吸收,因此初步选择蛋黄卵磷脂作为乳
化剂,筛选助乳化剂。将乳化剂和助乳化剂溶于油相中,油相:乳化剂:助乳化剂质量比为65:25:10,超声使其混合均匀,得到均一空白制剂。
[0072]
取200mg的oatg前药加入到1g上述空白制剂中,测定oatg前药在空白制剂中的平衡溶解度。
[0073]
此外,取30mg的oatg前药,加入1g上述空白制剂,超声使前药完全溶于其中,得到均一的脂质口服制剂,取500mg各含药制剂加到5ml模拟胃液中,搅拌5min以模拟胃蠕动,测定粒径和分布,不同助乳化剂的结果见表2。
[0074]
表2.不同助乳化剂制备的oatg脂质制剂性质
[0075][0076]
结果表明,加入不同的助乳化剂,制备的脂质制剂性能不同,而当助乳化剂为transcutol hp时,前药的溶解度最高,可以获得最佳的载药量,制备的oatg前药脂质制剂具有最佳的载药量、粒径和分布。
[0077]
3.2.3磷脂的选择
[0078]
以橄榄油为油相,以transcutol hp作为助乳化剂,对磷脂种类进行筛选,选择蛋黄卵磷脂,大豆卵磷脂,1,2-二辛酰基-sn-甘油-3-磷酸胆碱,1,2-二癸酰基-sn-甘油-3-磷酸胆碱,按照3.2.2的方法制备oatg脂质制剂,测定oatg前药在空白制剂中的平衡溶解度,并取500mg各含药制剂加到5ml模拟胃液中,搅拌5min以模拟胃蠕动,测定粒径和分布,不同磷脂所制备的脂质制剂的各指标均无显著差异,蛋黄卵磷脂相对较好。
[0079]
实例4 oatg前药脂质制剂的制备
[0080]
按照实例3中3.2.2的方法制备oatg前药脂质制剂,得到不同组方的前药脂质制剂,如表3所示。将500mg脂质制剂加到5ml模拟胃液中,搅拌5min模拟胃蠕动,测定粒径和分布,另取上述脂质制剂与模拟胃液混合物1ml与5ml胰液-胆汁混合,孵育2h后,低温离心(10000rpm,10min),取中间层液体,测定消化后粒径,以及oatg在中间层的占比,结果如表4所示:
[0081]
表3.不同组方的oatg脂质制剂
[0082]
[0083][0084]
表4.不同组方的oatg脂质制剂的性质
[0085][0086]
如表4所示,当橄榄油占比小于50%时(处方1-3),蛋黄卵磷脂和transcutolhp占比较高,对oatg的溶解度高,因此载药量高,此外,由于乳化剂和助乳化剂的强表面活性作用,经模拟胃液混合搅拌后,得到了粒子粒径小而均匀,这与基于mct的自微乳制剂体外结果相似,但是当与胰液-胆汁混合模拟消化后,得到的中间层的水相的粒径较大,且oatg前药在中间层的占比小于50%,对于类甘油三酯脂质制剂,最初的体外粒径并不能准确预测出体内的口服吸收,而经模拟消化后的中间水相相当于体内肠液的溶解相,其粒径越小,前药在中间水相中的占比越大,可能会有利于体内吸收。当橄榄油占比超过80%,蛋黄卵磷脂占比低于15%时,尽管体内模拟的结果仍在可接受范围,但是最初的模拟胃液乳化粒径大于300nm,这可能会延迟后续的肠道消化过程,进而影响口服吸收;当trancutolhp低于5%时,橄榄油与蛋黄卵磷脂不相容,无法形成均一的脂质制剂。
[0087]
当橄榄油在30%-85%,蛋黄卵磷脂在10%-45%,trancutolhp在5%-25%时,可以形成均一稳定的脂质制剂,其粒径均小于400nm。
[0088]
当橄榄油在55%-85%,蛋黄卵磷脂在10%-35%,trancutolhp在5%-10%时,可
以形成均一稳定的脂质制剂,其粒径均小于400nm,oatg在中间层占比大,有利于体内吸收。
[0089]
当橄榄油在55%-80%,蛋黄卵磷脂在15%-35%,trancutolhp在5%-10%时,可以形成均一稳定的脂质制剂,其粒径均小于300nm,且最有利于体内吸收。
[0090]
各组方的脂质制剂在15天时无分层沉淀,具有较好的稳定性。
[0091]
实例5 oatg前药脂质制剂的给药方案的优化
[0092]
称取300mg蛋黄卵磷脂溶于1g橄榄油中,同时加入100mg的二乙二醇单乙基醚,超声使其混合均匀,得到均一空白制剂。精确称取40mg的实例1所述前药,加入1g的空白制剂,超声使前药完全溶于其中,即得到均一的oatg前药脂质制剂。精密称取一定量的多西他赛母药,加入到上述空白制剂中,即得到多西他赛脂质制剂。
[0093]
该脂质制剂的载药量达到3.85%,相对现有技术中的纳米乳剂提高了14.6倍,在放置过程中始终为均一油性制剂,无分层沉淀现象出现。
[0094]
研究发现脱氧胆酸钠可以帮助改善类甘油三酯前药的口服吸收。按实例5所示处方,制备oatg和dtx的脂质制剂,并在sd大鼠给与上述制剂前,给与脱氧胆酸钠水溶液。图7显示了用脱氧胆酸钠溶液作为对oatg和dtx补充剂的基于lct的smedds给药后的口服血浆分布,表5给出了相应的药代动力学参数。与dtx相比,oatg前药同时给予一定脱氧胆酸钠溶液组的auc(0-24小时)显示提高了1.86倍。此外,oatg组(221.237
±
82.95ng ml-1)的c max与dtx组相比(31.984
±
13.852ng ml-1)提高了6.9倍。
[0095]
表5.基于lct的smedds制剂口服药动学参数
[0096][0097]
但是使用脱氧胆酸钠可能会导致胃肠道功能受损。因此,我们研究了制剂中脱氧胆酸钠的必要性。上述制剂去掉脱氧胆酸钠后oatg组和dtx组的sd大鼠的口服血浆曲线如图8。使用(633.916
±
164.122μg lh-1)和不使用(697.1
±
71.3μg l h-1)脱氧胆酸钠的oatg组的auc(0
–
24小时)并没有显著差异。oatg组的口服相对生物利用度(frel)为247.5%,口服绝对生物利用度(fab)为37%,与dtx组的15%形成了鲜明对比。
[0098]
实例6多西他赛-油酸甘油三酯前药脂质制剂的比格犬药动学实验
[0099]
以比格犬为模型,口服含有多西他赛-油酸甘油三酯前药脂质制剂(oatg lp)的硬胶囊及含有多西他赛脂质制剂(dtx lp)的硬胶囊,剂量为3mg/kg,定时眼眶采血,测定血浆中多西他赛母药的浓度,根据测定的浓度分别进行了药-时曲线的绘制及相应药动学参数的计算(表6)。为了计算绝对生物利用度,静脉给予多西他赛溶液剂,剂量为1mg/kg,测定血浆中多西他赛的含量。如图9和表6所示,相对于多西他赛脂质制剂,前药脂质制剂组的药-时曲线下面积(auc
0-24
)相对母药脂质制剂明显提高。由静脉多西他赛的数据计算前药的口
服生物利用度,前药组的绝对生物利用度达到41.08%。
[0100]
表6.多西他赛甘油三酯前药主要药动学参数
[0101][0102]
实例7多西他赛-甘油三酯oatg前药脂质制剂的胃肠道毒性
[0103]
采用mtt法考察oatg前药脂质制剂对人克隆结肠腺癌细胞(caco-2)的细胞毒性。以1000cells/孔的密度将细胞接种到96孔板中,置培养箱中孵育24h使其贴壁。待细胞贴壁后加入系列浓度的紫杉醇溶液剂、实例2制备的oatg纳米乳和实施例5中制备的oatg前药脂质制剂。加药12h后,mtt法测定细胞存活率。
[0104]
结果如图10所示,oatg前药脂质制剂对人克隆结肠腺癌细胞的细胞毒性都弱于多西他赛溶液剂和纳米乳剂,说明前药的脂质制剂对于胃肠道细胞无明显损伤,所用辅料及配比无胃肠道毒性。
[0105]
实例8多西他赛甘油三酯前药脂质制剂的药效学行为
[0106]
以4t1原位荷瘤balb/c小鼠模型,口服多西他赛甘油三酯脂质制剂、多西他赛脂质制剂、多西他赛溶液剂,给药剂量为10mg/kg(多西他赛等效剂量),每天给药;设置多西他赛溶液剂静注组为阳性对照,给药剂量为10mg/kg,每隔两天给一次药;设置pbs组为空白对照。结果如图11所示,多西他赛-甘油三酯前药脂质制剂组肿瘤体积是最小的,与对照组均有显著性差异,且无体重降低现象,表明其具有可靠的安全性且抗肿瘤效果好。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。