1.本发明属于钙钛矿量子点领域,涉及钙钛矿量子点/贵金属纳米颗粒复合材料制备方法。
背景技术:
2.1958年,moller等首次报道了全无机钙钛矿cspbx3(ipqds,x=cl,br,i)的晶体结构。2014年,kovalenko第一次用热注射法合成了cspbx3(x=cl,br,i)纳米晶。
3.cspbx3量子点作为一种新型的光电材料,具有优异的光致发光性能、超高量子产率、可调谐带隙和窄发光峰。由于钙钛矿晶格中卤化物快速运动,从而在溶液或固态中进行快速、活性的阴离子交换反应,这是钙钛矿量子点的一个独特性质。cspbx3量子点的量子限域效应较显著,可在室温下表现出强烈的荧光效应,其光致发光谱线宽度较窄,通常在12nm至42nm之间,量子产率可达50%至90%。与传统的cdse/cds-zns量子点相比,在量子产率方面,cspbbr3量子点可达90%左右,远高于65%的cdse/cds-zns量子点。全无机钙钛矿量子点的高效发光主要源于其高激子结合能、表面卤素自钝化、cspbx3/x类量子阱能带结构三者的协同效应。目前,主要通过两种方式来调控ipqds的发光波长:一是改变卤素组成,其发射光谱随cl
→
br
→
i逐渐红移并变宽。二是利用量子限域效应,通过调节量子点的尺寸来调控发光波长。高温法中,在合适温度范围内,量子点的尺寸随反应温度的升高而增大,而量子点的发光波长将随尺寸的增大而红移,这种光谱调节机制是ipqds所具有的独特优势。目前,已有多种方法可以制备cspbx3量子点,其中有代表性的方法包括:1)热注射,预加热的cs前驱体在特定温度下注射到pbx2溶液中形成纳米晶;2)室温法,将溶解在良溶剂中的csx和pbx2加入到不良溶剂中,溶解度的巨大下降立即产生高度过饱和状态,从而导致快速再结晶。
4.苏丹红iii是一种含有萘化合物的亲脂性偶氮染料。除了用于油漆、机油和布抛光剂的染色外,它还被非法用作食品添加剂。自1995年以来,由于苏丹红化学结构的性质,对人体肝肾器官有显著的毒性作用,具有致癌作用,从而被禁止作为食品添加剂。更重要的是,苏丹红染料通常是不溶于水的,一旦进入消化系统就难以去除。因此,开发一种可行、有效的苏丹红污染物降解方法具有重要意义。
5.cspbx3(x=cl,br,i)/au纳米颗粒复合材料具有良好转换发光以及半导体性质,作为转换机制主体和光生电子转移体,在可见光下其内置电场、小尺寸的金纳米颗粒和高比表面积可促进更高的光吸收和载流子分离,从而获得更好的光催化性能。本研究为利用卤化物钙钛矿材料在光催化降解污染物方面的应用开辟了新的途径。目前,由于ipqds光电应用主要集中在太阳能电池、发光器件、激光器、光电探测器等,光催化等其他应用很少被报道,这极大地限制了钙钛矿量子点在可见光下光降解苏丹红iii等污染物的潜在应用。
技术实现要素:
6.本发明的目的在于克服上述现有技术存在的缺陷与不足,本发明针对上述问题,
利用有机配体还原csx表面的aux,再与pb(oa)2溶液反应,得到cspbx3/au纳米颗粒复合材料,该方法未见专利或非专利文献报导。
7.开发一种钙钛矿量子点/au纳米颗粒复合材料的制备方法。
8.本发明提供的钙钛矿量子点/贵金属纳米颗粒复合材料制备方法包括下列步骤:
9.1)cs-油酸前驱体的制备:首先将cs2co3、油酸加入三口烧瓶中,接着加热搅拌并抽真空除去体系中的水,抽真空结束后通入氩气并继续加热,直至cs2co3完全溶解。
10.2)znx2溶液的制备:首先将znx2(x=cl,br,i)、油酸、十八烯和油胺加入到三口烧瓶中,接着加热搅拌并抽真空除去体系中的水和空气,抽真空结束后通入氩气并继续加热,直至znx2完全溶解。
11.3)pb(oa)2溶液的制备:首先将pbo、油酸加入到三口烧瓶中,直至pbo完全溶解。
12.4)csx纳米颗粒的制备:将步骤1)中得到的前驱体快速注入到步骤2)中(70-80℃),接着将注入前驱体的溶液置于冰水浴中冷却到室温,然后将冷却后的溶液离心,并将得到的沉淀分散到甲苯(或正己烷、氯苯)中,得到csx纳米颗粒分散液。
13.5)csx/au纳米颗粒的制备:取步骤4)得到的产物,在剧烈搅拌状态下加入aux颗粒进行溶解,反应结束后离心,并将得到的沉淀分散到甲苯中。
14.6)cspbx3纳米颗粒的制备:取步骤4)得到的产物,在剧烈搅拌状态下加入步骤3)得到的产物,反应结束后离心,并将得到的沉淀分散到甲苯(或正己烷、氯苯)中得到cspbx3纳米颗粒分散液。
15.7)cspbx3/au纳米颗粒的制备:取步骤5)得到的产物,在剧烈搅拌状态下加入步骤3)得到的产物,反应结束后离心,并将得到的沉淀分散到甲苯(或正己烷、氯苯)中得到cspbx3/au纳米颗粒。
16.所述步骤中对烧瓶的加热可以采用油浴加热。
17.优选的,cs2co3用量与油酸用量关系为0.1mmol-0.6mmol:5ml-50ml。
18.优选的,znx2溶液的制备中,znx2用量:十八烯用量:油酸用量:油胺用量=0.1mmol-0.6mmol:8ml-40ml:0.2ml-10ml:0.2ml-10ml,加热温度为50℃至160℃。
19.优选的,pb(oa)2溶液制备中pbo用量:油酸用量=0.1mmol-0.6mmol:5ml:30ml,加热温度为80℃至160℃。
20.优选的,csx/au纳米颗粒分散液制备中,aux质量为csx质量的0.04倍。
21.更具体的:
22.步骤1)中所述cs2co3用量优选为0.1mmol至0.6mmol,油酸用量优选为5ml至50ml。
23.所述步骤2)中十八烯用量优选为8ml至40ml,加热温度优选为50℃至160℃。
24.所述znx2用量优选为0.1mmol至0.6mmol。
25.所述油酸用量优选为0.2ml至10ml。
26.所述油胺用量优选为0.2ml至10ml。
27.所述步骤3)中pbo用量优选为0.1mmol至0.6mmol。
28.所述油酸用量优选为5ml至30ml。
29.所述加热温度优选为80℃至160℃。
30.所述步骤4)中的离心转速优选为3000rpm至13000rpm,离心时间优选为3min至30min;
31.所述前驱体用量优选0.5ml-3ml;
32.所述步骤5)中csx用量优选为0.01mmol-0.5mmol;
33.所述aux用量优选为csx用量的0.04倍;
34.所述离心转速优选为6000rpm-12000rpm,离心时间优选为3min至30min;
35.所述步骤6)和步骤7)中csx或csx/au用量优选为0.5ml-20ml;
36.所述pb(oa)2用量优选为0.2ml-8ml;
37.所述溶剂为能分散但不溶解csx、csx/au和钙钛矿纳米晶/au纳米颗粒复合材料的溶剂。
38.所述溶剂为但不限于正己烷、甲苯、氯苯,用量优选为4ml至40ml。
39.所述离心转速优选为3000rpm至10000rpm,离心时间优选为3min至30min。
40.本发明提出的与贵金属复合的钙钛矿量子点与现有同类钙钛矿量子点的区别在于:
41.1.cspbx3/au纳米颗粒具有更大的比表面积和更多的活性位点,增加了其光活性;
42.2.通过利用au沉积来形成cspbx3/au异质结构纳米颗粒,产生一个平衡的费米能级,在钙钛矿和金的界面上,存在一个从钙钛矿到金形态的“内置”电场,电场迅速清除自由载流子,从而建立耗尽区,由此可以增强光诱导空穴与电子的分离,抑制其重组,进一步提高其光催化性能。
附图说明
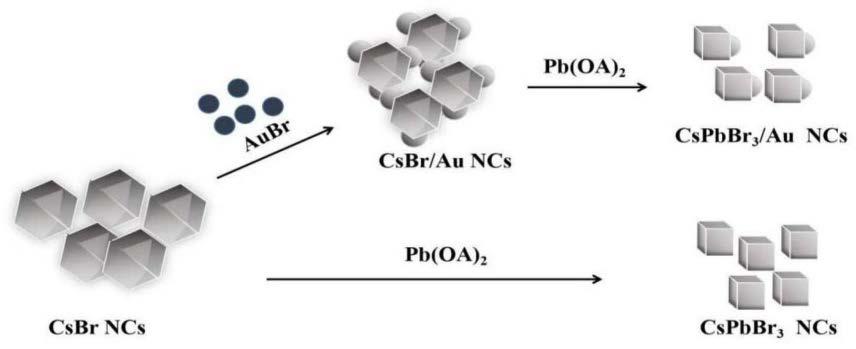
43.图1为csbr ncs形成cspbbr
3 ncs与au-cspbbr
3 ncs的原理示意图。
44.图2为au-cspbbr3的xrd图。
45.图3为csbr ncs、cspbbr
3 ncs、au-cspbbr
3 ncs的xrd图。
46.图4为csbr ncs(左)和au-csbr ncs(右)的tem形貌图。
47.图5为au-cspbbr
3 ncs的tem形貌图。
48.图6为可见光下加入不同催化剂的苏丹红ⅲ浓度-时间关系曲线。
具体实施方式
49.本发明不局限于下列具体实施方式,本领域一般技术人员根据本发明公开的内容,可以采用其他多种具体实施方式实施本发明的,或者凡是采用本发明的设计结构和思路,做简单变化或更改的,都落入本发明的保护范围。需要说明的是,在不冲突的情况下,本发明中的实施例及实施例中的特征可以相互组合。
50.本发明下面结合实施例作进一步详述:
51.对比实施例1
52.步骤1)称取cs2co
3 0.65g(aladdin,纯度99.99%,市售产品),将其溶解于含10ml油酸(oa)的三口烧瓶中,然后加热搅拌,并在120℃下抽真空1h,除去体系中的水,抽真空结束后通入氩气,直至cs2co3完全溶解,得cs-油酸前驱体溶液;
53.步骤2)称取znbr
2 0.1801g(aladdin,纯度99.99%,市售产品),将其溶解于含2ml oa、2ml油胺(oam)和4ml十八烯(ode)的三口烧瓶中,然后加热搅拌,并在80℃下抽真空1h,除去体系中的水,抽真空结束后通入氩气,直至znbr2完全溶解,得znbr2溶液。
54.步骤3)称取pbo 0.22g将其溶解于含10ml油酸的三口烧瓶中,接着加热至80℃直至pbo完全溶解,得pb(oa)2溶液。
55.步骤4)将1ml步骤1)制备的cs-油酸前驱体溶液迅速注入到步骤2)制备的znbr2溶液中(70-80℃),注射完成5min后置于冰水浴中,冷却至室温,然后进行后处理:将冷却至室温的溶液离心,离心转速为3800rpm,10min,取沉淀分散到12ml甲苯溶剂中,得到csbr纳米颗粒分散液,并调整其浓度为25mg/ml(如图4(左)所示)。
56.步骤5)取5ml步骤4)制得的csbr纳米颗粒分散液,加入2ml步骤3)制得的pb(oa)2溶液,搅拌30s,反应结束对溶液进行离心,离心转速为12000rpm,5min,最后取沉淀分散到甲苯中,得到最终产物cspbbr3量子点分散液,然后调整其浓度到10mg/ml。
57.步骤6)取5ml步骤5)制得的cspbbr3量子点分散液在搅拌条件下加入2mg aubr颗粒,反应10min,反应结束后对溶液进行离心,离心转速为12000rpm,5min,取沉淀分散到5ml甲苯中,获得最终产物au-cspbbr3量子点分散液,调整其浓度到10mg/ml。此方法会改变钙钛矿量子点的结构,出现cs2au2br6副产物(如图2所示)。
58.将最终产物用于光催化反应:配制80ml的苏丹红ⅲ溶液(10mg/l,溶剂为甲苯),取36ml注入到透明玻璃瓶中,并加入4ml对比实施例1的步骤(6)制备的au-cspbbr3分散液(10mg/ml);将混合后的溶液置于黑暗中搅拌12h以达到吸附-解附平衡;之后施加可见光照射(30mw/cm2),每间隔30min取样表征溶液中苏丹红ⅲ的浓度。结果表明,在au-cspbbr3的光催化作用下,光照6小时后,苏丹红ⅲ溶液浓度降低至初始浓度的81%。
59.对比实施例2
60.参照对比实施例1,制备cspbbr3量子点分散液,并调整其浓度到10mg/ml。与对比实施例1类似,将该产物用于光催化反应,在cspbbr3的光催化作用下,光照6小时后,苏丹红ⅲ溶液浓度降低至初始浓度的88%(图6)。
61.实施例1
62.首先,取10ml按照对比实施例1步骤4)制得的csbr纳米颗粒分散液,加入10mg aubr(经计算csbr纳米颗粒与aubr质量之比为25:1),反应1h后,取分散液进行离心,离心转速为7500rpm,5min,取沉淀分散到甲苯中,得到au-csbr纳米颗粒复合材料分散液(如图4(右)和3(右)所示)。
63.然后,取5ml au-csbr纳米颗粒复合材料分散液,加入2ml按照对比实施例1步骤3)制得的pb(oa)2溶液,搅拌30s,反应结束对溶液进行离心,离心转速为12000rpm,5min,最后取沉淀分散到甲苯中,得到最终产物au-cspbbr3量子点分散液,调整其浓度到10mg/ml(如图5(左)所示)。该au-cspbbr3量子点产物不含cs2au2br6副产物(图3(左))。
64.与对比实施例1类似,将最终产物用于光催化反应,在au-cspbbr3的光催化作用下,光照6小时后,苏丹红ⅲ溶液浓度降低至初始浓度的28%(图6)。
65.实施例2
66.首先,称取cs2co
3 0.65g(aladdin,纯度99.99%,市售产品),将其溶解于含10ml油酸(oa)的三口烧瓶中,然后加热搅拌,并在120℃下抽真空1h,除去体系中的水,抽真空结束后通入氩气,直至cs2co3完全溶解,得cs-油酸前驱体溶液;称取zncl
2 54.52mg(aladdin,纯度99.99%,市售产品),将其溶解于含2ml oa、2ml oam和4ml ode的三口烧瓶中,然后加热搅拌,并在80℃下抽真空1h,除去体系中的水,抽真空结束后通入氩气,直至zncl2完全溶
解,得zncl2溶液。
67.接着,将1ml本实施例制备的cs-油酸前驱体溶液迅速注入到zncl2溶液中(80-100℃),注射完成5min后置于冰水浴中,冷却至室温,然后按照对比实施例1步骤4)中的后处理方法得cscl纳米颗粒分散液,并调整其浓度为25mg/ml。
68.然后,在按照实施例1的制备方法,将csbr纳米颗粒替换为上述制备的cscl纳米颗粒,aubr替换为aucl,其余步骤与实施例1相同,制备得到au-cspbcl3量子点分散液。
69.最后,离心纯化并调整其浓度到10mg/ml。
70.与对比实施例1类似,将最终产物用于光催化反应,在au-cspbcl3的光催化作用下,光照6小时后,苏丹红ⅲ溶液浓度降低至初始浓度的73%。
71.实施例3
72.称取zni
2 0.128g(aladdin,纯度99.99%,市售产品)替代zncl2,其余步骤与实施例2相同,制备得到au-cspbi3量子点分散液,然后调整其浓度到10mg/ml。
73.与对比实施例1类似,将最终产物用于光催化反应,在au-cspbi3的光催化作用下,光照6小时后,苏丹红ⅲ溶液浓度降低至初始浓度的40%。
74.以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。