一种c-myc转录抑制剂及其制备方法和应用
技术领域
1.本发明涉及医药领域,具体涉及一种c-myc转录抑制剂及其制备方法和应用。
背景技术:
2.恶性肿瘤(又称为癌症)是当前危害人类生命健康的重大疾病之一,也是目前人类在医疗卫生领域面临的一大挑战。研究发现,90%以上的恶性肿瘤可观察到单个或者多种基因的持续变化。其中,癌基因的突变或过度表达是恶性肿瘤最常发生的细胞事件。原癌基因c-myc是目前研究最多的癌基因之一,也是激活最频繁的癌基因之一。c-myc属于myc家族,其表达直接和间接地调控细胞的增殖、生长、分化、代谢和凋亡等多个重要的生命过程,并与超过20%的恶性肿瘤的发生发展密切相关。已有大量研究证据表明,原癌基因c-myc位于许多生长促进信号转导通路的交叉点,c-myc的异常表达可诱导肿瘤的形成和维持肿瘤的生长。此外,c-myc的表达产物myc癌蛋白还可作为一种多功能的转录因子,单独或与其他转录因子结合一同参与多种基础生物过程相关基因的表达调控,促进肿瘤的发生与生长。目前已有研究表明通过降低c-myc的表达可显著抑制恶性肿瘤的发生发展,这为c-myc与myc蛋白靶向的抗肿瘤药物开发提供了理论依据。然而,myc蛋白内在的无序特性,使得设计特异性靶向的小分子变得困难。因此,目前控制myc的策略主要有两种:干扰myc的表达和功能,以及直接抑制癌基因c-myc的转录。直接抑制癌基因c-myc的转录是目前被广泛研究的肿瘤抑制策略。其中,c-myc基因上游调控元件是抑制c-myc转录的主要途径。
3.g-四链体是一种非典型的核酸二级结构,在基因转录过程中,若在c-myc的p1启动子区上游的核酸超敏元件nhe iii1区域形成g-四链体,可阻碍rna聚合酶的靠近,从而抑制c-myc的转录。特异性结合g-四链体的化合物可通过稳定c-myc启动子上形成的g-四链体结构,维持g-四链体的基因沉默功能,导致c-myc表达的下调。因此发展新型c-myc转录抑制剂在治疗癌症方面具有重大的意义和广泛的应用前景。
技术实现要素:
4.为了克服上述现有技术存在的问题,本发明的目的之一在于提供一种c-myc转录抑制剂,该c-myc转录抑制剂在体内外能够特异性的结合并稳定c-myc g-四链体,进而抑制人肝癌细胞hepg2 c-myc的转录并抑制人肝癌细胞hepg2的迁移,最终实现在体内抑制人肝癌细胞hepg2的生长。
5.本发明的目的之二在于提供上述c-myc转录抑制剂的制备方法。
6.本发明的目的之三在于提供一种g-四链体配体。
7.本发明的目的之四在于提供一种抗肿瘤药物。
8.本发明的目的之五在于提供上述c-myc转录抑制剂在制备预防或治疗肿瘤的药物中的应用。
9.为了实现上述目的,本发明所采取的技术方案是:
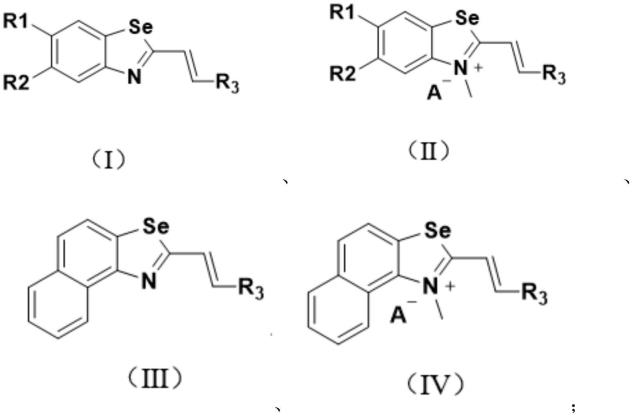
10.本发明的第一个方面在于提供一种c-myc转录抑制剂,包括式(i)~(iv)任一所示
的化合物或其药学上可接受的盐、异构体、溶剂化物;
[0011][0012]
其中,a-为n甲基化阴离子、碘离子、溴离子、对甲苯磺酸根、三氟乙酸根、高氯酸根、四氟硼酸根、甲基硫酸根或三氟甲磺酸根;
[0013]
r1、r2分别独立地选自氟、氯、溴、氢、氨基或胺类取代基;
[0014]
r3独立地选自芳香环、取代芳环或芳香杂环。
[0015]
优选地,式(i)~(iv)中:
[0016]
a-为碘离子、溴离子、对甲苯磺酸根、三氟乙酸根、高氯酸根、四氟硼酸根、甲基硫酸根或三氟甲磺酸根;
[0017]
r1、r2分别独立地选自氟、氢、氨基或胺类取代基;
[0018]
r3独立地选自芳香环、取代芳环或芳香杂环。
[0019]
优选地,式(i)~(iv)中:
[0020]
a-为碘离子、溴离子、对甲苯磺酸根、三氟乙酸根、高氯酸根、四氟硼酸根、甲基硫酸根或三氟甲磺酸根;
[0021]
r1、r2分别独立地选自氟、氢、氨基或胺类取代基;
[0022]
r3独立地选自
[0023]
优选地,式(i)~(iv)中:
[0024]
a-为碘离子;
[0025]
r1、r2均为氢;
[0026]
r3独立地选自
[0027]
本发明的第二个方面在于提供本发明第一个方面提供的c-myc转录抑制剂的制备方法,包括以下步骤:
[0028]
以为原料合成式(i)所示的化合物或式(ii)所示的化合物;以为原料合成式(iii)所示的化合物或式(iv)所示的化合物;
[0029]
所述式(i)所示的化合物的制备方法包括以下步骤:
[0030]
将与硒、溴化亚铜混合反应,制得然后将与次磷酸反应得到再与乙酰丙酮反应,制得然后将与反应,制得所述式(i)所示的化合物;
[0031]
或,
[0032]
所述式(iii)所示的化合物的制备方法包括以下步骤:
[0033]
将与硒、溴化亚铜混合反应,制得然后将与三丁基膦、乙酸反应,制得然后将与反应,制得所述式(iii)所示的化合物;
[0034]
或,
[0035]
所述式(ii)所示的化合物或式(iv)所示的化合物的制备方法包括以下步骤:
[0036]
将与甲基化试剂反应,制得
然后将与反应,制得式(ii)所示的化合物或式(iv)所示的化合物;
[0037]
或,所述式(ii)所示的化合物或式(iv)所示的化合物的制备方法包括以下步骤:
[0038]
将式(i)所示的化合物或式(iii)所示的化合物与甲基化试剂反应,制得式(ii)所示的化合物或式(iv)所示的化合物;
[0039]
其中,a-、r1、r2、r3如前述所定义。
[0040]
优选地,所述甲基化试剂为ch3a,所述a-为n甲基化阴离子、碘离子、溴离子、对甲苯磺酸根、三氟乙酸根、高氯酸根、四氟硼酸根、甲基硫酸根或三氟甲磺酸根。
[0041]
优选地,所述甲基化试剂为碘甲烷、溴甲烷、对甲苯磺酸甲酯、三氟甲磺酸甲酯、高氯酸甲酯、四氟硼酸甲酯、甲基硫酸甲酯或三氟甲磺酸甲酯。
[0042]
本发明的第三个方面在于提供一种g-四链体配体,包括式(i)~(iv)所示的化合物中的至少一种;
[0043][0044]
其中,a-、r1、r2、r3如上述所定义。
[0045]
本发明的第四个方面在于提供一种抗肿瘤药物,包括本发明第一个方面提供的c-myc转录抑制剂。
[0046]
优选地,所述肿瘤包括肝癌、结肠癌或结肠腺癌。
[0047]
优选地,所述抗肿瘤药物中,c-myc转录抑制剂的含量为0.05~50wt%;进一步优选地,所述抗肿瘤药物中,c-myc转录抑制剂的含量为1~50wt%;再进一步优选地,所述抗肿瘤药物中,c-myc转录抑制剂的含量为10~50wt%。
[0048]
本发明的第五个方面在于提供一种c-myc转录抑制剂在制备预防或治疗肿瘤的药物中的应用。
[0049]
优选地,所述肿瘤包括肝癌、结肠癌、结肠腺癌。
[0050]
本发明的有益效果是:本发明中的c-myc转录抑制剂稳定性好,便于储存,毒副作
用小,抗肿瘤效果好,具体而言,该c-myc转录抑制剂在体内外能够特异性的结合并稳定c-myc g-四链体,进而抑制人肝癌细胞hepg2 c-myc的转录并抑制人肝癌细胞hepg2的迁移,最终实现在体内抑制人肝癌细胞hepg2的生长。
[0051]
本发明中的c-myc转录抑制剂的制备方法制备简单、成本低廉,适用于大规模生产。
附图说明
[0052]
图1为本发明实施例6中的m-se3对c-myc g-四链体的稳定效果图。
[0053]
图2为本发明实施例1~8中的c-myc转录抑制剂对g-四链体的作用效果图。
[0054]
图3为本发明实施例6中的m-se3对c-myc基因的转录抑制效果图。
[0055]
图4为本发明实施例6中的m-se3在不同浓度时对hepg2细胞的增殖抑制效果图。
[0056]
图5为本发明实施例6中的m-se3在不同浓度时对hepg2细胞的迁移抑制效果图。
[0057]
图6为本发明实施例6中的m-se3体内抑制肝癌细胞hepg2生长图。
[0058]
图7为本发明实施例6中的m-se3对小鼠体重影响测试结果图。
具体实施方式
[0059]
以下结合附图和实例对本发明的具体实施作进一步详细说明,但本发明的实施和保护不限于此。需要指出的是,以下若有未特别详细说明之过程,均是本领域技术人员可参照现有技术实现或理解的。所用试剂或仪器未注明生产厂商者,视为可以通过市售购买得到的常规产品。
[0060]
实施例1
[0061]
本例中的c-myc转录抑制剂的结构式如下:
[0062][0063]
其中,r1为h,r2为h,r3为
[0064]
本例中的c-myc转录抑制剂采用以下制备方法制得,具体包括以下步骤:
[0065]
(1)双(2-氨基苯基)-二硒醚的合成
[0066]
反应式为:
[0067]
[0068]
将2-碘苯胺(5.0g,22.8mmol),硒粉(5.4g,68.5mmol),溴化亚铜(0.7g,4.6mmol)和氢氧化钾(2.6g,45.6mmol)加入三颈烧瓶中惰性气体ar的保护下加入10ml无水dmso,于油浴120℃反应48h。待原料2-碘苯胺反应完全,冷却至室温,加入过量饱和碳酸钠溶液稀释,乙酸乙酯萃取。有机相用无水硫酸钠干燥,旋蒸浓缩。硅胶柱层析法进行纯化(洗脱剂为石油醚和二氯甲烷的体积比为5:1的混合液),得到橙色油状液体,产率为25%。
[0069]1h nmr(400mhz,cdcl3):δ7.38(d,j=7.8hz,2h),7.12(t,j=7.6hz,2h),6.72(d,j=8.0hz,2h),6.65(t,j=7.5hz,2h),3.77(br,4h)。
[0070]
(2)2-甲基苯并硒唑的合成
[0071]
反应式:
[0072][0073]
将双(2-氨基苯基)-二硒醚(1.0g,2.9mmol)混悬于6ml甘油中,惰性气体ar气的保护下置于90℃油浴中。搅拌中滴加3ml次磷酸(50wt.%的水溶液),90℃下反应30min。待观察到黄色溶液变为无色,加入乙酰丙酮(0.6g,5.8mmol),90℃下反应6h。待中间体原料反应完全,加入过量水稀释,乙酸乙酯萃取。有机相用无水硫酸钠干燥,旋蒸浓缩。硅胶柱层析法进行纯化(洗脱剂为:石油醚和二氯甲烷的体积比为100:1的混合液),得到淡黄色油状液体,产率为89%。
[0074]1h nmr(400mhz,cdcl3)δ7.99(d,j=8.1hz,1h),7.86(d,j=7.9hz,1h),7.44(t,j=7.7hz,1h),7.29(t,j=7.8hz,1h),2.87(s,3h)。
[0075]
(3)c-myc转录抑制剂的合成
[0076]
反应式:
[0077][0078]
将2-甲基苯并硒唑(215.7mg,1.1mmol)和n-乙基咔唑-3-甲醛(50mg,0.22mmol)溶解在0.5ml dmso中,搅拌中缓慢滴加0.25ml 50%koh溶液,室温下反应24h。加入少量冰水,用1mol/l hcl溶液调节ph至7~8。减压抽滤,滤饼用冰乙醇洗涤三次,真空干燥后得到棕黄色固体(43.1mg,48.0%),即为本例中的c-myc转录抑制剂,记为se1。经hplc测定的纯度为95.2%,hrms(esi)m/z:calcd for c
23h18
n2se,[m h]
403.0709,发现403.0698的质谱峰。
[0079]1h nmr(400mhz,cdcl3)δ8.31(s,1h),8.13(d,j=7.7hz,1h),8.03(d,j=8.0hz,1h),7.90(d,j=7.8hz,1h),7.75(dd,j=8.5,1.2hz,1h),7.61(d,j=15.9hz,1h),7.47(dt,j=18.2,8.3hz,5h),7.29(t,j=7.4hz,2h),4.39(q,j=7.2hz,2h),1.46(t,j=7.2hz,3h);
[0080]
13
c nmr(126mhz,cdcl3)δ172.19,155.69,140.88,140.84,140.57,136.97,126.66,126.45,126.35,125.29,125.15,124.85,124.29,123.57,122.98,122.40,120.70,120.61,119.67,109.14,108.97,37.88,13.99。
[0081]
实施例2
[0082]
本例中的c-myc转录抑制剂的结构式如下:
[0083][0084]
其中,r1为h,r2为h,r3为
[0085]
本例中的c-myc转录抑制剂采用以下制备方法制得,具体包括以下步骤:
[0086]
(1)n-(3-(n,n-二甲氨基)丙基)-咔唑-3-甲醛的合成
[0087]
在冰浴环境下,将咔唑(500mg,2.99mmol)溶于5ml无水dmf加入15ml耐压管中,随后加入nah(120mg,4.95mmol)搅拌1h,接着逐滴加入n,n-二甲基-3-氯丙胺(542.37mg,4.48mmol)搅拌10min。随后升温到60℃反应16h,tlc(薄层色谱)检测反应完全后,用20ml冰水混合物淬灭,乙酸乙酯(ea)萃取,硅胶柱层析法进行纯化得到n-(3-(n,n-二甲氨基)丙基)-咔唑(632mg,80%)。
[0088]
在冰浴环境下,将无水dmf(400mg,5.4mmol)加入三颈瓶中,ar保护,随后逐滴加入pocl3(700mg,4.56mmol)搅拌30min,移至室温反应1h。接着将n-(3-(n,n-二甲氨基)丙基)-咔唑(486.7mg,1.93mmol)加入混合体系中室温反应30min,随后升温至60℃反应16h,tlc检测反应完全后,冷却至室温加入20ml冰水淬灭,调ph至中性,ea萃取,硅胶柱层析法进行纯化得到n-(3-(n,n-二甲氨基)丙基)-咔唑-3-甲醛(178mg,35%)。
[0089]
1h nmr(400mhz,dmso-d6)δ10.06(s,1h),8.76(d,j=1.2hz,1h),8.29(d,j=7.7hz,1h),8.00(dd,j=8.6,1.5hz,1h),7.78(d,j=8.6hz,1h),7.70(d,j=8.2hz,1h),7.57
–
7.52(m,2h),7.34
–
7.29(m,2h),4.48(t,j=6.8hz,2h),2.18(t,j=6.8hz,3h),2.11(s,6h),1.92(p,j=6.8hz,4h)。
[0090]
(2)c-myc转录抑制剂的合成
[0091]
将2-甲基苯并硒唑(350mg,1.78mmol)和n-(3-(n,n-二甲氨基)丙基)-咔唑-3-甲醛(100mg,0.36mmol)溶解在0.5ml dmso中,搅拌中缓慢滴加0.25ml 50%koh溶液,室温下反应24h。加入少量冰水,用1mol/l hcl溶液调节ph至7~8,旋干。hplc纯化,得到黄色固体(6.1mg,3.7%),制得本例中的c-myc转录抑制剂,记为se2。经hplc测得的纯度为95.7%,hrms(esi)m/z:calcd for c
26h25
n3se,[m h]
460.1288,发现460.1289的质谱峰。
[0092]1h nmr(400mhz,dmso-d6)δ8.65(s,1h),8.24(d,j=7.8hz,1h),8.13(d,j=7.9hz,1h),7.96(d,j=8.6hz,2h),7.79
–
7.66(m,4h),7.56
–
7.47(m,2h),7.37
–
7.28(m,2h),4.55
–
4.49(m,2h),3.26
–
3.10(m,2h),2.76(s,6h),2.18(dd,j=8.5,6.3hz,2h);
[0093]
13
c nmr(101mhz,dmso-d6)δ153.79,140.51,140.35,140.31,139.41,137.88,126.95,126.57,126.46,125.61,125.28,124.07,123.86,122.94,122.25,121.32,120.70,
119.90,119.71,109.99,109.66,54.60,42.48,42.48,40.15,23.92。
[0094]
实施例3
[0095]
本例中的c-myc转录抑制剂的结构式如下:
[0096][0097]
其中,r1为h,r2为h,r3为
[0098]
本例中的c-myc转录抑制剂采用以下制备方法制得,具体包括以下步骤:
[0099]
将2-甲基苯并硒唑(176.5mg,0.9mmol)和n-苄基咔唑-3-甲醛(50mg,0.18mmol)溶解在0.5ml dmso中,搅拌中缓慢滴加0.25ml 50%koh溶液,室温下反应24h。加入少量冰水,用1mol/l的hcl溶液调节ph至7~8。减压抽滤,滤饼用冰乙醇洗涤三次,真空干燥后得到土黄色固体(65.8mg,81.0%),即为本例中的c-myc转录抑制剂,记为se3。通过hplc测得的产品的纯度为95.6%;hrms(esi)m/z:calcd for c
28h20
n2se,[m h]
465.0866,发现465.0874的质谱峰。
[0100]1h nmr(400mhz,cdcl3)δ8.33(s,1h),8.15(d,j=7.7hz,1h),8.01(d,j=8.0hz,1h),7.90(d,j=7.8hz,1h),7.70(d,j=8.6hz,1h),7.59(d,j=15.9hz,1h),7.46(dt,j=8.3,4.1hz,3h),7.40(t,j=9.4hz,3h),7.33
–
7.27(m,4h),7.15(d,j=6.2hz,2h),5.53(s,2h);
[0101]
13
c nmr(126mhz,cdcl3)δ172.07,155.75,141.58,141.32,140.60,137.05,136.82,129.01,129.01,127.79,127.17,126.60,126.52,126.52,126.46,125.39,125.33,124.86,124.36,123.73,123.07,122.71,120.68,120.51,120.10,109.64,109.45,46.88。
[0102]
实施例4
[0103]
本例中的c-myc转录抑制剂的结构式如下:
[0104][0105]
其中,a为i,r1为h,r2为h,r3为
[0106]
本例中的c-myc转录抑制剂采用以下制备方法制得,具体包括以下步骤:
[0107]
(1)n-甲基2-甲基苯并硒唑碘化物的合成
[0108]
将2-甲基苯并硒唑(480mg,2.45mmol)溶于200μl乙腈中,加入碘甲烷(855μl,13.7mmol),避光,于80℃下回流反应24h。薄层色谱法监测反应。待体系中无原料剩余,冷却至室温。待过量碘甲烷挥发,加入过量无水乙醇,超声使固体均匀分散于乙醇中,减压抽滤,滤饼用无水乙醇洗涤三次,真空干燥后得到n-甲基2-甲基苯并硒唑碘化物。
[0109]
1h nmr(400mhz,methanol-d4)δ8.36(d,j=8.1hz,1h),8.24(d,j=8.5hz,1h),7.89(t,j=7.9hz,1h),7.76(t,j=7.7hz,1h),4.26(s,3h),3.24(s,3h)。
[0110]
(2)c-myc转录抑制剂的合成
[0111]
将n-甲基2-甲基苯并硒唑碘化物(100mg,0.47mmol)与1.1倍当量的n-乙基咔唑-3-甲醛溶于0.5ml乙醇中,80℃下反应24h。薄层色谱法监测反应。待反应体系无中间体原料剩余,冷却至室温,有固体析出。减压抽滤,滤饼用冰乙醇洗涤三次,真空干燥后得到红色固体(127mg,56.0%),即为本例中的c-myc转录抑制剂,记为m-se1。经hplc测定的纯度为99.6%,hrms(esi)m/z:calcd for c
24h21
n2se
,[m-i]
417.0866,发现417.0866的质谱峰。
[0112]1h nmr(400mhz,dmso-d6)δ8.95(s,1h),8.45(dd,j=15.6,11.7hz,2h),8.23(d,j=8.2hz,2h),8.16(d,j=8.4hz,1h),8.04(d,j=15.4hz,1h),7.83
–
7.77(m,2h),7.71(d,j=8.2hz,1h),7.67(t,j=7.8hz,1h),7.56(t,j=7.7hz,1h),7.34(t,j=7.5hz,1h),4.52(q,j=6.9hz,2h),4.34(s,3h),1.37(t,j=7.1hz,3h);
[0113]
13
c nmr(126mhz,dmso-d6)δ180.75,152.45,143.30,142.17,140.27,128.96,128.64,128.24,127.65,127.02,126.81,125.55,123.80,122.92,122.21,120.62,120.26,117.95,113.01,110.05,110.05,37.41,37.41,13.83。
[0114]
实施例5
[0115]
本例中的c-myc转录抑制剂的结构式如下:
[0116][0117]
其中,a为i,r1为h,r2为h,r3为
[0118]
本例中的c-myc转录抑制剂采用以下制备方法制得,具体包括以下步骤:
[0119]
将n-甲基2-甲基苯并硒唑碘化物(100mg,0.47mmol)与1.1倍当量的n-(3-(n,n-二甲氨基)丙基)-咔唑-3-甲醛溶于0.5ml乙醇中,80℃下反应24h。薄层色谱法监测反应。待反应体系无中间体原料剩余,冷却至室温,有固体析出。减压抽滤,滤饼用冰乙醇洗涤三次,真空干燥后得到红色固体,即为本例中的c-myc转录抑制剂,记为m-se2。经hplc测定的纯度为99.1%,hrms(esi)m/z:calcd for c
27h28
n3se ,[m-i] 474.1445,发现474.1428的质谱峰。
[0120]1h nmr(400mhz,dmso-d6)δ8.97(s,1h),8.42(dd,j=11.5,2.8hz,2h),8.24(t,j
=7.5hz,3h),8.06(d,j=15.9hz,1h),7.91
–
7.82(m,2h),7.78(t,j=8.0hz,2h),7.63
–
7.55(m,1h),7.38(t,j=7.3hz,1h),4.61
–
4.51(m,2h),4.38(s,3h),3.16
–
3.05(m,2h),2.70(s,6h),2.21
–
2.11(m,2h);
[0121]
13
c nmr(101mhz,dmso-d6)δ172.01,150.46,142.54,142.05,140.63,129.33,128.39,128.15,127.44,127.00,125.54,124.52,124.12,123.67,123.08,122.33,120.82,120.55,116.82,116.58,110.45,54.23,42.21,42.21,40.15,38.97,23.71。
[0122]
实施例6
[0123]
本例中的c-myc转录抑制剂的结构式如下:
[0124][0125]
其中,a为i,r1为h,r2为h,r3为
[0126]
本例中的c-myc转录抑制剂采用以下制备方法制得,具体包括以下步骤:
[0127]
将n-甲基-2-甲基苯并硒唑碘化物(100mg,0.47mmol)与1.1倍当量n-苄基咔唑-3-甲醛溶于0.5ml乙醇中,80℃下反应24h。薄层色谱法监测反应。待反应体系无中间体原料剩余,冷却至室温,有固体析出。减压抽滤,滤饼用冰乙醇洗涤,得到红色固体(126.7mg,55.9%),即为本例中的c-myc转录抑制剂,记为m-se3。经hplc检测的纯度为98.6%;hrms(esi)m/z:calcd for c
29h23
n2se
,[m-i]
479.1023,发现479.1009的质谱峰。
[0128]1h nmr(400mhz,dmso-d6)δ8.98(s,1h),8.46(dd,j=15.2,11.8hz,2h),8.28
–
8.15(m,3h),8.06(d,j=15.4hz,1h),7.87(d,j=8.7hz,1h),7.83
–
7.77(m,1h),7.70(dd,j=18.3,8.0hz,2h),7.53(t,j=7.7hz,1h),7.36(t,j=7.4hz,1h),7.33
–
7.21(m,5h),5.76(s,2h),4.34(s,3h);
[0129]
13
c nmr(101mhz,dmso-d6)δ180.89,152.28,143.36,142.83,140.88,137.20,129.07,128.71,128.69,128.69,128.32,127.76,127.49,127.04,126.96,126.82,126.82,125.96,123.71,123.05,122.31,120.62,120.57,118.07,113.38,110.58,110.55,45.93,37.43。
[0130]
实施例7
[0131]
本例中的c-myc转录抑制剂的结构式如下:
[0132]
[0133]
其中,r3为
[0134]
本例中的c-myc转录抑制剂采用以下制备方法制得,具体包括以下步骤:
[0135]
(1)双(1-氨基-2-萘基)二硒醚的合成
[0136]
反应式:
[0137][0138]
将1-氨基-2-溴萘(5.0g,22.5mmol),硒粉(5.3g,67.5mmol),溴化亚铜(0.6g,4.5mmol)和氢氧化钾(2.5g,45.0mmol)溶解在15ml无水dmso中,惰性气体保护下于油浴120℃反应48h。薄层色谱法监测反应情况。待原料1-氨基-2-溴萘反应完全,冷却至室温,加入过量饱和碳酸钠溶液稀释,乙酸乙酯萃取。有机相用无水硫酸钠干燥,旋蒸浓缩。硅胶柱层析法进行纯化(洗脱剂为:石油醚和二氯甲烷的体积比为30:1的混合液),得到黑紫色固体,产率为19%。
[0139]1h nmr(400mhz,cdcl3)δ7.81
–
7.71(m,4h),7.49(d,j=8.6hz,2h),7.45(dq,j=6.7,3.5hz,4h),7.15(d,j=8.5hz,2h),4.86(br,4h)。
[0140]
(2)2-甲基-β-萘并硒唑的合成
[0141]
反应式:
[0142][0143]
将双(1-氨基-2-萘基)二硒醚(500mg,1.1mmol)溶解于6.8ml无水甲苯中,惰性气体保护下加入三丁基膦(724mg,3.3mmol),室温搅拌5min。加入乙酸(204mg,3.3mmol),微波辐射(100℃,100w)下反应2h。加入过量饱和碳酸钠溶液稀释,二氯甲烷萃取三次。有机相用无水硫酸钠干燥,旋蒸浓缩。硅胶柱层析法进行纯化(洗脱剂为石油醚和二氯甲烷的体积比为50:1的混合液),得到黄色油状液体,产率为84%。
[0144]1h nmr(400mhz,cdcl3)δ8.81(d,j=8.0hz,1h),7.92(dd,j=7.8,6.4hz,2h),7.75(d,j=8.7hz,1h),7.67
–
7.61(m,1h),7.59
–
7.54(m,1h),2.98(s,3h)。
[0145]
(3)c-myc转录抑制剂的合成
[0146]
反应式:
[0147][0148]
将2-甲基-β-萘并硒唑(270.8mg,1.1mmol)和n-乙基咔唑-3-甲醛(50mg,0.22mmol)溶解在0.5ml dmso中,搅拌中缓慢滴加0.25ml 50%koh溶液,室温下反应24h。加入少量冰水,用1mol/l的hcl溶液调节ph至7~8。减压抽滤,滤饼用冰乙醇洗涤三次,真空干燥后得到黄色固体(50.2mg,54.7%),记为p-se1。经hplc测试的纯度为96.0%,hrms(esi)m/z:calcd for c
27h20
n2se,[m h]
453.0866,发现453.0858的质谱峰。
[0149]1h nmr(400mhz,cdcl3)δ8.88(d,j=8.2hz,1h),8.35(s,1h),8.14(d,j=7.6hz,1h),7.96
–
7.91(m,2h),7.76(dd,j=12.7,8.8hz,2h),7.67(d,j=7.5hz,1h),7.65(s,1h),7.59(s,1h),7.56(d,j=6.8hz,1h),7.54
–
7.48(m,2h),7.43(d,j=8.4hz,2h),7.30(d,j=7.4hz,1h),4.39(dd,j=14.1,7.0hz,2h),1.47(t,j=7.1hz,3h);
[0150]
13
c nmr(126mhz,cdcl3)δ172.08,151.44,140.80,140.60,139.59,134.39,132.32,129.81,128.14,126.97,126.32,125.97,125.80,125.23,124.90,124.89,123.62,123.05,122.87,122.06,120.72,120.40,119.64,109.11,108.97,37.89,14.00。
[0151]
实施例8
[0152]
本例中的c-myc转录抑制剂的结构式如下:
[0153][0154]
其中,a为i,r3为
[0155]
本例中的c-myc转录抑制剂采用以下制备方法制得,具体包括以下步骤:
[0156]
将p-se1(50mg,0.11mmol)溶解于0.5ml乙腈中,快速加入碘甲烷(62.4mg,0.44mmol),于80℃下回流反应24h。冷却至室温,待过量碘甲烷挥发,减压抽滤,滤饼用乙醚洗涤三次,冰乙醇洗涤一次,真空干燥后,得到红褐色固体(28.9mg,56.0%),记为m-p-se1。经hplc测试所得的纯度为99.3%;hrms(esi)m/z:calcd for c
28h23
n2se
,[m-i]
467.1023,发现467.0998的质谱峰。
[0157]
反应式为:
[0158]
[0159]1h nmr(400mhz,dmso-d6)δ8.92(s,1h),8.90(d,j=8.6hz,1h),8.47
–
8.40(m,2h),8.26
–
8.20(m,3h),8.17(d,j=8.7hz,1h),8.09(d,j=15.3hz,1h),7.88
–
7.76(m,3h),7.69(d,j=8.3hz,1h),7.54(t,j=7.6hz,1h),7.34(t,j=7.4hz,1h),4.75(s,3h),4.49(dd,j=13.7,6.6hz,2h),1.36(t,j=7.0hz,3h);
[0160]
13
c nmr(101mhz,dmso-d6)δ180.59,151.31,142.00,140.26,138.87,133.71,130.27,129.85,129.05,128.07,127.93,127.29,126.77,125.72,123.90,123.48,123.13,122.91,122.64,122.21,120.59,120.19,113.48,110.01,110.00,43.65,37.38,13.81。
[0161]
性能测试:
[0162]
(1)对c-myc g-四链体的稳定作用
[0163]
分别用g-四链体缓冲液(10mmol/l tris-hcl缓冲液,ph为7.4,2mmol/l kcl)将dna溶液(c-myc pu22核酸溶液)稀释至2μmol/l,得到两组c-myc pu22核酸溶液溶液,其中一组作为对照组,另一组加入m-se3使其浓度为10μmol/l,作为实验组,然后将两组溶液分别加入石英比色皿中并置于圆二色谱仪内。设置仪器程序,以0.5s/nm扫描速度,在230~330nm范围内采集光谱,带宽(bandwidth)为1nm,步长(step size)1nm,每点时间(time per point)为0.5s,升温范围为25~95℃,升温速度为3℃/min。每隔5℃采集一次cd光谱,用origin 9.0软件进行数据分析,测试结果见图1。由图1可以看出,与对照组(单独c-myc pu22核酸溶液)相比,加入c-myc转录抑制剂m-se3后实验组的c-myc g-四链体热稳定曲线明显右移,tm位移值为11.4℃;表明m-se3化合物能够在体外稳定c-myc g-四链体。
[0164]
取本发明实施例1~8中的c-myc转录抑制剂用g-四链体缓冲液配制成工作浓度为1μmol/l的溶液100μl,加入到3
×
3mm的石英比色皿中并置于荧光光谱仪内。设置c-myc转录抑制剂的相应的激发波长和发射波长范围,激发和发射狭缝为5nm,采集c-myc转录抑制剂溶液的荧光光谱。加入核酸储液到c-myc转录抑制剂溶液中至工作浓度2μmol/l。使c-myc转录抑制剂和核酸的浓度比例为1:2。充分混匀静置30s后,与上述同样条件下采集c-myc转录抑制剂和核酸溶液的荧光光谱,测试结果如图2所示,其中,图2中的pu22(c-myc)、pu22mut、c-kit1、hras、htg22均为g-四链体序列;图2(a)为se1对c-myc g-四链体的作用效果图;图2(b)为se2对c-myc g-四链体的作用效果图;图2(c)为se3对c-myc g-四链体的作用效果图;图2(d)为m-se1对c-myc g-四链体的作用效果图;图2(e)为m-se2对c-myc g-四链体的作用效果图;图2(f)为m-se3对c-myc g-四链体的作用效果图;图2(g)为p-se1对c-myc g-四链体的作用效果图;图2(h)为m-p-se1对c-myc g-四链体的作用效果图。从图2可知,c-myc转录抑制剂m-se1和m-se3对c-myc g-四链体的选择性最强。
[0165]
(2)对人肝癌细胞hepg2 c-myc基因转录与表达的抑制作用
[0166]
为了探究m-se3下调c-myc转录的同时,是否也会对其他的癌基因的表达水平产生影响,我们通过rt-qpcr实验进行验证。选择了三种启动子区可形成g-四链体的癌基因vegf、kras和c-kit,以gapdh为内参,并通过各癌基因的特异性引物进行相应cdna片段的扩增。取对数生长期的hepg2细胞以每孔20万个接种在6孔板中,置于含5%co2的37℃培养箱中培养24h。取m-se3,用含10%胎牛血清的培养基配制成2.5μmol/l的工作浓度。弃去6孔板的培养基,每孔加入含2.5μmol/l m-se3的培养基2ml,control组为不含化合物的培养基2ml。置于含5%co2的37℃培养箱中培养48h。弃去培养基,用pbs洗涤细胞,再加入pbs,用ep管离心收集细胞。吸去pbs,随后进行rna的提取。rna样品置于-80℃冰箱保存。再除去rna样
品中的基因组dna,以减少基因组dna对后续逆转录得到cdna 的定量分析造成干扰,随后放置于4℃冰箱储存,得到反应液。然后按照逆转录试剂盒说明书,配制rt反应液,将rt反应液与上述反应液混匀后于pcr仪37℃反应15min,85℃反应5s,然后冷却至4℃放置。接着将cdna与pcr体系加入pcr板中,封上pcr封板膜,离心后放置于实时荧光定量pcr仪中,按95℃30s、95℃5s、62℃1min、40℃30s进行四十个循环扩增,结束后放置于4℃,通过ct值计算rna水平的相对变化,计算结果如图3所示。由图3中的rt-pcr结果表明,本发明中的m-se3对c-myc基因的转录有较强的抑制作用,且具有高选择性。综合可见,m-se3可选择性抑制c-myc转录,并且均对其他癌基因转录的影响较小。
[0167]
(3)对人肝癌细胞hepg2的增殖和迁移的抑制作用
[0168]
取对数生长期的hepg2细胞以每孔500个接种在6孔板中,置于含5%co2的37℃培养箱中培养24h。弃去6孔板的培养基,每孔加入含不同浓度m-se3(浓度分别为:0μmol/l,0.125μmol/l,0.25μmol/l,0.5μmol/l,1μmol/l,2μmol/l)的培养基2ml。置于含5%co2的37℃培养箱中培养14天,每隔3天更换一次培养基。弃去培养基,使用0.01mol/l的pbs缓冲溶液洗涤细胞3次。用4%多聚甲醛固定细胞20min,弃去多聚甲醛。用1%结晶紫溶液染色15min。用水洗涤细胞除去多余染料,晾干,拍照,结果如图4所示,其中,图4(a)为m-se3的浓度为0μmol/l时对hepg2细胞的增殖抑制效果图,图4(b)为m-se3的浓度为0.125μmol/l时对hepg2细胞的增殖抑制效果图;图4(c)为m-se3的浓度为0.25μmol/l时对hepg2细胞的增殖抑制效果图;图4(d)为m-se3的浓度为0.5μmol/l时对hepg2细胞的增殖抑制效果图;图4(e)为m-se3的浓度为1μmol/l时对hepg2细胞的增殖抑制效果图;图4(f)为m-se3的浓度为2μmol/l时对hepg2细胞的增殖抑制效果图。由图4可知,随着m-se3浓度上升,m-se3以剂量依赖性的方式显著抑制了hepg2细胞的克隆形成,高浓度的m-se3对hepg2细胞的克隆的抑制作用更强。
[0169]
取对数生长期的hepg2细胞以每孔50万个接种在6孔板中,置于含5%co
2 37℃培养箱中培养24h。吸去培养基。用微量枪头在单层细胞中央划开一条等宽直线。用灭菌0.01mol/l的pbs缓冲溶液洗涤划痕,除去划痕处细胞。显微镜下观察并拍照记录细胞迁移0h的划痕情况,具体见图5(a)~图5(f),其中,图5(a)至图5(f)均为未加入m-se3时的细胞迁移0h的显微图。接着加入浓度梯度m-se3化合物工作液(浓度分别为:0μmol/l,0.3μmol/l,0.6μmol/l,1.2μmol/l,2.5μmol/l,5μmol/l)2ml。置于含5%co2的37℃培养箱中培养48h。显微镜下观察并拍照记录细胞迁移48h的划痕情况。结果如图5(g)~图5(l)所示,其中,图5(g)为m-se3的浓度为0μmol/l时对hepg2细胞迁移48h的抑制效果图;图5(h)为m-se3的浓度为0.3μmol/l时对hepg2细胞迁移48h的抑制效果图;图5(i)为m-se3的浓度为0.6μmol/l时对hepg2细胞迁移48h的抑制效果图;图5(j)为m-se3的浓度为1.2μmol/l时对hepg2细胞迁移48h的抑制效果图;图5(k)为m-se3的浓度为2.5μmol/l时对hepg2细胞迁移48h的抑制效果图;图5(l)为m-se3的浓度为5μmol/l时对hepg2细胞迁移48h的抑制效果图。由图5可知,培养48h后,未给药组细胞间间距明显缩小,发生迁移。而随着m-se3浓度上升,细胞间间距缩小幅度明显降低,划痕明显,细胞迁移减少,表明本发明中的m-se3能够明显抑制hepg2细胞的迁移。
[0170]
(4)m-se3体内抑制人肝癌细胞的生长
[0171]
收集对数生长期hepg2细胞,将1
×
108个/100μl的细胞悬液皮下注射至4-5周龄的
balb/c-nu/nu雄鼠(免疫缺陷小鼠,此小鼠没有胸腺)腋下。待肿瘤体积生长超过50mm3后,将小鼠随机分成3组,每组5只,3组中设置一个给药组、一个溶剂组和一个对照组,其中,给药组给予m-se3药物,给药剂量为15mg/kg;溶剂组给予相同用量的生理盐水溶液,对照组给予化疗药物阿霉素(doxorubicin,1mg/kg),我们按小鼠重量100μl/10g腹腔注射进行给药。每两天给药一次,并测量记录小鼠重量和肿瘤体积,给药处理28天后,对小鼠进行脱臼处死,m-se3体内抑制肝癌细胞hepg2生长图见图6所示;小鼠体重测试结果见图7所示。由图6和图7可知,与溶剂组相比,给药组的肿瘤平均体积明显减小,可见m-se3能够显著抑制肿瘤的生长,且不会造成小鼠体重降低。
[0172]
(5)癌细胞抑制活性
[0173]
取对数生长期的人肝癌细胞hepg2、人结肠癌细胞hct116、人结肠腺癌细胞rko共三株癌细胞和一株人正常结肠上皮细胞ncm460分别以每孔3000个接种在96孔板中。置于含5%co2的37℃培养箱中培养24h。取本发明实施例1~8中的c-myc转录抑制剂用含10%胎牛血清的培养基分别配制成0μmol/l,3.125μmol/l,6.25μmol/l,12.5μmol/l,25μmol/l,50μmol/l,100μmol/l的工作浓度。弃去96孔板的培养基,每孔加入含上述不同浓度化合物的培养基100μl,每个浓度设置三个复孔。置于含5%co2的37℃培养箱中培养48h。取mtt溶液(3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐,噻唑蓝),与无血清的培养基按2:8比例配制混匀。弃去96孔板的培养基,每孔加入含mtt的培养基100μl,于含5%co2的37℃培养箱中孵育4h。弃去mtt和培养基混合溶液,每孔加入dmso 100μl,轻柔混匀。用酶标仪分别检测570nm和490nm波长处各孔的吸光值。计算c-myc转录抑制剂对不同细胞的ic
50
值,结果如表1所示。
[0174]
表1本发明实施例1~8中的c-myc转录抑制剂对不同细胞的ic
50
值
[0175][0176][0177]
从表1可知,c-myc转录抑制剂的癌细胞抑制活性与其c-myc g-四链体的结合能力和选择性显著相关。本发明中的c-myc转录抑制剂对癌细胞有明显增殖抑制毒性,其原因可能是对c-myc g-四链体结合增强的结果。综合来看,在癌细胞增殖抑制中表现较好的化合物有m-se1、m-se3和m-p-se1,在c-myc g-四链体特异性结合和癌细胞增殖抑制这两方面中综合表现较好的两个苯并硒唑类小分子分别为m-se1和m-se3。
[0178]
上面对本发明实施例作了详细说明,但是本发明不限于上述实施例,在所属技术
领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下做出各种变化。此外,在不冲突的情况下,本发明的实施例及实施例中的特征可以相互组合。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。