用于构建lmna基因突变的扩张型心肌病模型猪核移植供体细胞的基因编辑系统及其应用
技术领域:
1.本发明属于基因编辑技术领域,具体涉及crispr/cas9系统及ssodn同源重组技术在构建lmna基因点突变的扩张型心肌病模型猪核移植供体细胞中的应用。
背景技术:
2.扩张型心肌病(dcm,dilated cardiomyopathy,又称充血性心肌病)是一种原因未明的原发性心肌疾病。病征为左或右心室或双侧心室扩大,并伴有心室收缩功能减退,伴或不伴充血性心力衰竭,室性或房性心律失常多见。病情呈进行性加重,死亡可发生于疾病的任何阶段。
3.临床表现患者以中年人居多。起病多缓慢,有时可达10年以上。症状以充血性心力衰竭为主,其中以气短和水肿最为常见。此外,尚可有脑、肾、肺等处的栓塞。已经在30%至40%的dcm患者中发现了遗传病因,但其中只有50%与已知的致病基因变异相关。在所有dcm病例中,约有6%是由lmna基因(定位于人1q21.2)突变引起的。lamin a/c(层粘蛋白a和c)是lmna基因的两个可变剪接产物,在所有体细胞中均有表达。lmna中的突变多以组织特有的方式发生,大多数引起骨骼肌受累的心肌病。超过200种不同的lmna突变已被证实与遗传性心肌病有关,主要是dcm,且主要以常染色体显性方式遗传,但也有报道常染色体隐性病例,已经确认可致病的点突变包括r60g、l85r、n195k、e203k、e203g、r225x、r571s、r644c等数十种。dcm在出现心室扩张的证据之前,可能与传导系统疾病有关,而lmna突变引起的心脏传导系统疾病包括窦房结疾病、房性心律不齐、房室心脏传导阻滞和室性心律失常等。
4.dcm动物模型的构建将为研究及治疗人类dcm提供有力的实验工具。猪作为大动物,是人类长期以来主要的肉食供应动物,易于大规模繁殖饲养,而且在伦理道德及动物保护等方面要求较低,且猪体型大小和器官功能与人类近似,是理想的人类疾病模型动物。因此,研发出人类dcm猪模型用于进行药物筛选、药效检测、疾病病理、基因治疗及细胞治疗等研究,能够为进一步的临床应用提供有效的实验数据,也为成功治疗人类dcm提供有力的实验手段。
5.基因编辑是近年来不断取得重大发展的一种生物技术,其包括从基于同源重组的基因编辑到基于核酸酶的zfn、talen、crispr/cas9等编辑技术,其中crispr/cas9技术是当前最先进的基因编辑技术。目前,基因编辑技术被越来越多地应用到动物模型的制作上。
技术实现要素:
6.本发明的目的是针对现有技术的上述不足,提供用于构建lmna基因突变的基因编辑系统。
7.本发明的另一目的是提供基因编辑系统的应用。
8.本发明的又一目的是提供一种重组细胞及其应用。
9.本发明的目的可通过以下技术方案实现:用于构建lmna基因突变的基因编辑系
bl21(de3)菌株为目的蛋白表达菌株,该菌株可高效表达克隆于含有噬菌体t7启动子的表达载体(如pet-32a)的外源基因。同时,对于cas9蛋白的密码子,本发明进行了密码子优化,使之完全适应表达菌株的密码子偏好,从而提高目的蛋白的表达水平。另外,本发明在细菌生长至一定数量后,再用iptg在低温下诱导目的蛋白的表达,可避免目的蛋白过早表达对宿主菌生长的影响,低温下诱导表达也显著提高所表达的目的蛋白的可溶性。经过上述各项优化设计及实验实施,所得到的cas9蛋白活性比商品cas9蛋白有了极显著的提高。
23.(3)采用本发明构建并表达的cas9高效蛋白联合体外转录的grna进行基因编辑,并对cas9和grna的最佳摩尔配比进行了优化,配合合成的ssodn作为donor,最终获得靶位点点突变的效率高达17.5%,远高于常规的点突变效率(《5%)。
24.(4)利用本发明所得到的靶基因突变单细胞克隆株进行体细胞核移植动物克隆可直接得到含靶标基因纯合突变的克隆猪,并且该纯合突变可稳定遗传。
25.在小鼠模型制作中采用的受精卵显微注射基因编辑材料后再进行胚胎移植的方法,因其直接获得纯合突变后代的概率非常低(低于5%),需要进行后代的杂交选育,这不太适用于妊娠期较长的大动物(如猪)模型制作。因此,本发明采用技术难度大、挑战性高的原代细胞体外编辑并筛选阳性编辑单细胞克隆的方法,后期再通过体细胞核移植动物克隆技术直接获得相应疾病模型猪,可大大缩短模型猪制作周期并节省人力、物力、财力。
26.本发明为通过基因编辑手段获得扩张型心肌病猪模型奠定了坚实的基础,将有助于研究并揭示dcm发病机制,也可用于进行药物筛选、药效检测、疾病病理、基因治疗及细胞治疗等研究,能够为进一步的临床应用提供有效的实验数据,进而为成功治疗人类dcm提供有力的实验手段。本发明对于扩张型心肌病药物的研发及揭示该病的发病机制具有重大应用价值。
附图说明
27.图1为质粒px330的结构示意图。
28.图2为质粒pkg-ge3的结构示意图。
29.图3为质粒pkg-u6grna的结构图谱。
30.图4为将20bp左右的dna分子(用于转录形成grna的靶序列结合区)插入质粒pkg-u6grna的示意图。
31.图5为质粒pet-32a的结构图谱。
32.图6为质粒pkg-ge4的结构示意图。
33.图7为实施例3中步骤3.3.3的电泳图。
34.图8为实施例3中步骤3.4.3的电泳图。
35.图9为实施例4中步骤4.2.3的电泳图。
36.图10为实施例4中步骤4.2.4的电泳图。
37.图11为实施例4中步骤4.6.4的电泳图。
38.图12为实施例5中步骤5.1.3的电泳图。
39.图13为实施例5中步骤5.6.3的电泳图。
40.图14为实施例5中步骤5.6.4判定为野生型的示例性测序峰图。
41.图15为实施例5中步骤5.6.4判定为杂合突变型的示例性测序峰图。
42.图16为实施例5中步骤5.6.4判定为双等位基因不同变异的纯合突变型示例性测序峰图。
43.图17为实施例5中步骤5.6.4判定为双等位基因相同变异的纯合突变型示例性测序峰图。
44.图18为实施例5中步骤5.6.4判定为靶标位点点突变的杂合突变型示例性测序峰图。
45.图19为实施例5中步骤5.6.4判定为靶标位点点突变的纯合突变型示例性测序峰图。
具体实施方式
46.实施例1、质粒的构建
47.出发质粒为px330-u6-chimeric_bb-cbh-hspcas9(简称质粒px330),序列如seq id no.1所示,结构示意图见图1。seq id no.1中,第440-725位核苷酸组成cmv增强子,第727-1208位核苷酸组成chickenβ-actin启动子,第1304-1324位核苷酸编码sv40核定位信号(nls),第1325-5449位核苷酸编码cas9蛋白,第5450-5497位核苷酸编码nucleoplasmin核定位信号(nls)。
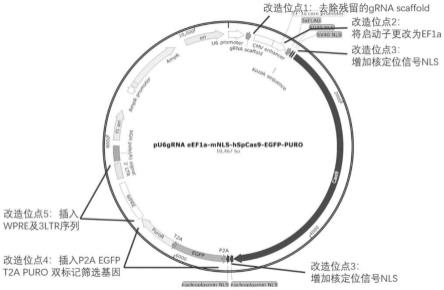
48.构建质粒pu6grna-eef1a-mnls-hspcas9-egfp-puro(简称质粒pkg-ge3),序列如seq id no.2所示,结构示意图见图2。seq id no.2中,第395-680位核苷酸组成cmv增强子,第682-890位核苷酸组成ef1a启动子,第986-1006位核苷酸编码核定位信号(nls),第1016-1036位核苷酸编码核定位信号(nls),第1037-5161位核苷酸编码cas9蛋白,第5162-5209位核苷酸编码核定位信号(nls),第5219-5266位核苷酸编码核定位信号(nls),第5276-5332位核苷酸编码自剪切多肽p2a(自剪切多肽p2a的氨基酸序列为“atnfsllkqagdveenpgp”,发生自剪切的断裂位置为c端开始第一个氨基酸残基和第二个氨基酸残基之间),第5333-6046位核苷酸编码egfp蛋白,第6056-6109位核苷酸编码自裂解多肽t2a(自裂解多肽t2a的氨基酸序列为“egrgslltcgdveenpgp”,发生自裂解的断裂位置为c端开始第一个氨基酸残基和第二个氨基酸残基之间),第6110-6703位核苷酸编码puromycin蛋白(简称puro蛋白),第6722-7310位核苷酸组成wpre序列元件,第7382-7615位核苷酸组成3’ltr序列元件,第7647-7871位核苷酸组成bgh poly(a)signal序列元件。seq id no.2中,第911-6706形成融合基因,表达融合蛋白。由于自剪切多肽p2a和自裂解多肽t2a的存在,融合蛋白自发形成如下三个蛋白:具有cas9蛋白的蛋白、具有egfp蛋白的蛋白和具有puro蛋白的蛋白。
49.与质粒px330相比,所构建的质粒pkg-ge3主要进行了如下改造:
①
去除残留的grna骨架序列(gttttagagctagaaatagcaagttaaaataaggctagtccgtttt),降低干扰;
②
将原有chickenβ-actin启动子改造为具更高表达活性的ef1a启动子,增加cas9基因的蛋白表达能力;
③
在cas9基因的上游和下游均增加核定位信号编码基因(nls),增加cas9蛋白的核定位能力;
④
原质粒无任何真核细胞筛选标记,不利于阳性转化细胞的筛选和富集,依次在cas9基因的下游插入p2a-egfp-t2a-puro编码基因,赋予载体荧光和真核细胞抗性筛选能力;
⑤
插入wpre元件和3’ltr序列元件,增强cas9基因的蛋白翻译能力。
50.构建质粒pkg-u6grna,序列如seq id no.3所示,结构示意图见图3。seq id no.3中,第2280-2539位核苷酸组成hu6启动子,第2558-2637位核苷酸用于转录形成grna骨架。
使用时,将20bp左右的dna分子(用于转录形成grna的靶序列结合区)插入质粒pkg-u6grna,形成重组质粒,示意图见图4,在细胞中重组质粒转录得到grna。
51.质粒pkg-ge4是以质粒pet-32a作为骨架进行改造而来,主要改造如下:
①
保留了trxa蛋白的编码区域,可以帮助所表达的目的蛋白形成二硫键,增加目的蛋白溶解性及活性,但是在此序列之前加入碱性磷酸酶(phoa)的信号肽(sp)序列,该sp可以引导所表达的目的蛋白分泌至细菌的膜周质腔中,并可被原核周质信号肽酶酶切;
②
在trxa蛋白编码序列之后增加his-tag标签基团,可用于所表达的目的蛋白的富集;
③
在his-tag标签下游增加肠激酶(ek)酶切位点ddddk(asp-asp-asp-asp-lys),纯化出的蛋白将在肠激酶作用下去除his-tag标签和上游所融合的trxa蛋白。
④
插入cas9蛋白的编码序列,同时在该基因的上游和下游均增加核定位信号编码序列(nls),增加后期纯化出的cas9蛋白的核定位能力。
52.构建质粒pet32a-t7lac-phoa:sp-trxa-his-ek-nls-spcas9-nls-t7ter(简称质粒pkg-ge4),序列如seq id no.4所示,结构示意图见图6。seq id no.5中,第5121-5139位核苷酸组成t7启动子,第5140-5164位核苷酸编码lac operator信号,第5209-5271位核苷酸编码phoa(碱性磷酸酶)信号肽(signal peptide,sp),第5272-5598位核苷酸编码trxa蛋白,第5620-5637位核苷酸编码his-tag标签,第5638-5652位核苷酸编码肠激酶酶切位点,第5656-5670位核苷酸编码sv40核定位信号(nls),第5701-9801位核苷酸编码spcas9蛋白(其密码子已优化为适宜在大肠杆菌bl21(de3)菌株中进行表达),第9802-9849位核苷酸编码nucleoplasmin核定位信号(nls),第9902-9949位核苷酸编码t7终止子。
53.pkg-ge4质粒构建方法如下:
54.(1)骨架载体的制备
55.质粒pet-32a(结构示意图见图5)用xbai、xhoi酶切,回收载体片段(约5329bp左右)。
56.(2)全基因合成插入序列
57.全基因合成如seq id no.5所示插入序列,依次包含前述的phoa signal peptide序列、trxa蛋白编码序列、his-tag标签基团、ek酶切位点、spcas9蛋白编码序列及其两端的nls序列,在全基因合成的n端和c端分别包含与骨架载体序列同源的25个碱基对。
58.(3)全基因合成插入片段与骨架载体连接
59.将步骤(1)回收的骨架载体和步骤(2)全基因合成的序列重组得到pet32a-t7lac-phoa:sp-trxa-his-ek-nls-spcas9-nls-t7ter载体,简称pkg-ge4,质粒图谱如图6,核苷酸序列见seq id no.4。
60.实施例2 pkg-ge4质粒转化e.coli bl21(de3)表达菌株及cas9蛋白的表达
61.2.1融合蛋白trxa-his-ek-nls-spcas9-nls的诱导表达
62.将鉴定正确的pkg-ge4质粒转化入到大肠杆菌表达宿主bl21(de3)(武汉灵淼生物公司)中,涂氨苄抗性(ampr)平板,培养过夜后,挑选单菌落,接种到含100μg/ml氨苄青霉素的lb液体培养基中,在37℃、200转/min条件下培养过夜,然后将过夜培养的菌液接种到500ml lb培养基中,接种比例为1:200,30℃、230转/min条件下培养至od600达到1.0左右,加入终浓度为0.5mm的异丙基硫代半乳糖苷(iptg)诱导bl21(de3)菌株表达目的蛋白,然后25℃培养12小时进行目的蛋白的低温诱导可溶性表达。4℃,10000g离心15分钟收集菌体,用pbs洗涤菌体并离心收集菌体沉淀。
63.2.2融合蛋白trxa-his-ek-nls-spcas9-nls的纯化
64.2.2.1融合蛋白的粗提
65.粗提缓冲液为20mm tris-hcl ph 8.0,0.5m nacl,5mm imidazole,1mm pmsf。粗提方法:按每克湿菌加入10ml上述缓冲液,悬浮细菌,均质机破碎,1000par循环三次。然后4℃,15000g离心细菌悬液30min,收集上清,经0.22μm滤膜过滤用于下一步的亲和层析蛋白纯化。
66.2.2.2融合蛋白的纯化
67.采用ni-nta琼脂糖柱(金斯瑞,l00250/l00250-c)进行融合蛋白的纯化。首先用平衡液(20mm tris-hcl ph 8.0,0.5m nacl,5mm imidazole)对ni柱进行平衡,然后将上述过滤后的菌液上清上样到平衡后的ni柱,再用平衡液洗涤ni柱,然后用缓冲液(20mm tris-hcl ph 8.0,0.5m nacl,50mm imidazole)洗去杂蛋白,最后用洗脱液(20mm tris-hcl ph 8.0,0.5m nacl,500mm imidazole)洗脱目的蛋白。
68.2.3融合蛋白(trxa-his-ek-nls-spcas9-nls)的酶切与cas9蛋白的纯化
69.使用amicon超滤管(sigma,ufc9100)将上述ni柱纯化后的融合蛋白溶液浓缩至200μl并用25mm tris-hcl ph 8.0稀释至1ml。然后将商品来源的带his标签的重组牛肠激酶(生工生物,c620031)加入到25mm tris-hcl ph 8.0稀释后的融合蛋白液中,进行酶切反应。酶切用量为每50μg融合蛋白用肠激酶2个单位,酶切缓冲体系为25mm tris-hcl ph 8.0,酶切温度25℃,酶切时间16小时。
70.酶切结束后,将酶切后溶液与80μl的ni-nta树脂混匀,在室温下剧烈搅拌15min,然后7000g离心3min,上清与树脂分离,取上清即为酶切后去除trxa-his的nls-spcas9-nls目的蛋白。酶切后的trxa-his多肽片段及带his标签的肠激酶ek均结合在ni-nta树脂上,从而将上清中的cas9蛋白分离纯化。最后将纯化后的cas9蛋白(命名为pkg-ge4-cas9蛋白)浓缩后用50%的甘油于-80℃保存。
71.实施例3 pkg-ge4-cas9与商品cas9蛋白的切割效率比较及与grna最佳摩尔配比摸索
72.3.1 ttn基因靶点grna设计及转录
73.3.1.1使用benchling对ttn基因进行grna靶点设计,经预筛确定选用以下两条grna:
74.ttn-grna1:agagcacagtcagcctggcg
75.ttn-grna2:cttccagaattggatctccg
76.3.1.2 grna分子不同区段序列的合成(由基因合成公司合成)
77.t7-grna1:ggcttgtcggactcttcgctattacgccagctggcgaagggggat
78.t7-grna2:tgggaaaaccctggcgttacccaacttaatcgccttgcagcacatcccccttcgccagc
79.t7-grna3:acgccagggttttcccagtcacgacgttaggaaattaatacgactcactatagg
80.ttn-g1t7-grna4:ttctagctctaaaaccgccaggctgactgtgctctcctatagtgagtcgtattaatttc
81.ttn-g1t7-grna5:cctggcggttttagagctagaaatagcaagttaaaataaggctagtccgttatcaactt
82.ttn-g2t7-grna4:ttctagctctaaaaccggagatccaattctggaagcctatagtgagtcgtatt
aatttc
83.ttn-g2t7-grna5:atctccggttttagagctagaaatagcaagttaaaataaggctagtccgttatcaactt
84.t7-grna6:aaaagcaccgactcggtgccactttttcaagttgataacggactagccttat(seq id no.16)
85.3.1.3设计用于鉴定包含ttn grna靶点片段的引物
86.ttn-f55:tacggaattggggagccagcgga
87.ttn-r560:caaagttaactctctgtgtct
88.3.1.4转录模板的扩增
89.ttn-t7-grna1转录模板序列如下所示:
90.ggcttgtcggactcttcgctattacgccagctggcgaagggggatgtgctgcaaggcgattaagttgggtaacgccagggttttcccagtcacgacgttaggaaattaatacgactcactataggagagcacagtcagcctggcggttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttt。
91.上述ttn-t7-grna1转录模板是使用t7-grna1、t7-grna2、t7-grna3、ttn-g1t7-grna4、ttn-g1t7-grna5、t7-grna6共计6条合成引物采用重叠延伸pcr扩增技术制作的,该序列中含有t7启动子,可启动相关序列的转录。扩增后将目的条带切胶后按照fast pure gel dna extraction mini kit(vazyme,dc301)说明书进行操作,回收产物作为转录模板。ttn-t7-grna2转录模板序列如下所示:ggcttgtcggactcttcgctattacgccagctggcgaagggggatgtgctgcaaggcgattaagttgggtaacgccagggttttcccagtcacgacgttaggaaattaatacgactcactataggcttccagaattggatctccggttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgctttt。
92.上述ttn-t7-grna2转录模板是使用t7-grna1、t7-grna2、t7-grna3、ttn-g2t7-grna4、ttn-g2t7-grna5、t7-grna6共计6条合成引物采用重叠延伸pcr扩增技术制作的,该序列中含有t7启动子,可启动相关序列的转录。扩增后将目的条带切胶后按照fast pure gel dna extraction mini kit(vazyme,dc301)说明书进行操作,回收产物作为转录模板。
93.3.1.5 grna的转录
94.以步骤3.1.4制备的转录模板按照transcript aid t7 high yield transcription kit(fermentas,k0441)和mega clear
tm transcription clean-up kit(thermo,am1908)说明书进行操作,所获产物即为可用于细胞电转的grna。
95.3.2猪原代成纤维细胞制备
96.3.2.1取刚出生从江香猪耳组织0.5g,去除毛发及骨组织,75﹪酒精浸泡30-40s;
97.3.2.2用含5%p/s(gibco penicillin-streptomycin)的pbs洗涤5次,不含p/s的pbs洗一次;
98.其中5%p/s的pbs配方为:5%p/s(gibco penicillin-streptomycin) 95%pbs,5%、95%为体积百分比。
99.3.2.3用剪刀将组织剪碎,加入5ml 0.1%的胶原酶(sigma)溶液,37℃摇床消化1h;
100.3.2.4 500g离心5min,去上清,将沉淀用1ml完全培养基重悬,铺入含10ml完全培
streptomycin) 1%hepes(solarbio));
118.6)混匀后放置于37℃,5%co2、5%o2的恒温培养箱中进行培养;
119.7)电转后12-18h换液,36-48h用0.25%(gibco)的胰蛋白酶消化并收集细胞于1.5ml离心管中。
120.3.3.3基因编辑效率分析
121.提取3.3.2中收集的细胞基因组dna,采用ttn-f55和ttn-r560组成的引物对进行pcr扩增,然后进行1%琼脂糖凝胶电泳(见图7)。505bp条带为野生型条带(wt),254bp左右(条带505bp理论缺失251bp)为缺失突变条带(mt)。
122.基因缺失突变效率=(mt灰度/mt条带bp数)/(wt灰度/wt条带bp数 mt灰度/mt条带bp数)
×
100%。据此计算,第一组基因缺失突变效率为19.9%,第二组基因缺失突变效率为39.9%,第三组基因缺失突变效率为79.9%,第四组基因缺失突变效率为44.3%。
123.结果表明,当两个grna与pkg-ge4-cas9蛋白的质量比为1:1:4,实际用量为1μg:1μg:4μg时基因编辑效率最高,确定两个grna与pkg-ge4-cas9蛋白的最适用量为1μg:1μg:4μg。
124.3.4.1共转染分组情况
125.cas9-a组:将转录的ttn-t7-grna1、ttn-t7-grna2和商品cas9-a蛋白共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:1μg ttn-t7-grna1:1μg ttn-t7-grna2:4μg cas9-a。
126.pkg-ge4组:将转录的ttn-t7-grna1、ttn-t7-grna2和pkg-ge4-cas9蛋白共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:1μg ttn-t7-grna1:1μg ttn-t7-grna2:4μg pkg-ge4-cas9。
127.cas9-b组:将转录的ttn-t7-grna1、ttn-t7-grna2和商品cas9-b蛋白共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:1μg ttn-t7-grna1:1μg ttn-t7-grna2:4μg cas9-b。
128.control组:将转录的ttn-t7-grna1、ttn-t7-grna2共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:1μg ttn-t7-grna1:1μg ttn-t7-grna2。
129.3.4.2共转染操作方法
130.同本实施例中步骤3.3.2。
131.3.4.3基因编辑效率分析
132.提取3.4.2中收集的细胞基因组dna,采用ttn-f55和ttn-r560组成的引物对进行pcr扩增,然后进行1%琼脂糖凝胶电泳(见图8)。505bp条带为野生型条带(wt),254bp条带(条带505bp理论缺失251bp)为缺失突变条带(mt)。
133.基因缺失突变效率=(mt灰度/mt条带bp数)/(wt灰度/wt条带bp数 mt灰度/mt条带bp数)
×
100%。据此计算,采用商品cas9-a蛋白的基因缺失突变效率为28.5%,采用pkg-ge4-cas9蛋白的基因缺失突变效率为85.6%,采用商品cas9-b蛋白的基因缺失突变效率为16.6%。
134.结果表明,与采用商品的cas9蛋白相比,采用本发明制备的pkg-ge4-cas9蛋白使得基因编辑效率显著提高。
135.实施例4 lmna基因靶点grna的筛选
136.4.1基因组dna的提取
137.使用vazyme公司fastpure cell/tissue dna isolation mini kit(vazyme cat.dc102-01)分别进行18只猪(雄性a、b、c、d、e、f、g、h雌性1、2、3、4、5、6、7、8、9、10)耳组织的基因组dna的柱式提取,使用nanodrop进行定量,-20℃保存备用。
138.4.2 lmna基因预点突变位点及邻近基因组序列保守性分析
139.4.2.1猪lmna基因信息
140.编码lamin a/c蛋白;位于4号染色体;geneid为100126859,sus scrofa。猪lmna基因编码的氨基酸序列如seq id no.6所示。基因组dna中,猪lmna基因具有16个外显子,而与人类dcm相关的lmna突变研究中n195k和e203k均位于人lmna基因第3外显子上,对应猪第7外显子(猪lmna基因第6至第8外显子,含第6和第7内含子序列如seq id no.7所示)。
141.4.2.2 lmna基因预设点突变位点外显子及邻近基因组序列pcr扩增引物设计
142.根据查到的猪lmna基因组序列
143.(https://www.ncbi.nlm.nih.gov/nuccore/nc_010446.5?report=genbank&from=93899019&to=93927255&strand=true),设计引物扩增前述18只猪基因组样品lmna基因外显子7的位点。
144.使用oligo7进行引物设计,设计结果如下:
145.lmna-e7grna-jdf1:agtcgttcctgccagggagtg
146.lmna-e7grna-jdr1:cgtctcatggcggcgcttggt
147.lmna-e7grna-jdf2:accacagcaggaacgccagtc
148.lmna-e7grna-jdr2:tctgccagccggctctcaaac
149.4.2.3 lmna基因组pcr扩增引物筛选
150.使用猪(雌性1、2、3)耳朵组织提取的基因组为模板,分别采用lmna-e7grna-jdf1和lmna-e7grna-jdr1组成的引物对或lmna-e7grna-jdf2和lmna-e7grna-jdr2组成的引物对进行pcr扩增,然后进行电泳,结果见图8。lmna-e7grna-jdf1和lmna-e7grna-jdr1组成的引物对效果更好。
151.4.2.4 18只猪lmna基因片段pcr扩增
152.分别以18个基因组模板(雄性a、b、c、d、e、f、g、h雌性1、2、3、4、5、6、7、8、9、10),引物lmna-e7grna-jdf1/lmna-e7grna-jdr1,max酶进行lmna基因组片段的扩增,产物进行1%琼脂糖凝胶电泳,结果如图9。
153.4.2.5 lmna基因序列保守性分析
154.以上pcr扩增产物,使用扩增引物进行测序(通用生物公司测序),将测序结果与公共数据库中的lmna基因序列进行比对分析。根据比对结果,可采用lmna-e7grna-jdf1/lmna-e7grna-jdr1引物检测突变(引物本身无可能的突变位点)。
155.4.3 grna靶点设计及构建
156.4.3.1使用benchling进行靶点grna设计
157.设计靶点已避开可能的突变位点,使用benchling进行靶点grna设计:
158.https://benchling.com/
159.lmna基因敲除靶点设计如下:
160.lmna-e7-grna1:ggatgagatgctgcgccgag(seq id no.8)
161.lmna-e7-grna2:caggctgcagaccctgaagg(seq id no.9)
162.lmna-e7-grna3:gaacaggctgcagaccctga(seq id no.10)
163.lmna-e7-grna4:tgaggccaagaaacaacttc(seq id no.11)
164.合成的lmna基因共4个靶点的插入序列互补dna oligo如下:
165.lmna-e7-grna1-s:caccggatgagatgctgcgccgag(seq id no.12)
166.lmna-e7-grna1-a:aaacctcggcgcagcatctcatcc(seq id no.13)
167.lmna-e7-grna2-s:caccgcaggctgcagaccctgaagg(seq id no.14)
168.lmna-e7-grna2-a:aaacccttcagggtctgcagcctgc(seq id no.15)
169.lmna-e7-grna3-s:caccgaacaggctgcagaccctga(seq id no.16)
170.lmna-e7-grna3-a:aaactcagggtctgcagcctgttc(seq id no.17)
171.lmna-e7-grna4-s:caccgtgaggccaagaaacaacttc(seq id no.18)
172.lmna-e7-grna4-a:aaacgaagttgtttcttggcctcac(seq id no.19)
173.lmna-e7-grna1-s、lmna-e7-grna1-a、lmna-e7-grna2-s、lmna-e7-grna2-a、lmna-e7-grna3-s、lmna-e7-grna3-a、lmna-e7-grna4-s、lmna-e7-grna4-a均为单链dna分子。
174.4.3.2将grna序列克隆到pkg-u6grna骨架载体上的方法
175.同实施例2中2.1.4。
176.4.3.3 grna载体构建
177.1)将合成的lmna-e7-grna1-s和lmna-e7-grna1-a混合并进行退火,得到具有粘性末端的双链dna分子。将具有粘性末端的双链dna分子和载体骨架连接,得到质粒pkg-u6grna(lmna-e7-grna1)。质粒pkg-u6grna(lmna-e7-grna1)将会在所转染的细胞中转录出与lmna-e7-grna1序列所对应的grna。
178.2)将合成的lmna-e7-grna2-s和lmna-e7-grna2-a混合并进行退火,得到具有粘性末端的双链dna分子。将具有粘性末端的双链dna分子和载体骨架连接,得到质粒pkg-u6grna(lmna-e7-grna2)。质粒pkg-u6grna(lmna-e7-grna2)将会在所转染的细胞中转录出与lmna-e7-grna2序列所对应的的grna。
179.3)将合成的lmna-e7-grna3-s和lmna-e7-grna3-a混合并进行退火,得到具有粘性末端的双链dna分子。将具有粘性末端的双链dna分子和载体骨架连接,得到质粒pkg-u6grna(lmna-e7-grna3)。质粒pkg-u6grna(lmna-e7-grna3)将会在所转染的细胞中转录出与lmna-e7-grna3序列所对应的grna。
180.4)将合成的lmna-e7-grna4-s和lmna-e7-grna4-a混合并进行退火,得到具有粘性末端的双链dna分子。将具有粘性末端的双链dna分子和载体骨架连接,得到质粒pkg-u6grna(lmna-e7-grna4)。质粒pkg-u6grna(lmna-e7-grna4)将会在所转染的细胞中转录出与lmna-e7-grna4序列所对应的grna。
181.4.3.3 grna载体鉴定
182.从lb平板上挑取单克隆置入加有相应抗生素的lb培养液内,37℃恒温摇床内培养12-16h后小提质粒后送通用公司测序,经序列比对确认pkg-u6grna(lmna-e7-grna1)、pkg-u6grna(lmna-e7-grna2)、pkg-u6grna(lmna-e7-grna3)和pkg-u6grna(lmna-e7-grna4)载体均构建成功。
183.4.4猪原代成纤维细胞制备
184.同实施例3中3.2。
185.4.5使用构建好的grna质粒、质粒(pkg-ge3)共转染猪原代成纤维细胞
186.4.5.1共转染分组情况
187.第一组:将质粒pkg-u6grna(lmna-e7-grna1)、质粒pkg-ge3共转染猪原代成纤维细胞。配比:约20万个猪原代成纤维细胞:0.92μg质粒pkg-u6grna(lmna-e7-grna1):1.08μg质粒pkg-ge3。
188.第二组:将质粒pkg-u6grna(lmna-e7-grna2)、质粒pkg-ge3共转染猪原代成纤维细胞。配比:约20万个猪原代成纤维细胞:0.92μg质粒pkg-u6grna(lmna-e7-grna2):1.08μg质粒pkg-ge3。
189.第三组:将质粒pkg-u6grna(lmna-e7-grna3)、质粒pkg-ge3共转染猪原代成纤维细胞。配比:约20万个猪原代成纤维细胞:0.92μg质粒pkg-u6grna(lmna-e7-grna3):1.08μg质粒pkg-ge3。
190.第四组:将质粒pkg-u6grna(lmna-e7-grna4)、质粒pkg-ge3共转染猪原代成纤维细胞。配比:约20万个猪原代成纤维细胞:0.92μg质粒pkg-u6grna(lmna-e7-grna4):1.08μg质粒pkg-ge3。
191.第五组:猪原代成纤维细胞,同等电转参数不加质粒进行电转染操作。
192.4.5.2共转染操作方法
193.同实施例3中3.3.2。
194.4.6 lmna基因不同grna靶点的编辑效率分析
195.4.6.1分别向步骤4.5.2中收集在1.5ml离心管中的5组细胞加入10μl kapa2g裂解液裂解细胞,得到释放出基因组dna的细胞裂解液
196.kapa2g裂解液配制体系如下:
197.10
×
extract buffer
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
1μl
198.enzyme
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
0.2μl
199.ddh2o
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
8.8μl
200.75℃15min—95℃5min—4℃,反应结束后细胞裂解液于-20℃保存;
201.4.6.2采用前述针对lmna基因e7的引物对lmna-e7grna-jdf1/lmna-e7grna-jdr1,并以上述细胞裂解液为dna模板,进行pcr扩增lmna基因靶点区域,检测细胞靶基因突变情况,目的pcr产物长度为619bp;
202.4.6.3使用常规pcr反应扩增lmna靶点基因;
203.4.6.4将pcr反应产物进行1%琼脂糖凝胶电泳,如图10,将目的产物及其附近产物切胶回收后送测序公司进行测序,然后将测序结果利用网页版synthego ice工具分析测序峰图得出lmna-e7-grna1、lmna-e7-grna2、lmna-e7-grna3、lmna-e7-grna4不同靶点的编辑效率依次为82%、39%、74%、25%。结果表明,lmna-e7-grna1和lmna-e7-grna3编辑效率较高。
204.实施例5制备lmna基因突变的从江香猪单细胞克隆
205.5.1 lmna基因高效靶点grna模板制备及转录
206.5.1.1使用实施例4中筛选的两个靶点grna
207.lmna-e7-grna1:ggatgagatgctgcgccgag(seq id no.8)
208.lmna-e7-grna3:gaacaggctgcagaccctga(seq id no.10)
209.5.1.2设计靶点grna转录模板的不同区段序列(由基因合成公司合成)
210.t7-grna1、t7-grna2、t7-grna3、t7-grna6序列同实施例3中步骤3.1.2;
211.lmna-g1t7-grna4:ttctagctctaaaacctcggcgcagcatctcatcccctatagtgagtcgtattaatttc(seq id no.20)
212.lmna-g1t7-grna5:cgccgaggttttagagctagaaatagcaagttaaaataaggctagtccgttatcaactt(seq id no.21)
213.lmna-g3t7-grna4:ttctagctctaaaactcagggtctgcagcctgttccctatagtgagtcgtattaatttc(seq id no.22)
214.lmna-g3t7-grna5:accctgagttttagagctagaaatagcaagttaaaataaggctagtccgttatcaactt(seq id no.23)
215.5.1.3转录模板的扩增
216.lmna-t7-grna1转录模板序列如seq id no.24所示,是使用t7-grna1、t7-grna2、t7-grna3、lmna-g1t7-grna4、lmna-g1t7-grna5、t7-grna6共计6条合成引物采用重叠延伸pcr扩增技术制作的,该序列中含有t7启动子,可启动相关序列的转录。扩增结果如图11所示,将目的条带切胶后按照fast pure gel dna extraction mini kit(vazyme,dc301)说明书进行操作,回收产物作为转录模板。
217.lmna-t7-grna3转录模板序列如seq id no.25所示,是使用t7-grna1、t7-grna2、t7-grna3、lmna-g3t7-grna4、lmna-g3t7-grna5、t7-grna6共计6条合成引物采用重叠延伸pcr扩增技术制作的,该序列中含有t7启动子,可启动相关序列的转录。扩增结果如图11所示,将目的条带切胶后按照fast pure gel dna extraction mini kit(vazyme,dc301)说明书进行操作,回收产物作为转录模板。
218.5.1.4高效grna的转录
219.以步骤5.1.3制备的转录模板按照transcript aid t7 high yield transcription kit(fermentas,k0441)和mega clear
tm transcription clean-up kit(thermo,am1908)说明书进行操作,所获产物即为可用于细胞电转的grna。
220.5.2合成含有lmna突变位点的单链dna
221.合成对应人lmna n195k和e203k氨基酸突变的单链dna作为donor dna,该单链dna除靶位点突变外还含有lmna-e7-grna1和lmna-e7-grna3靶点pam的同义突变,命名为lmna-mutant-ss130,序列如seq id no.26所示。
222.5.3猪原代成纤维细胞制备
223.同实施例3中3.2。
224.5.4转染猪原代成纤维细胞
225.使用转录的lmna-t7-grna1和lmna-t7-grna3、pkg-ge4-cas9蛋白、lmna-mutant-ss130共转染猪原代成纤维细胞。配比:约10万个猪原代成纤维细胞:1.77μg lmna-t7-grna1:1.77μg lmna-e7-grna3:2.46μg pkg-ge4-cas9蛋白:2μg lmna-mutant-ss130。共转染操作方法同实施例3中3.3.2。
226.5.5筛选lmna-mutant-ss130发生同源重组(hdr)的单细胞克隆株
227.5.5.1将步骤5.4所得电转48h的群体细胞,使用胰蛋白酶进行消化,完全培养基中
和,500g离心5min,去上清,将沉淀用200μl完全培养基重悬,并适当稀释,用口吸管挑取单细胞转移到每孔含100μl完全培养基的96孔板中,每孔放置一个细胞。
228.5.5.2 37℃、5%co2、5%o2的恒温培养箱中进行培养,每2~3天换一次细胞培养基,期间用显微镜观察每孔细胞生长情况,排除无细胞及非单细胞克隆的孔;
229.5.5.3待96孔板的孔中细胞长满孔底,使用胰蛋白酶消化并收集细胞,其中2/3细胞接种到含有完全培养基的6孔板中,剩余的1/3的细胞收集在1.5ml离心管中备后续基因型测定用;
230.5.5.4待6孔板长至80%汇合度时使用0.25%(gibco)的胰蛋白酶消化并收集细胞,使用细胞冻存液(90%完全培养基 10%dmso,体积比)将细胞冻存。
231.5.6单细胞克隆的鉴定
232.5.6.1在步骤5.5.3收集在1.5ml离心管中得到的细胞中加入10μl kapa2g裂解液裂解细胞,得到释放出基因组dna的细胞裂解液。
233.kapa2g裂解液配制体系如下:
234.10
×
extract buffer
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
1μl
235.enzyme
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
0.2μl
236.ddh2o
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
8.8μl
237.75℃15min—95℃5min—4℃,反应结束后细胞裂解液于-20℃保存;
238.5.6.2采用前述针对lmna基因e7的引物对lmna-e7grna-jdf1/lmna-e7grna-jdr1,并以上述细胞裂解液为dna模板,进行pcr扩增lmna靶点基因,检测单细胞克隆的靶基因突变情况,目的pcr产物长度为619bp;
239.5.6.3将pcr产物进行电泳,电泳结果如图12,泳道编号与单细胞克隆编号一致。回收pcr扩增产物并测序。
240.5.6.4将测序结果与lmna靶标位点突变序列信息进行比对,从而判断该单细胞克隆株是否为靶标位点成功突变株。
241.编号为1、2、6、16、19、26、32、37的单细胞克隆的基因型为野生型。编号为4、5、9、15、17、22、24、25、28、29、36、38、39的单细胞克隆的基因型为杂合突变型。编号为7、10、12、18、20、27、30、31、35的单细胞克隆的基因型为双等位基因不同变异的纯合突变型。编号为3、8、11、13、14、21、23、33、34、40的单细胞克隆的基因型为双等位基因相同变异的纯合突变型。其中,15、17的单细胞克隆为靶标位点点突变的杂合突变型,3、11、14、23、40的单细胞克隆为靶标位点点突变的纯合突变型。得到lmna基因编辑单细胞克隆的比率为80%,得到靶标位点点突变的单细胞克隆的比率为17.5%。
242.示例性的测序比对结果如图13至图17,其中图13是克隆号为lmna-ss130-1的正向测序与靶标位点突变序列的比对结果,为野生型;图14是克隆号为lmna-ss130-9的正向测序与靶标位点突变序列的比对结果,为杂合突变型;图15是克隆号为lmna-ss130-20的正向测序和反向测序同时与靶标位点突变序列的比对结果,为双等位基因不同变异的纯合突变型;图16是克隆号为lmna-ss130-8的正向测序与靶标位点突变序列的比对结果,为双等位基因相同变异的纯合突变型;图17是克隆号为lmna-ss130-15的正向测序与靶标位点突变序列的比对结果,为靶标位点点突变的杂合突变型;图18是克隆号为lmna-ss130-3的正向测序与靶标位点突变序列的比对结果,为靶位点点突变的纯合突变型。
243.通过具体序列的分析,lmna各单细胞克隆基因型如表1:
244.表1 lmna基因点突变单细胞克隆的基因型测定结果
245.246.[0247][0248]
以上对本发明进行了详述。对于本领域技术人员来说,在不脱离本发明的宗旨和
范围,以及无需进行不必要的实验情况下,可在等同参数、浓度和条件下,在较宽范围内实施本发明。虽然本发明给出了特殊的实施例,应该理解为,可以对本发明作进一步的改进。总之,按本发明的原理,本技术欲包括任何变更、用途或对本发明的改进,包括脱离了本技术中已公开范围,而用本领域已知的常规技术进行的改变。按以下附带的权利要求的范围,可以进行一些基本特征的应用。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。