1.本发明涉及再利用技术领域,具体涉及一种由废弃聚双环戊二烯制备色素炭黑的方法。
背景技术:
2.聚双环戊二烯是一种新型热固性工程塑料,拥有高的抗冲击强度、高的模量等优异的力学性能,以及良好的成型工艺进行成型性,特别适宜制成高强度、大面积的超薄制件。因此在交通车辆、工程机械、化工环保、国防军工、医疗和体育器材等领域得到了广泛的应用。但是,聚双环戊二烯废弃后难以再生利用,会对环境造成严重的不良影响。随着聚双环戊二烯材料的广泛地应用,其废弃后的再利用已经引起人们的广泛关注。
3.目前,对于废弃聚双环戊二烯制品的再利用主要采用热裂解技术使其转变成可燃的气体和液体,残渣为炭黑。然而,其成分非常复杂而难以成为普通的燃料使用,因而有可能成为潜在的二次污染源;残渣炭黑由于质量不佳而只能作为低端场景使用。对消除废弃聚双环戊二烯对环境的不良影响,并将其转变成具有更高附加值的产品的研究具有重要的意义。
4.聚双环戊二烯是含碳元素较高的聚合物,点燃时因不完全燃烧会产生大量絮状炭黑,因此可以根据这个特性将其转变成高质量的色素炭黑。色素炭黑与普通炭黑相比具有粒径小、黑度高的特点,在油墨、油漆、涂料、塑料等制品中作着色颜料使用,因而具有更高的价值。色素炭黑生产工艺主要是油炉法和槽法,前者原料为高芳烃油料,如乙烯焦油和蒽油等,后者为天然气、焦炉气或重质液态烃,生产工艺复杂,设备庞大复杂,成本高。若以废弃聚双环戊二烯为原料,采用不完全燃烧工艺制备高档色素炭黑在技术上是可行的,具有很大的成本优势。
5.但是,利用聚双环戊二烯不完全燃烧生产色素炭黑需要将炭黑气流经过过滤袋过滤得到,炭黑气流携带大量的热量,而现有的过滤袋的耐热性能不佳,易于被炭黑气流携带的热量损坏变形,导致过滤效果差,炭黑收率低,而且易于堵塞,若是为了防堵则需要增加滤网的孔径,导致过滤效果差且需要多次过滤,成本高。
6.如何改善现有的废弃聚双环戊二烯制备色素炭黑的方法中使用的过滤袋的耐热性能不佳,易于堵塞,导致过滤效果差,炭黑收率低,而且需要多次过滤,成本高是本发明的关键,因此,亟需一种由废弃聚双环戊二烯制备色素炭黑的方法来解决以上问题。
技术实现要素:
7.为了克服上述的技术问题,本发明的目的在于提供一种由废弃聚双环戊二烯制备色素炭黑的方法:通过将氮气、二氧化碳以及空气混合,得到混合燃气,将混合燃气通入燃烧室,将废弃聚双环戊二烯放进通入含有混合气体的燃烧室中并点燃,在缺氧状态下燃烧,产生的絮状炭黑,将含有絮状炭黑气流引入设有耐温过滤袋的通道,将其收集,得到色素炭黑,解决了现有的废弃聚双环戊二烯制备色素炭黑的方法中使用的过滤袋的耐热性能不
佳,易于堵塞,导致过滤效果差,炭黑收率低,而且需要多次过滤,成本高的问题。
8.本发明的目的可以通过以下技术方案实现:
9.一种由废弃聚双环戊二烯制备色素炭黑的方法,包括以下步骤:
10.步骤一:将氮气、二氧化碳以及空气混合,得到混合燃气,将混合燃气通入燃烧室;
11.步骤二:将废弃聚双环戊二烯放进通入含有混合气体的燃烧室中并点燃,在缺氧状态下燃烧,产生的絮状炭黑;
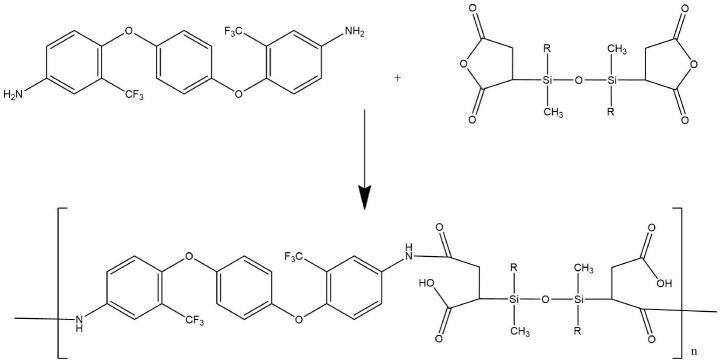
12.步骤三:将含有絮状炭黑气流引入设有耐温过滤袋的通道,将其收集,得到色素炭黑,产生的燃烧废气含有少量的二氧化碳和甲烷,再将燃烧废气引入二次燃烧室中混合空气进行二次燃烧,尾气引入活性炭吸附装置进行吸附后排入大气。
13.作为本发明进一步的方案:所述耐温过滤袋由以下步骤制备得到:
14.s1:将自制二酐单体、自制二胺单体放置于真空干燥箱,在温度为80-85℃的条件下干燥5-6h,之后将自制二酐单体和n,n-二甲基乙酰胺加入至安装有搅拌器、温度计的三口烧瓶中,在温度为25-30℃,搅拌速率为300-500r/min的条件下搅拌20-30min,之后将自制二胺单体分3-5次加入至三口烧瓶中,每次加入量相同,加入完毕后继续搅拌反应5-6h,之后降温至5-10℃,得到耐温纺丝液;
15.反应过程如下:
[0016][0017]
s2:将纺丝液静电纺丝,形成耐温纤维膜,将耐温纤维膜放置于真空干燥箱中,在温度为80-85℃的条件下干燥3-4h,之后放置于马沸炉中,依次在温度为100℃、200℃、300℃以及350℃的条件下保温热处理30-40min,耐温纤维膜分子内部的氨基与羧基脱水在分子内部闭环,得到耐温过滤材料;
[0018]
反应过程如下:
[0019][0020]
s3:将耐温过滤材料经过裁切、缝制,得到耐温过滤袋。
[0021]
作为本发明进一步的方案:所述自制二酐单体、自制二胺单体以及n,n-二甲基乙酰胺的用量比为0.11mol:0.1mol:200-250ml;所述氮气、二氧化碳以及空气的流量比为1-2.5:1-2.5:5-8。
[0022]
作为本发明进一步的方案:所述自制二酐单体的制备方法包括以下步骤:
[0023]
a1:将1,2,3-三氯苯、环丁砜、氟化钾以及四丁基溴化铵加入至安装有搅拌器、温度计以及导气管的三口烧瓶中,通入氮气保护,在温度为170-175℃,压力为2-2.2mpa,搅拌速率为300-400r/min的条件下搅拌反应3-5h,反应结束后将反应产物蒸馏去除环丁砜,之后冷却至室温,得到中间体1;
[0024]
反应过程如下:
[0025][0026]
a2:将中间体1、二氯乙烷加入至安装有搅拌器、温度计以及恒压滴液漏斗的三口烧瓶中,在温度为5-10℃,搅拌速率为300-400r/min的条件下搅拌20-30min,之后加入溴化钠、磷酸二氢钠以及去离子水,之后边搅拌边逐滴加入次氯酸钠溶液,控制滴加速率为1-2滴/s,滴加完毕后在温度为10-13℃的条件下继续搅拌反应3-5h,反应结束后将反应产物静置分层,将有机相用蒸馏水洗涤2-3次,之后用无水硫酸镁干燥,之后旋转蒸发去除溶剂,得到中间体2;
[0027]
反应过程如下:
[0028][0029]
a3:将中间体2、无水四氢呋喃以及镁屑加入至安装有搅拌器、温度计、回流冷凝管以及恒压滴液漏斗的四口烧瓶中,在温度为70-75℃,搅拌速率为300-400r/min的条件下搅拌反应2-3h,之后降温至5-10℃的条件下边搅拌边逐滴加入1,3,5,7-四甲基环四硅氧烷,控制滴加速率为1-2滴/s,滴加完毕后继续搅拌反应1-2h,之后边搅拌边逐滴加入盐酸溶液,控制滴加速率为1-2滴/s,滴加完毕后继续搅拌反应1-2h,反应结束后将反应产物静置分层,将有机相旋转蒸发去除溶剂,得到中间体3;
[0030]
反应过程如下:
[0031][0032]
a4:将马来酸酐、甲醇以及对甲基苯磺酸加入至安装有搅拌器、温度计以及回流冷凝管的三口烧瓶中,在搅拌速率为200-300r/min的条件下边搅拌边升温至回流并搅拌反应10-15h,反应结束后将反应产物蒸馏去除未反应的甲醇,得到中间体4;
[0033]
反应过程如下:
[0034][0035]
a5:将中间体4、无水甲苯、氯铂酸溶液加入至安装有搅拌器、温度计、导气管以及恒压滴液漏斗的三口烧瓶中,通入氮气保护,在温度为85-90℃,搅拌速率为300-400r/min的条件下搅拌5-10min,之后边搅拌边逐滴加入中间体3溶液,控制滴加速率为1-2滴/s,滴加完毕后继续搅拌反应10-15h,反应结束后将反应产物冷却至室温,得到中间体5溶液,之后向中间体5溶液中加入二氯甲烷、无水甲醇以及氢氧化钠,在温度为25-30℃的条件下搅拌反应20-30h,反应结束后将反应产物加入至蒸馏水中静置分层,将水相降温至0℃,之后用浓盐酸调节ph为0.5-1,之后静置20-30h,真空抽滤,将滤饼用蒸馏水洗涤2-3次,之后放置于真空干燥箱中,在温度为50-60℃条件下干燥10-15h,得到中间体6;
[0036]
反应过程如下:
[0037][0038]
a6:将中间体6、乙酸酐以及二甲苯加入至安装有搅拌器、温度计以及回流冷凝管的三口烧瓶中,在温度为110-115℃,搅拌速率为300-350r/min的条件下搅拌反应20-30h,反应结束后加入活性炭,在温度为60-70℃的条件下搅拌2-3h,之后真空抽滤,将滤液加入至无水乙醚中析出沉淀,真空抽滤,将滤饼用石油醚洗涤2-3次,之后放置于真空干燥箱中,在温度为120-130℃条件下干燥10-15h,得到自制二酐单体。
[0039]
反应过程如下:
[0040][0041]
作为本发明进一步的方案:步骤a1中的所述1,2,3-三氯苯、环丁砜、氟化钾以及四丁基溴化铵的用量比为0.1mol:20-30ml:0.35-0.4mol:0.2-0.3g。
[0042]
作为本发明进一步的方案:步骤a2中的所述中间体1、二氯乙烷、溴化钠、磷酸二氢钠、去离子水以及次氯酸钠溶液的用量比为0.1mol:25-30ml:0.1mol:0.1mol:30-40ml:90-100g,所述次氯酸钠溶液的质量分数为10%。
[0043]
作为本发明进一步的方案:步骤a3中的所述中间体2、无水四氢呋喃、镁屑、1,3,5,7-四甲基环四硅氧烷以及盐酸溶液的用量比为0.1mol:30-40ml:0.11-0.13mol:0.25-0.3mol:18-22ml,所述盐酸溶液的质量分数为20-25%。
[0044]
作为本发明进一步的方案:步骤a4中的所述马来酸酐、甲醇以及对甲基苯磺酸的用量比为0.1mol:20-25ml:0.8-1.2g。
[0045]
作为本发明进一步的方案:步骤a5中的所述中间体4、无水甲苯、氯铂酸溶液、中间体3溶液、二氯甲烷、无水甲醇以及氢氧化钠的用量比为0.02mol:15-20ml:2-3ml:10-15ml:
150-200ml:25-30ml:2.5-4.0g,所述氯铂酸溶液为氯铂酸按照1g:50ml溶解于无水乙醇所形成的溶液,所述中间体3溶液为中间体3按照0.01ml:10ml溶解于无水基本所形成的溶液,所述浓盐酸的质量分数为35-38%。
[0046]
作为本发明进一步的方案:步骤a6中的所述中间体6、乙酸酐、二甲苯以及活性炭的用量比为0.1mol:60-80ml:30-40ml:3-5g。
[0047]
作为本发明进一步的方案:所述自制二胺单体的制备方法包括以下步骤:
[0048]
b1:将对苯二酚、n,n-二甲基乙酰胺加入至安装有搅拌器、温度计、回流冷凝管以及恒压滴液漏斗的四口烧瓶中,在温度为120-130℃,搅拌速率为350-450r/min的条件下搅拌10-15min,之后加入无水碳酸钾继续搅拌10-15min,之后边搅拌边逐滴加入2-氯-5-硝基三氟甲苯溶液,控制滴加速率为1-2滴/s,滴加完毕后升温至150-155℃的条件下继续搅拌反应20-30h,反应结束后将反应产物冷却至室温,真空抽滤,将滤液加入至乙醇溶液中析出沉淀,真空抽滤,将滤饼放置于真空干燥箱中,在温度为60-70℃的条件下干燥8-10h,得到中间体7;
[0049]
反应过程如下:
[0050][0051]
b2:将还原铁粉、乙醇溶液以及浓盐酸加入至安装有搅拌器、温度计以及回流冷凝管的三口烧瓶中,在温度为75-80℃,搅拌速率为300-400r/min的条件下搅拌25-30min,之后降温至55-65℃的条件下加入中间体7,之后边搅拌边升温至80-85℃,控制升温速率为2-3℃/min,之后继续搅拌反应7-9h,反应结束后将反应产物真空抽滤,将滤液用氢氧化钠溶液调节ph为8-8.5,静置8-10h,之后真空抽滤,将滤液加入至蒸馏水中析出沉淀,真空抽滤,将滤饼放置于真空干燥箱中,在温度为60-70℃的条件下干燥5-7h,得到自制二胺单体。
[0052]
反应过程如下:
[0053][0054]
作为本发明进一步的方案:步骤b1中的所述对苯二酚、n,n-二甲基乙酰胺、无水碳酸钾以及2-氯-5-硝基三氟甲苯溶液的用量比为0.1mol:80-100ml:0.11-0.12mol:0.22-0.24mol,所述乙醇溶液为无水乙醇与去离子水按照体积比为1:1的混合物。
[0055]
作为本发明进一步的方案:步骤b2中的所述还原铁粉、乙醇溶液、浓盐酸以及中间体7的用量比为0.4-0.5mol:100-120ml:4-6ml:0.2mol,所述乙醇溶液的体积分数80%,所述浓盐酸的质量分数为36-38%,所述氢氧化钠溶液的摩尔浓度为1-2mol/l。
[0056]
本发明的有益效果:
[0057]
本发明的一种由废弃聚双环戊二烯制备色素炭黑的方法,通过将氮气、二氧化碳以及空气混合,得到混合燃气,将混合燃气通入燃烧室,将废弃聚双环戊二烯放进通入含有混合气体的燃烧室中并点燃,在缺氧状态下燃烧,产生的絮状炭黑,将含有絮状炭黑气流引
入设有耐温过滤袋的通道,将其收集,得到色素炭黑;该方法中利用氮气、二氧化碳与空气混合,提供燃烧所需要的氧气的同时增加不燃烧的氮气、二氧化碳,从而使得聚双环戊二烯能够点燃但燃烧不完全,从而会产生大量絮状炭黑,然后利用耐温过滤袋进行过滤,耐温过滤袋的过滤效果好且耐高温,同时具有不易粘附的优点,从而能够将炭黑充分收集,且不会堵塞耐温过滤袋,从而长效保持优良的过滤效果;
[0058]
在制备色素炭黑的过程中也制备了一种耐温过滤袋,首先利用氟化钾将1,2,3-三氯苯进行氟化,引入大量的f原子,得到中间体1,之后利用溴化钠对中间体1进行溴化,从而引入溴原子,得到中间体2,之后中间体2发生格氏反应从而引入si-o-si键以及si-h键,得到中间体3,前者赋予中间3一定的柔性以及低表面能,后者与马来酸酐与甲醇的产物中间体4进行硅氢加成反应生成中间体5,之后酯基水解形成羧基,得到中间体6,之后中间体6脱水形成酸酐,得到自制二酐单体,然后利用对苯二酚上的羟基与2-氯-5-硝基三氟甲苯上的氯原子发生亲核取代反应,从而引入两个硝基,得到中间体7,之后利用还原铁粉将中间体7上的硝基还原成氨基,得到自制二胺单体,最后利用自制二酐单体、自制二胺单体脱水形成酰胺类聚合物,形成纺丝液,最后将纺丝液纺丝形成纤维膜,再将纤维膜热处理使得酰胺类聚合物分子内部的氨基与羧基脱水在分子内部闭环,得到耐温过滤材料;该耐温过滤材料的分子链上含有大量的环状结构,包括苯环以及含氮杂环,环状结构稳定性高,赋予了耐温过滤材料一定的耐高温性能,而且该耐温过滤材料的分子上含有大量的si-o-si键以及c-f键,键能大,而且si-o-si键以及c-f键极性大,能够将连接的烃基基团起到保护屏蔽作用,进一步的提升耐温过滤材料的耐高温性能,而且氟原子核对其核外电子和成键电子云具有较强的束缚能力,c-f键可极化性低,含有碳氟键的聚合物分子之间作用力小,从而使得含氟的聚合物表面张力小,具有较强的疏水拒油的能力,从而赋予了该耐温过滤材料良好的抗粘附性能,从而使得炭黑不易粘附在耐温过滤材料上,且不易堵塞耐温过滤材料;因此,在大量的环状结构、si-o-si键以及c-f键的协同作用下赋予了耐温过滤材料良好的耐高温性能以及防堵塞效果。
具体实施方式
[0059]
下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
[0060]
实施例1:
[0061]
本实施例为一种自制二酐单体的制备方法,包括以下步骤:
[0062]
a1:将0.1mol 1,2,3-三氯苯、20ml环丁砜、0.35mol氟化钾以及0.2g四丁基溴化铵加入至安装有搅拌器、温度计以及导气管的三口烧瓶中,通入氮气保护,在温度为170℃,压力为2mpa,搅拌速率为300r/min的条件下搅拌反应3h,反应结束后将反应产物蒸馏去除环丁砜,之后冷却至室温,得到中间体1;
[0063]
a2:将0.1mol中间体1、25ml二氯乙烷加入至安装有搅拌器、温度计以及恒压滴液漏斗的三口烧瓶中,在温度为5℃,搅拌速率为300r/min的条件下搅拌20min,之后加入0.1mol溴化钠、0.1mol磷酸二氢钠以及30ml去离子水,之后边搅拌边逐滴加入90g质量分数
为10%的次氯酸钠溶液,控制滴加速率为1滴/s,滴加完毕后在温度为10℃的条件下继续搅拌反应3h,反应结束后将反应产物静置分层,将有机相用蒸馏水洗涤2次,之后用无水硫酸镁干燥,之后旋转蒸发去除溶剂,得到中间体2;
[0064]
a3:将0.1mol中间体2、30ml无水四氢呋喃以及0.11mol镁屑加入至安装有搅拌器、温度计、回流冷凝管以及恒压滴液漏斗的四口烧瓶中,在温度为70℃,搅拌速率为300r/min的条件下搅拌反应2h,之后降温至5℃的条件下边搅拌边逐滴加入0.25mol 1,3,5,7-四甲基环四硅氧烷,控制滴加速率为1滴/s,滴加完毕后继续搅拌反应1h,之后边搅拌边逐滴加入18ml质量分数为20%的盐酸溶液,控制滴加速率为1滴/s,滴加完毕后继续搅拌反应1h,反应结束后将反应产物静置分层,将有机相旋转蒸发去除溶剂,得到中间体3;
[0065]
a4:将0.1mol马来酸酐、20ml甲醇以及0.8g对甲基苯磺酸加入至安装有搅拌器、温度计以及回流冷凝管的三口烧瓶中,在搅拌速率为200r/min的条件下边搅拌边升温至回流并搅拌反应10h,反应结束后将反应产物蒸馏去除未反应的甲醇,得到中间体4;
[0066]
a5:将0.02mol中间体4、15ml无水甲苯、2ml氯铂酸按照1g:50ml溶解于无水乙醇所形成的氯铂酸溶液加入至安装有搅拌器、温度计、导气管以及恒压滴液漏斗的三口烧瓶中,通入氮气保护,在温度为85℃,搅拌速率为300r/min的条件下搅拌5min,之后边搅拌边逐滴加入10ml中间体3按照0.01ml:10ml溶解于无水基本所形成的中间体3溶液,控制滴加速率为1滴/s,滴加完毕后继续搅拌反应10h,反应结束后将反应产物冷却至室温,得到中间体5溶液,之后向中间体5溶液中加入150ml二氯甲烷、25ml无水甲醇以及2.5g氢氧化钠,在温度为25℃的条件下搅拌反应20h,反应结束后将反应产物加入至蒸馏水中静置分层,将水相降温至0℃,之后用质量分数为35%的浓盐酸调节ph为0.5,之后静置20h,真空抽滤,将滤饼用蒸馏水洗涤2次,之后放置于真空干燥箱中,在温度为50℃条件下干燥10h,得到中间体6;
[0067]
a6:将0.1mol中间体6、60ml乙酸酐以及30ml二甲苯加入至安装有搅拌器、温度计以及回流冷凝管的三口烧瓶中,在温度为110℃,搅拌速率为300r/min的条件下搅拌反应20h,反应结束后加入3g活性炭,在温度为60℃的条件下搅拌2h,之后真空抽滤,将滤液加入至无水乙醚中析出沉淀,真空抽滤,将滤饼用石油醚洗涤2次,之后放置于真空干燥箱中,在温度为120℃条件下干燥10h,得到自制二酐单体。
[0068]
实施例2:
[0069]
本实施例为一种自制二酐单体的制备方法,包括以下步骤:
[0070]
a1:将0.1mol 1,2,3-三氯苯、30ml环丁砜、0.4mol氟化钾以及0.3g四丁基溴化铵加入至安装有搅拌器、温度计以及导气管的三口烧瓶中,通入氮气保护,在温度为175℃,压力为2.2mpa,搅拌速率为400r/min的条件下搅拌反应5h,反应结束后将反应产物蒸馏去除环丁砜,之后冷却至室温,得到中间体1;
[0071]
a2:将0.1mol中间体1、30ml二氯乙烷加入至安装有搅拌器、温度计以及恒压滴液漏斗的三口烧瓶中,在温度为10℃,搅拌速率为400r/min的条件下搅拌30min,之后加入0.1mol溴化钠、0.1mol磷酸二氢钠以及40ml去离子水,之后边搅拌边逐滴加入100g质量分数为10%的次氯酸钠溶液,控制滴加速率为2滴/s,滴加完毕后在温度为13℃的条件下继续搅拌反应5h,反应结束后将反应产物静置分层,将有机相用蒸馏水洗涤3次,之后用无水硫酸镁干燥,之后旋转蒸发去除溶剂,得到中间体2;
[0072]
a3:将0.1mol中间体2、40ml无水四氢呋喃以及0.13mol镁屑加入至安装有搅拌器、
温度计、回流冷凝管以及恒压滴液漏斗的四口烧瓶中,在温度为75℃,搅拌速率为400r/min的条件下搅拌反应3h,之后降温至10℃的条件下边搅拌边逐滴加入0.3mol 1,3,5,7-四甲基环四硅氧烷,控制滴加速率为2滴/s,滴加完毕后继续搅拌反应2h,之后边搅拌边逐滴加入22ml质量分数为25%的盐酸溶液,控制滴加速率为2滴/s,滴加完毕后继续搅拌反应2h,反应结束后将反应产物静置分层,将有机相旋转蒸发去除溶剂,得到中间体3;
[0073]
a4:将0.1mol马来酸酐、25ml甲醇以及1.2g对甲基苯磺酸加入至安装有搅拌器、温度计以及回流冷凝管的三口烧瓶中,在搅拌速率为300r/min的条件下边搅拌边升温至回流并搅拌反应15h,反应结束后将反应产物蒸馏去除未反应的甲醇,得到中间体4;
[0074]
a5:将0.02mol中间体4、20ml无水甲苯、3ml氯铂酸按照1g:50ml溶解于无水乙醇所形成的氯铂酸溶液加入至安装有搅拌器、温度计、导气管以及恒压滴液漏斗的三口烧瓶中,通入氮气保护,在温度为90℃,搅拌速率为400r/min的条件下搅拌10min,之后边搅拌边逐滴加入15ml中间体3按照0.01ml:10ml溶解于无水基本所形成的中间体3溶液,控制滴加速率为2滴/s,滴加完毕后继续搅拌反应15h,反应结束后将反应产物冷却至室温,得到中间体5溶液,之后向中间体5溶液中加入200ml二氯甲烷、30ml无水甲醇以及4.0g氢氧化钠,在温度为30℃的条件下搅拌反应30h,反应结束后将反应产物加入至蒸馏水中静置分层,将水相降温至0℃,之后用质量分数为38%的浓盐酸调节ph为1,之后静置30h,真空抽滤,将滤饼用蒸馏水洗涤3次,之后放置于真空干燥箱中,在温度为60℃条件下干燥15h,得到中间体6;
[0075]
a6:将0.1mol中间体6、80ml乙酸酐以及40ml二甲苯加入至安装有搅拌器、温度计以及回流冷凝管的三口烧瓶中,在温度为115℃,搅拌速率为350r/min的条件下搅拌反应30h,反应结束后加入5g活性炭,在温度为0℃的条件下搅拌3h,之后真空抽滤,将滤液加入至无水乙醚中析出沉淀,真空抽滤,将滤饼用石油醚洗涤3次,之后放置于真空干燥箱中,在温度为130℃条件下干燥15h,得到自制二酐单体。
[0076]
实施例3:
[0077]
本实施例为一种自制二胺单体的制备方法,包括以下步骤:
[0078]
b1:将0.1mol对苯二酚、80mln,n-二甲基乙酰胺加入至安装有搅拌器、温度计、回流冷凝管以及恒压滴液漏斗的四口烧瓶中,在温度为120℃,搅拌速率为350r/min的条件下搅拌10min,之后加入0.11mol无水碳酸钾继续搅拌10min,之后边搅拌边逐滴加入0.22mol 2-氯-5-硝基三氟甲苯溶液,控制滴加速率为1滴/s,滴加完毕后升温至150℃的条件下继续搅拌反应20h,反应结束后将反应产物冷却至室温,真空抽滤,将滤液加入至无水乙醇与去离子水按照体积比为1:1混合而成的乙醇溶液中析出沉淀,真空抽滤,将滤饼放置于真空干燥箱中,在温度为60℃的条件下干燥8h,得到中间体7;
[0079]
b2:将0.4mol还原铁粉、100ml体积分数80%的乙醇溶液以及4ml质量分数为36%的浓盐酸加入至安装有搅拌器、温度计以及回流冷凝管的三口烧瓶中,在温度为75℃,搅拌速率为300r/min的条件下搅拌25min,之后降温至55℃的条件下加入0.2mol中间体7,之后边搅拌边升温至80℃,控制升温速率为2℃/min,之后继续搅拌反应7h,反应结束后将反应产物真空抽滤,将滤液用摩尔浓度为1mol/l的氢氧化钠溶液调节ph为8,静置8h,之后真空抽滤,将滤液加入至蒸馏水中析出沉淀,真空抽滤,将滤饼放置于真空干燥箱中,在温度为60℃的条件下干燥5h,得到自制二胺单体。
[0080]
实施例4:
[0081]
本实施例为一种自制二胺单体的制备方法,包括以下步骤:
[0082]
b1:将0.1mol对苯二酚、100mln,n-二甲基乙酰胺加入至安装有搅拌器、温度计、回流冷凝管以及恒压滴液漏斗的四口烧瓶中,在温度为130℃,搅拌速率为450r/min的条件下搅拌15min,之后加入0.12mol无水碳酸钾继续搅拌15min,之后边搅拌边逐滴加入0.24mol 2-氯-5-硝基三氟甲苯溶液,控制滴加速率为2滴/s,滴加完毕后升温至155℃的条件下继续搅拌反应30h,反应结束后将反应产物冷却至室温,真空抽滤,将滤液加入至无水乙醇与去离子水按照体积比为1:1混合而成的乙醇溶液中析出沉淀,真空抽滤,将滤饼放置于真空干燥箱中,在温度为70℃的条件下干燥10h,得到中间体7;
[0083]
b2:将0.5mol还原铁粉、120ml体积分数80%的乙醇溶液以及6ml质量分数为38%的浓盐酸加入至安装有搅拌器、温度计以及回流冷凝管的三口烧瓶中,在温度为80℃,搅拌速率为400r/min的条件下搅拌30min,之后降温至65℃的条件下加入0.2mol中间体7,之后边搅拌边升温至85℃,控制升温速率为3℃/min,之后继续搅拌反应9h,反应结束后将反应产物真空抽滤,将滤液用摩尔浓度为2mol/l的氢氧化钠溶液调节ph为8.5,静置10h,之后真空抽滤,将滤液加入至蒸馏水中析出沉淀,真空抽滤,将滤饼放置于真空干燥箱中,在温度为70℃的条件下干燥7h,得到自制二胺单体。
[0084]
实施例5:
[0085]
本实施例为所述耐温过滤袋由以下步骤制备得到:
[0086]
s1:将0.11mol来自于实施例3中的自制二酐单体、0.1mol来自于实施例5中的自制二胺单体放置于真空干燥箱,在温度为80℃的条件下干燥5h,之后将自制二酐单体和250mln,n-二甲基乙酰胺加入至安装有搅拌器、温度计的三口烧瓶中,在温度为25℃,搅拌速率为300r/min的条件下搅拌20min,之后将自制二胺单体分3次加入至三口烧瓶中,每次加入量相同,加入完毕后继续搅拌反应5h,之后降温至5℃,得到耐温纺丝液;
[0087]
s2:将纺丝液静电纺丝,形成耐温纤维膜,将耐温纤维膜放置于真空干燥箱中,在温度为80℃的条件下干燥3h,之后放置于马沸炉中,依次在温度为100℃、200℃、300℃以及350℃的条件下保温热处理30min,耐温纤维膜分子内部的氨基与羧基脱水在分子内部闭环,得到耐温过滤材料;
[0088]
s3:将耐温过滤材料经过裁切、缝制,得到耐温过滤袋。
[0089]
实施例6:
[0090]
本实施例为所述耐温过滤袋由以下步骤制备得到:
[0091]
s1:将0.11mol来自于实施例2中的自制二酐单体、0.1mol来自于实施例4中的自制二胺单体放置于真空干燥箱,在温度为85℃的条件下干燥6h,之后将自制二酐单体和200mln,n-二甲基乙酰胺加入至安装有搅拌器、温度计的三口烧瓶中,在温度为30℃,搅拌速率为500r/min的条件下搅拌30min,之后将自制二胺单体分5次加入至三口烧瓶中,每次加入量相同,加入完毕后继续搅拌反应6h,之后降温至10℃,得到耐温纺丝液;
[0092]
s2:将纺丝液静电纺丝,形成耐温纤维膜,将耐温纤维膜放置于真空干燥箱中,在温度为85℃的条件下干燥4h,之后放置于马沸炉中,依次在温度为100℃、200℃、300℃以及350℃的条件下保温热处理40min,耐温纤维膜分子内部的氨基与羧基脱水在分子内部闭环,得到耐温过滤材料;
[0093]
s3:将耐温过滤材料经过裁切、缝制,得到耐温过滤袋。
[0094]
将实施例5-6的耐温过滤袋的性能进行检测,检测结果如下表所示:
[0095][0096]
参阅上表数据,可以得知,本发明中的耐温过滤袋的过滤效率高,过滤阻力低,表示该耐温过滤袋过滤效率高同时透气性好,从而表示耐温过滤袋的过滤效果好,而且该耐温过滤袋的表面能低,表示该耐温过滤袋防粘性好,能够使得炭黑不易粘附,从而收率高且不易于堵塞,同时该耐温过滤袋的起始失重温度高,且300℃、600℃失重低,表示该耐温过滤袋具有良好的耐高温性能。
[0097]
实施例7:
[0098]
本实施例为一种由废弃聚双环戊二烯制备色素炭黑的方法,包括以下步骤:
[0099]
步骤一:将氮气、二氧化碳以及空气按照流量比为1.5:1.5:7混合,得到混合燃气,将混合燃气通入燃烧室;
[0100]
步骤二:将废弃聚双环戊二烯放进通入含有混合气体的燃烧室中并点燃,在缺氧状态下燃烧,产生的絮状炭黑;
[0101]
步骤三:将含有絮状炭黑气流引入设有来自于实施例6中的耐温过滤袋的通道,将其收集,得到色素炭黑,产生的燃烧废气含有少量的二氧化碳和甲烷,再将燃烧废气引入二次燃烧室中混合空气进行二次燃烧,尾气引入活性炭吸附装置进行吸附后排入大气。
[0102]
在本说明书的描述中,参考术语“一个实施例”、“示例”、“具体示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
[0103]
以上内容仅仅是对本发明所作的举例和说明,所属本技术领域的技术人员对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,只要不偏离发明或者超越本权利要求书所定义的范围,均应属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。