一种pqq的制备方法
技术领域
1.本发明涉及一种化合物的制备方法,特别涉及一种pqq的制备方法。
背景技术:
2.pqq的中文简称为吡咯喹啉醌,化学名称为4,5-二氧代-1h-吡咯[2,3-f]喹啉-2,7,9-三甲酸,是20世纪70年代末发现的一种氧化还原酶的新辅酶,是继黄素核苷酸和烟酰胺核苷酸之后发现的第三种辅酶。作为一种氧化还原酶辅基,pqq几乎存在于所有生物组织中,是一种新型的水溶性b族维生素。研究表明pqq具有刺激机体生长、促进神经生长因子合成、防护肝损伤、调解机体自由基水平、提高细菌对毒性和辐射等极端条件耐受性等多种功能,也作为动物生长发育的必需因子。美国和欧盟已经将吡咯喹啉醌钠盐列为高安全性的新型膳食补充剂。
[0003]
pqq为含有吡咯并喹啉醌结构的三元羧酸,结构比较复杂。自从1979年pqq分子结构被确定以来,其全合成就成为了有机合成的热点之一。文献j.org.chem.1981,46(21),4317-4319首次报道了pqq的全合成,虽然这一全合成路线需要经过12步反应,总收率仅有2%,但为以后的pqq全合成奠定了基础。文献j.am.chem.soc.1981,103,18,5599-5600通过10步反应,以20%的总产率得到了高纯的pqq。但该方法只适合毫克级别的样品的制备。随后,多篇文献如j.org.chem.1985,50,10,1688-1695,j.am.chem.soc.1985,107,5555,tetrahedron lett.1988,29,3709;chim.acta 1993,76,988;helvetica chimica acta 1996,79,658;tetrahedron,2005,61,12330等也通过不同的策略全合成了pqq。这些方法也都存在工艺路线长,总收率低,使用昂贵的试剂、成本高等缺陷。近年来科技工作者对pqq的合成方法也不断地研究,如文献cn 104557921 a以2-甲氧基-5-硝基苯胺盐酸盐为原料,经10步合成了pqq;文献cn 108329313 a以丙酮酸乙酯为起始原料,经8步合成了pqq;cn 110981873 a以4-甲基-5-硝基-2-氟苯胺为原料,经5步合成了pqq等。这些方法虽进行了一些改进,但仍存在工艺路线长,总收率低,工艺操作复杂、使用昂贵的试剂、成本高,难以实现规模生产等缺陷。
技术实现要素:
[0004]
发明目的:本发明旨在提供一种原料廉价,工艺路线为五步反应的pqq的制备方法。
[0005]
技术方案:本发明所述的pqq的制备方法,包括以下步骤:
[0006]
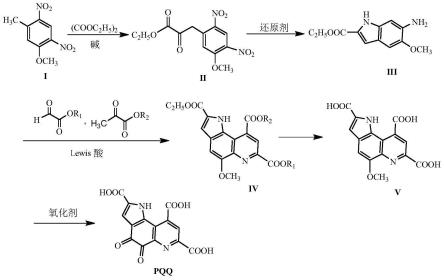
(1)化合物i 5-甲基-2,4-二硝基苯甲醚与草酸二乙酯在碱的作用下发生缩合反应,制得化合物ii 3-(5-甲氧基-2,4-二硝基)苯基丙酮酸乙酯;
[0007]
(2)化合物ii在还原剂作用下发生还原反应关环,制得化合物iii 6-氨基-5-甲氧基-1h-吲哚-2-甲酸乙酯;
[0008]
(3)化合物iii与乙醛酸酯和丙酮酸酯在lewis酸催化剂作用下在氧气氛围下发生三组分一锅反应,制得化合物iv 5-甲氧基-1h-吡咯[2,3-f]喹啉-2,7,9-三甲酸酯;
[0009]
(4)化合物iv发生酯水解反应,制得化合物v 5-甲氧基-1h-吡咯[2,3-f]喹啉-2,7,9-三甲酸;
[0010]
(5)化合物v在氧化剂ce(nh4)2(no3)6作用下发生氧化反应即制得pqq;
[0011]
具体的合成路线如下:
[0012][0013]
其中,r1、r2为c1~c6的烷基或c3~c6的环烷基。
[0014]
优选的,步骤(1)中,草酸二乙酯相对于化合物i过量,溶剂为乙醇,回流反应;碱选自甲醇钠、乙醇钠、异丙醇钠、叔丁醇钠、氢化钠、氨基钠或三苯甲基钠。
[0015]
优选的,步骤(2)中,所述还原剂为水合肼/raney ni,回流反应至反应结束,反应过程采用tlc[展开剂:v(石油醚):v(乙酸乙酯)=1:1]跟踪反应进程。
[0016]
优选的,步骤(2)中,所述还原剂为水合肼-活性炭/fecl3 alcl3,反应温度为70~80℃;所述还原剂中三氯化铁、三氯化铝与化合物ii的物质的量的比为0.1~0.2:0.01~0.02:1;所述水合肼浓度为质量分数80%;反应过程采用tlc[展开剂:v(石油醚):v(乙酸乙酯)=1:1]跟踪反应进程。
[0017]
优选的,步骤(2)中,所述还原剂为10%pd-c/h2、raney ni/h2、10%pd-c/甲酸铵、铁粉或锌粉时,溶剂为乙醇,反应温度为55~65℃,还包括添加剂;所述添加剂为浓盐酸、醋酸、浓硫酸或对甲苯磺酸;所述浓盐酸的浓度为质量分数36~38%的盐酸;所述浓硫酸为质量分数98%的硫酸。
[0018]
优选的,步骤(3)中,反应温度为40~100℃。
[0019]
优选的,步骤(3)中,化合物iii与丙酮酸酯和乙醛酸酯的摩尔比为1~1.5:1~1.2:1。
[0020]
优选的,步骤(3)中,溶剂为乙腈、甲苯、苯、n,n-二甲基甲酰胺(dmf)或二甲基亚砜(dmso)。
[0021]
优选的,步骤(3)中,所述lewis酸催化剂与乙醛酸酯物质的量之比0.005~0.15:1;所述lewis酸催化剂为cucl2、cubr2、cui2、cu(otf)2、cu(oocch3)2、zn(otf)2、zncl2、znbr2、
fecl3.6h2o或febr3·
6h2o。
[0022]
有益效果:与现有技术相比,本发明具有如下显著优点:(1)本方法以廉价的5-甲基-2,4-二硝基苯甲醚为起始原料,通过五步即制备了pqq,总收率最高达39%;(2)反应条件温和,操作简单,反应原料廉价、易得。
附图说明
[0023]
图1为本发明合成路线;
[0024]
图2为合成的pqq二钠盐的hplc图;
[0025]
图3为合成的pqq二钠盐的的核磁氢谱图。
具体实施方式
[0026]
下面结合实施例对本发明的技术方案作进一步说明。
[0027]
实施例1
[0028]
化合物ii 3-(5-甲氧基-2,4-二硝基)苯基丙酮酸乙酯的制备:将110mmol乙醇钠和100ml乙醇加入反应瓶中,搅拌混合均匀。将反应体系温度降至10℃左右,将100mmol化合物i加入,保温搅拌反应0.5h,再将105mmol草酸二乙酯滴入,滴完后将反应体系温度升至回流,并保温搅拌反应1h,停止反应,减压蒸去溶剂,加入100ml蒸馏水和100ml乙酸乙酯,搅拌后静置,分去水层,用乙酸乙酯洗涤水层,合并有机层,用无水硫酸钠干燥后减压蒸去溶剂,得粗产物,该粗产物不经纯化,直接进行下一步实验。
[0029]
这里乙醇钠可以用甲醇钠、异丙醇钠、叔丁醇钠、氢化钠、氨基钠和三苯甲基钠代替。
[0030]
实施例2
[0031]
化合物iii 6-氨基-5-甲氧基-1h-吲哚-2-甲酸乙酯的制备:向实施例1得到的化合物ii中加入300ml乙醇和6g raneyni,搅拌均匀后缓慢滴加600mmol质量分数80%水合肼,滴完后将反应体系温度升至回流,并保温搅拌反应2h,tlc[展开剂:v(石油醚):v(乙酸乙酯)=1:1]跟踪反应进程,反应结束后,冷却反应液至室温,过滤,滤渣用乙醇洗涤,合并滤液和洗液,减压蒸除乙醇,残留物用150ml乙酸乙酯分三次萃取,合并有机层,再用100ml水分两次洗涤后再用50ml饱和食盐水洗涤,无水硫酸镁干燥后减压蒸去大部分溶剂,加入少量石油醚,置于冰箱内重结晶,抽滤析出的固体,干燥得式(iii)化合物,收率70%(以化合物i计)。
[0032]
实施例3
[0033]
化合物ⅲ6-氨基-5-甲氧基-1h-吲哚-2-甲酸乙酯的制备:向实施例1得到的化合物ii中加入300ml乙醇和25g铁粉,搅拌后加入100ml浓盐酸(质量分数36~38%),将反应体系温度升至回流,并保温反应5~6h,tlc[展开剂:v(石油醚):v(乙酸乙酯)=1:1]跟踪反应进程,反应结束后,冷却反应液至室温,过滤,减压蒸去乙醇,残留物中加入20%na2co3,调节溶液ph值至9左右,用150ml乙酸乙酯分三次萃取,合并有机层,再用100ml水分两次洗涤后再用50ml饱和食盐水洗涤,无水硫酸镁干燥后减压蒸去大部分溶剂,加入少量石油醚,置于冰箱内重结晶,抽滤析出的固体,干燥得化合物ⅲ,收率60%(以化合物i计)。
[0034]
这里铁粉可以用锌粉代替,添加剂浓盐酸可以用醋酸、硫酸、对甲苯磺酸等酸代
替,得到的化合物ⅲ的收率为50~61%。
[0035]
实施例4
[0036]
化合物iii 6-氨基-5-甲氧基-1h-吲哚-2-甲酸乙酯的制备:向实施例1得到的化合物ii中加入150乙醇和150ml水,将反应体系温度升至50℃,搅拌下加入8.0g活性炭、20mmol六水合三氯化铁、2mmol六水合三氯化铝,升温至75℃,缓慢滴加600mmol80%水合肼。滴毕再继续保温反应2h,tlc[展开剂:v(石油醚):v(乙酸乙酯)=1:1]跟踪反应进程,反应结束后,冷却反应液至室温,过滤,滤渣用乙醇洗涤,合并滤液和洗液,减压蒸除乙醇,残留物用150ml乙酸乙酯分三次萃取,合并有机层,再用100ml水分两次洗涤后再用50ml饱和食盐水洗涤,无水硫酸镁干燥后减压蒸去大部分溶剂,加入少量石油醚,置于冰箱内重结晶,抽滤析出的固体,干燥得化合物ⅲ,收率55%(以化合物i计)。
[0037]
实施例5
[0038]
化合物iii 6-氨基-5-甲氧基-1h-吲哚-2-甲酸乙酯的制备:向实施例1得到的化合物ii中加入300ml乙醇,搅拌均匀后加入100ml浓盐酸(质量分数36~38%)和8.5g 10%钯碳,真空置换氢气3次,加压至0.15~0.2mpa,将反应体系温度升至60℃,并保温反应16~20h,反应结束,反应液冷却至室温后,用硅藻土滤去钯碳,加入质量分数20%na2co3水溶液,调节溶液ph值至9左右,减压蒸去乙醇,残留物用150ml乙酸乙酯分三次萃取,合并有机层,再用100ml水分两次洗涤后再用50ml饱和食盐水洗涤,无水硫酸镁干燥后减压蒸去大部分溶剂,加入少量石油醚,置于冰箱内重结晶,抽滤析出的固体,干燥得化合物ⅲ,收率66%(以化合物i计)。
[0039]
这里还原剂10%钯碳/h2可以用10%钯碳/甲酸铵和raneyni/h2代替,添加剂浓盐酸可以用醋酸、硫酸、对甲苯磺酸等酸代替。
[0040]
实施例6
[0041]
化合物iv 5-甲氧基-1h-吡咯[2,3-f]喹啉-2,7,9-三羧酸-2,7-二乙酯-9-甲酯的制备:将20mmol化合物ⅲ、20mmol乙醛酸乙酯、20mmol丙酮酸甲酯和30ml乙腈加入反应瓶中,搅拌溶解后,混合液在80℃氧气氛围中搅拌反应过夜,停止反应,冷却至室温,减压蒸除溶剂,加入50ml水搅拌,再用60ml乙酸乙酯分三次萃取,合并有机层,用无水硫酸钠干燥后减压蒸去大部分溶剂,加入30ml石油醚,置于冰箱内重结晶,抽滤析出的固体,干燥得化合物iv,收率17%。
[0042]
实施例7
[0043]
化合物iv 5-甲氧基-1h-吡咯[2,3-f]喹啉-2,7,9-三羧酸-2,7-二乙酯-9-甲酯的制备:将20mmol化合物ⅲ、20mmol乙醛酸乙酯、20mmol丙酮酸甲酯和30ml乙腈加入反应瓶中,搅拌溶解后,再加入2mmolcubr2。混合液在80℃氧气氛围中搅拌反应过夜,停止反应,冷却至室温,减压蒸除溶剂,加入50ml水搅拌,再用60ml乙酸乙酯分三次萃取,合并有机层,用无水硫酸钠干燥后减压蒸去大部分溶剂,加入30ml石油醚,置于冰箱内重结晶,抽滤析出的固体,干燥得化合物iv,收率70%。
[0044]
实施例8
[0045]
化合物iv 5-甲氧基-1h-吡咯[2,3-f]喹啉-2,7,9-三羧酸-2,7-二乙酯-9-甲酯的制备:将30mmol化合物ⅲ、20mmol乙醛酸乙酯、20mmol丙酮酸甲酯和30ml乙腈加入反应瓶中,搅拌溶解后,再加入2mmol cubr2。混合液在80℃氧气氛围中搅拌反应过夜(24h),停止
反应,冷却至室温,减压蒸除溶剂,加入50ml水搅拌,再用60ml乙酸乙酯分三次萃取,合并有机层,用无水硫酸钠干燥后减压蒸去大部分溶剂,加入30ml石油醚,置于冰箱内重结晶,抽滤析出的固体,干燥得化合物iv,收率80%。
[0046]
实施例9
[0047]
化合物iv 5-甲氧基-1h-吡咯[2,3-f]喹啉-2,7,9-三羧酸-2,7-二乙酯-9-甲酯的制备:将30mmol化合物ⅲ、20mmol乙醛酸乙酯、24mmol丙酮酸甲酯和30ml乙腈加入反应瓶中,搅拌溶解后,再加入2mmolcubr2。混合液在80℃氧气氛围中搅拌反应过夜(24h),停止反应,冷却至室温,减压蒸除溶剂,加入50ml水搅拌,再用60ml乙酸乙酯分三次萃取,合并有机层,用无水硫酸钠干燥后减压蒸去大部分溶剂,加入30ml石油醚,置于冰箱内重结晶,抽滤析出的固体,干燥得化合物iv,收率86%。
[0048]
在实施例9的基础上,乙醛酸乙酯可以用乙醛酸甲酯、乙醛酸环戊酯、乙醛酸环己酯代替;丙酮酸甲酯可以用丙酮酸乙酯、丙酮酸丙酯、丙酮酸环戊酯代替,得到系列5-甲氧基-1h-吡咯[2,3-f]喹啉-2,7,9-三甲酸2-乙酯7,9二烷基酯;催化剂可以用cucl2、cui2、cu(otf)2、cu(oocch3)2、zn(otf)2、zncl2、znbr2、fecl3.6h2o、febr3.6h2o等代替;溶剂可以用甲苯、苯、dmf、dmso等代替;反应温度可以在40~100℃之间调整;其他条件不变,反应的实验结果见下表。
[0049]
表1实施例10-30的制备条件和结果
[0050][0051][0052]
实施例31
[0053]
化合物v 5-甲氧基-1h-吡咯[2,3-f]喹啉-2,7,9-三甲酸的制备:将25mmol 5-甲氧基-1h-吡咯[2,3-f]喹啉-2,7,9-三羧酸-2,7-二乙酯-9-甲酯和200ml乙醇加入反应瓶中,搅拌后,将反应体系温度降到10℃,滴加1n氢氧化钠溶液100ml,滴完后将反应体系温度升至室温,并搅拌过夜(24h)。减压蒸去乙醇,将反应液置于冰水浴中,滴入2n的盐酸调节ph至5左右,析出大量固体,过滤,滤饼用蒸馏水洗涤,得化合物
ⅴ
5-甲氧基-1h-吡咯[2,3-f]喹啉-2,7,9-三甲酸的粗固体,该粗产物不经纯化,直接进行下一步实验。
[0054]
这里,5-甲氧基-1h-吡咯[2,3-f]喹啉-2,7,9-三羧酸-2,7-二乙酯-9-甲酯可以用实施例10~30合成的系列5-甲氧基-1h-吡咯[2,3-f]喹啉-2,7,9-三羧酸-2-乙酯-7,9-二烷基酯代替。
[0055]
实施例32
[0056]
pqq的制备:向实施例31制得的化合物
ⅴ
中加入200mlthf,将反应体系温度降到-10℃,搅拌下滴加含有100mmol ce(nh4)2(no3)6的100ml水溶液,滴完后保温反应1h,将200ml冷水加入,搅拌0.5h,过滤,滤液用300ml二氯甲烷萃取3次,干燥后减压蒸去溶剂,向残留物中加入30ml乙酸乙酯和石油醚的混合溶液打浆,过滤,收集固体,再向固体中加入300ml二氯甲烷和4g硅胶,室温搅拌1h,过滤,用硅藻土滤去硅胶,减压蒸去溶剂,固体干燥后得pqq,收率63%(以化合物iv 5-甲氧基-1h-吡咯[2,3-f]喹啉-2,7,9-三羧酸-2,7-二乙酯-9-甲酯计)。
[0057]
结构表征
[0058]
将制备的pqq 10g加入500ml水中,搅拌成悬浊液,加入1n氢氧化钠调节ph至8.5,搅拌至全溶,过滤,搅拌下向得到的滤液中缓慢加入盐酸调节ph至3.5左右;通过截留分子量150-300d的纳滤膜纳滤脱盐,纯化水洗涤至透出液电导率≤50μs/cm,减压浓缩结晶;抽滤析出的红色固体,50℃真空干燥,得晶体10.9g。对所得晶体进行结构检测和表征,hplc测定其纯度为99.83%;阳离子色谱检测na离子含量,结合hplc外标法测得pqq含量,求出该晶体所含pqq与na的物质量比为pqq∶na=1∶1.98,表明该晶体为pqq二钠盐。对该二钠盐晶体进行了氢谱测定。氢谱数据为:1h nmr(500mhz,dmso-d6)δ:8.62(s,1h),7.09(s,1h)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。