新型偶氮苯衍生物、其制备方法及其在与电离辐射联合的治疗性治疗中的用途
1.本发明涉及通过可被电离辐射激活的化合物对病理学(例如癌症)的治疗性治疗。
2.氧化还原和光敏系统已经开发了一个多世纪,并且用于触发复杂的动作,例如特定的键裂解或构型转换,可用于影响活性分子的释放、控制蛋白质活性和基因表达等。不幸的是,这些敏感系统的临床应用仍仅限于局部或眼科治疗,因为除了使用侵入性电极或光纤外,目前缺乏能够在体表以下大于数百微米深度激活这些系统的技术。事实上,迄今为止开发的所有光敏分子仅对紫外线、可见光或近红外辐射敏感,其对组织的渗透本质上是有限的。
3.wo 2011/158189中提出了通过电离辐射触发的化合物的活化。然而,疗效极其有限,并且需要的剂量太高使得无法在临上床应用。
4.因此,希望提供可以通过辐射激活而不限制靶标生物组织的深度的新化合物。电离辐射可用于有效到达深层组织,在那里辐射的高能量将被局部转换以特定激活氧化还原/光敏治疗系统。
5.根据本发明,将分子能量转换器(金属螯合物)与光敏开关(偶氮苯部分)结合以提供可通过与临床应用相容的中等剂量的高穿透光激活的光敏系统。
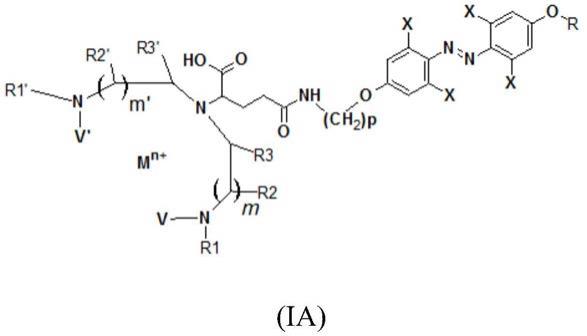
6.根据第一个目的,本发明因此涉及式(i)的化合物或其药学上可接受的盐:
7.[化学式1]
[0008][0009]
其中:
[0010]
m表示选自ce(iii)、pr(iii)、nd(iii)、sm(iii)、eu(iii)、gd(iii)、tb(iii)、dy(iii)、ho(iii)、er(iii)、tm(iii)、yb(iii)、mg(ii)、ca(ii)、mn(ii)、fe(ii)、fe(iii)、cu(ii)、zn(ii)、ga(iii)、y(iii)、zr(iii)、tc(iv)、tc(vi)、tc(vii)、ru(ii)、ru(iii)、ru(iv)、pd(ii)、ag(i)、in(iii)、hf(iv)、re(vi)、w(ii)、w(iii)、w(iv)、w(v)、w(vi)、os(iii)、os(iv)、ir(iii)、ir(iv)、pt(ii)、au(i)、au(iii)、tl(iii)、zr(iv)、nb(iii)、bi(iii)中的金属原子;
[0011]
n是1、2、3、4、5、6或7;
[0012]
v和v'可以相同或不同,是氢原子或直链或支链的c1
‑
c10烷基或烷氧基链,或连接在一起以形成环的c1
‑
c10烷基链,该c1
‑
c10烷基链包含一个或多个选自n、o或s的杂原子,
并且任选地被一个或多个独立地选自卤素原子和腈、硝基、硫基、氨基、酰胺基、芳基、杂芳基、羟基、羧酸或羧基的取代基取代;
[0013]
r1、r1'、r2、r2'可以相同或不同,是氢原子或任选被一个或多个独立地选自卤素原子和腈、硝基、硫基、氨基、酰胺基、芳基、杂芳基、羟基、羧酸或羧基的取代基取代的直链或支链的c1
‑
c10烷基或烷氧基链;
[0014]
r3和r3'可以相同或不同,是氢原子,或任选地被一个或多个独立地选自卤素原子和腈、硝基、硫基、氨基、酰胺基、芳基、杂芳基、羟基、羧酸或羧基的取代基取代的直链或支链的c1
‑
c10烷基或烷氧基链,或r3和r3'连接在一起形成5元至14元的杂环或杂芳基;
[0015]
m和m'可以相同或不同,等于1或2;
[0016]
x、x'、x”、x”'、y、y'、y”、y”'可以相同或不同,独立地选自h;卤素原子;烷氧基、烷基或环烷基,其任选地被一个或多个杂原子或基团cooh、conh2、cosh、oh、nh2、sh中断或取代;5元至12元芳基或杂芳基,其任选地被一个或多个cooh、conh2、cosh基团;cooh或nh2基团取代;
[0017]
z表示任选地被一个或多个杂原子或cooh、conh2、cosh、oh、nh2、sh基团中断或取代的烷基;任选地被一个或多个cooh、conh2、cosh基团取代的5元至12元芳基或杂芳基;或cooh或nh2基团;
[0018]
w和w'可以相同或不同,独立地表示ch2基团;或芳基或环烷基;氧或氮原子(仲(secondary)或叔(ternary));酰胺键;酯键;硫醚键;
[0019]
u和u'可以相同或不同,表示ch或nh基团,应理解双键u=u'为顺式或反式;
[0020]
t表示ch2基团;
‑
c(=o)nh基团;烷氧基、烷基或环烷基,其任选地被一个或多个杂原子或cooh、con2、cosh、oh、nh2、sh基团中断或取代;含有一个或多个杂原子和/或任选地被一个或多个选自coo烷基、conh烷基、cosh烷基的基团取代的5元至12元芳基或杂芳基;
[0021]
r表示h,或直链或支链的c1
‑
c18烷基或烷氧基链,所述直链或支链的c1
‑
c18烷基或烷氧基链任选地被一个或多个独立地选自卤素原子、腈、硝基、硫基、氨基、酰胺基、芳基、杂芳基、羟基、羧酸或羧基的取代基取代;
[0022]
p和q是0至6的整数,特别是1至6的整数;
[0023]
可以理解的是,为了确保分子的中性,m的n个阳离子电荷可以通过0至n个在r1、r1'、r2、r2'、r3、r3'、v、v'上任选取代的羧基(
‑
cooh)中和,和/或如果需要,通过0至n个存在于溶液中的抗衡离子中和;
[0024]
所述式(i)的化合物为以顺式和/或反式异构体及其混合物的形式。
[0025]
当被电离辐射激活时,根据本发明的化合物能够诱导癌细胞的通透性和死亡。在引入非活性(顺式)形式后,应用电离辐射照射靶标部位。这种辐射诱导分子转化为其活性形式(反式),导致由于分子亲脂平衡的变化而引起的细胞通透性和死亡。用于激活分子的射线不受深度限制地穿透生物组织,使激活技术非常适合临床应用,不同于传统光敏工具的激活仅限于紫外线至近红外照射(穿透生物组织小于1厘米)。
[0026]
根据本发明的化合物是辐射敏感的,其中它们的作用是由电离辐射触发的。因此,它们表示了一种用于实时控制和调节治疗作用的有前景的工具。
[0027]
尺寸为z=20至z=83的金属原子,特别是以下金属fe、cu、zn、ga、y、zr、tc、ru、pd、ag、in、eu、gd、tb、dy、ho、yb、hf、w、os、ir、pt、au、bi。
[0028]
在上述通式(i)中,单独地或以其任意组合地设想了以下实施方式:
[0029]
‑
v和v'通过包含2个氮原子的c1
‑
c9烷基链连接在一起,其任选地被2个羧基取代;和/或
[0030]
‑
v和v'相连形成链:[化学式2]和/或
[0031]
‑
z表示
‑
ch(cooh)
‑
基团;和/或
[0032]
‑
m=m'=1;和/或
[0033]
‑
r2=r3=r2'=r3'=h;和/或
[0034]
r1=r1'=(ch2)cooh;和/或
[0035]
‑
p=q=2;和/或
[0036]
‑
y=y'=y”=y”'=h;
[0037]
‑
w=w'=o;
[0038]
‑
u=u'=nh。
[0039]
根据一种实施方式,式(i)的化合物具有通式(ia)或药学上可接受的盐:
[0040]
[化学式3]
[0041][0042]
其中:
[0043]
m、n、r1、r2、r3、v、m、r1'、r2'、r3'、v'、m'、p如式(i)中定义且
[0044]
x选自氢和卤素原子;
[0045]
r是直链或支链的c1
‑
c12烷基;
[0046]
n=n双键呈顺式或反式;
[0047]
应当理解的是,为了确保分子的中性,m的n个阳离子电荷可以通过0至n个在r1、r1'、r2、r2'、r3、r3'、v、v'上取代的羧基(
‑
cooh)中和,和/或如果需要,通过0至n个存在于溶液中的抗衡离子中和。
[0048]
根据一种实施方式,式(i)的化合物具有通式(ib)或药学上可接受的盐:
[0049]
[化学式4]
[0050][0051]
其中:
[0052]
m、n、p、x和r如上定义。
[0053]
根据一种实施方式,式(i)的化合物具有通式(ic)或药学上可接受的盐:
[0054]
[化学式5]
[0055][0056]
其中:
[0057]
m、n、x如上定义。
[0058]
根据一种实施方式,在式(i)、(ia)或(ib)中:
[0059]
‑
x选自h和f;和/或
[0060]
‑
r表示直链c4
‑
c8烷基;
[0061]
‑
p=2;
[0062]
‑
m是选自cu、ga、y、in、eu、gd、yb、bi的金属;
[0063]
‑
n=2或3。
[0064]
在上述和下述内容中:
[0065]“烷基”是指链中具有约1至约20个碳原子的可以是直链或支链的脂族烃基。优选的烷基链中具有1至约12个碳原子。支链是指一个或多个低级烷基,例如甲基、乙基或丙基,与直链烷基链相连。
[0066]“烷氧基”是指烷基
‑
o
‑
基团,其中烷基如上所述。烷氧基的典型实例包括甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基和庚氧基。
[0067]“环烷基”是指约3至约10个碳原子,优选约5至约10个碳原子的非芳族单或多环环系统。环系统的优选环尺寸包括约5至约6个环原子。示例性单环环烷基包括环戊基、环己基和环庚基。
[0068]“羧基”是指ho(o)c
‑
基团(羧酸)。
[0069]“芳基”是指具有约6至约14个碳原子,优选具有约6至约10个碳原子的芳族单环或多环系统。芳基的典型实例包括苯基和萘基。
[0070]“杂芳基”是指约5至约14个碳原子,优选约5至约10个碳原子的芳族单环或多环环
系统,其中环系统中的一个或多个碳原子是碳以外的杂元素,例如氮、氧或硫。环系统的优选环尺寸包括约5至约6个环原子。示例性杂芳基包括吡嗪基、噻吩基、异噻唑基、噁唑基、吡唑基、呋咱基(furazanyl)、吡咯基、1,2,4
‑
噻二唑基、哒嗪基、喹喔啉基、酞嗪基、咪唑并[1,2
‑
a]吡啶、咪唑并[2,1
‑
b]噻唑基、苯并呋咱基、氮杂吲哚基、苯并咪唑基、苯并噻吩基、噻吩并吡啶基、噻吩并嘧啶基、吡咯并吡啶基、咪唑并吡啶基、苯并氮杂吲哚、1,2,4
‑
三嗪基、苯并噻唑基、呋喃基、咪唑基、吲哚基、吲哚啉嗪基、异噁唑基、异喹啉基、异噻唑基、噁二唑基、吡嗪基、哒嗪基、吡唑基、吡啶基、嘧啶基、吡咯基、喹唑啉基、喹啉基、1,3,4
‑
噻二唑基、噻唑基、噻吩基和三唑基。
[0071]“卤素”是指氟、氯、溴或碘。优选氟、氯或溴,尤其是氟。
[0072]
本发明还涉及根据本发明的化合物的治疗用途。
[0073]
根据另一个目的,本发明涉及包含主要呈顺式形式的式(i)的化合物和至少一种药学上可接受的赋形剂的药物组合物。
[0074]
根据另一个目的,本发明涉及顺式和/或反式形式的式(i)的化合物用于治疗癌症的用途。
[0075]
下面的式(i)的化合物通过电离辐射活化,顺式双键转变为细胞毒性反式形式。
[0076]
因此,所述用途包括给药主要呈顺式形式的式(i)的化合物并对靶位点施加辐射以原位形成反式化合物。
[0077]
所述用途还包括通过体内mri、pet、x射线或spect成像监测治疗。
[0078]
根据一种实施方式,所述用途包括向有需要的患者给药有效量的顺式形式的式(i)化合物。
[0079]
根据一种实施方式,所述式(i)的化合物的给药通常可以肠胃外进行。
[0080]
所考虑的电离辐射通常对应于放射疗法治疗中使用的辐射。这种由外部刺激引起的作用允许时间和空间控制,并且可以根据需要实时调整以达到最佳治疗效果。这些包括电离辐射,例如光子(能量范围为1kev至25mev的x射线和γ射线)、电子和强子(例如能量范围为60至300mev的质子或碳),尤其是x射线(1
‑
200kev)、伽马射线(662kev)和电子束(4500kev)。
[0081]
这种辐射允许在2至20gy,特别是2至10gy,更特别是2至5gy的低剂量下实现期望的激活。
[0082]
由于本发明化合物的作用是通过细胞通透性发挥的,因此所有癌症都可以被本发明的化合物潜在地治疗,因为治疗方法不限于特定的细胞表面特征。这些包括肺癌、胰腺癌、肝癌、脾癌、小细胞肺癌、前列腺癌、横纹肌肉瘤、胃癌、胃肠癌、结直肠癌、肾癌、乳腺癌、卵巢癌、睾丸癌、甲状腺癌、头颈癌、皮肤癌、软组织肉瘤、膀胱癌、骨癌、骨髓瘤、浆细胞瘤、生殖细胞癌、子宫癌、白血病、淋巴瘤、神经母细胞瘤、骨肉瘤、视网膜母细胞瘤、中枢神经系统癌症、维尔姆斯氏瘤,尤其是胰腺癌或白血病。
[0083]
在一种实施方式中,由于本发明化合物的分子结构中金属原子的存在,该用途还允许通过mri在体内检测化合物的存在和可能的其分子转换。
[0084]
本发明化合物的mri检测允许以非侵入性方式定位和监测所述化合物的治疗作用,从而促进治疗方案的开发和监测,从而允许治疗诊断方法。
[0085]
根据一种实施方式,本发明的化合物可以与一种或多种抗癌剂组合给药,例如化
疗或免疫治疗剂,如抗ctla4、抗pd
‑
l1和抗pd1等免疫检查点抑制剂。
[0086]“有效量”是指有效产生所需治疗效果的根据本发明的化合物/组合物的量。
[0087]“适合肠胃外给药的制剂”是指适合于肠胃外给药至患者的形式的制剂。制剂是无菌的,包括乳液、悬浮液、水性和非水性注射液,其中可以含有悬浮剂和增稠剂以及抗氧化剂(在检查这些潜在的辐射防护效果不会阻止辐射下的预期效果后)、缓冲液、抑菌剂和使制剂等渗的溶质,并具有适当调节的ph值,与所需的受体血液。
[0088]
该制剂优选通过注射给药,包括经肌肉、静脉内、腹膜内和皮下给药。对于注射,有用的根据本发明的化合物可以配制在液体溶液中,优选在生理相容的缓冲液中,例如汉克氏(hank's)溶液或林格氏(ringer's)溶液。此外,化合物可以配制成固体形式并在使用前立即重新溶解或悬浮。冻干形式也包括在内。
[0089]
溶媒的选择和溶媒中活性物质的含量通常由活性化合物的溶解度和化学性质、特定的给药方式和药物实践中要遵守的规定确定。
[0090]
制剂可以通过药学领域众所周知的任何方法制备成单独的药物形式。这些方法包括将活性成分与构成一种或多种辅助成分的载体结合的步骤。通常,通过将活性成分和液体载体或精细分级的固体载体或两者均匀且紧密地结合,然后,如果需要,使产品成型来制备制剂。
[0091]
本发明的组合物中活性成分的实际剂量水平可以变化以获得有效量的活性成分以实现特定组合物和给药方式的所需治疗响应。因此,选择的剂量水平取决于所需的治疗效果、给药方式、所需的治疗持续时间和其他因素。
[0092]
剂量单位组合物可含有可用于构成日剂量的此类约数的量。然而,应当理解,任何特定患者的特定剂量水平将取决于多种因素,包括体重、一般健康状况、性别、饮食、给药时间和方法,吸收和排泄率,与其他药物的组合以及所治疗疾病的严重程度。
[0093]
每种成分的给药量由治疗医师在考虑疾病的病因和严重程度、患者的状况和年龄、每种成分的效力和其他因素后确定。
[0094]
制剂可以装在单剂量或多剂量容器中,例如用弹性密封件密封的安瓿和小瓶,可以以冻干形式储存,只需要在使用前添加无菌液体载体,例如注射用水。注射剂和混悬剂中的临时制备的溶液可由上述类型的无菌粉末、颗粒和片剂制备。
[0095]“液体剂型”是指待给予患者的活性化合物的剂量是液体形式,例如乳液、溶液、悬浮液、糖浆和药学上可接受的酏剂。除活性化合物外,液体剂型可含有本领域常用的惰性稀释剂,例如水或其他溶剂、增溶剂和乳化剂,例如乙醇、异丙醇、碳酸乙酯、乙酸乙酯、苯甲醇、苯甲酸苄酯、丙二醇、1,3
‑
丁二醇、二甲基甲酰胺、油(在检查这些潜在的辐射防护效果不会阻止辐射下的预期效果后),特别是棉籽油、花生油、玉米胚芽油、橄榄油、蓖麻油和芝麻油、甘油、四氢糠醇、聚乙二醇和脱水山梨糖醇脂肪酸酯或这些物质的混合物等。
[0096]“患者”包括人和其他哺乳动物。
[0097]“药物组合物”是指包含式i化合物和至少一种选自由药学上可接受的载体、稀释剂、佐剂、赋形剂或溶媒组成的组中的组分(如防腐剂、填充剂、润湿剂、乳化剂、悬浮剂、抗细菌剂、抗真菌剂、润滑剂和分散剂,取决于给药方式和剂型的性质)的组合物。悬浮剂的实例包括乙氧基化异硬脂醇、聚氧乙烯山梨糖醇和山梨糖醇酯、微晶纤维素、偏氢氧化铝、膨润土、琼脂和黄蓍胶,或这些物质的混合物。可以通过各种抗细菌剂和抗真菌剂(例如对羟
基苯甲酸酯、氯丁醇、苯酚、山梨酸等)来防止微生物的作用。还可能需要包括等渗剂,例如糖、氯化钠等。合适的载体、稀释剂、溶剂或溶媒的实例包括水、乙醇、多元醇、它们的合适混合物、植物油(例如橄榄油)和可注射的有机酯例如油酸乙酯。
[0098]“药学上可接受的”是指在合理的医学判断范围内,适用于与人体和低等动物细胞接触使用,而无过度毒性、刺激性、过敏反应等,并具有相对合理的风险/效益比。
[0099]
术语“药学上可接受的盐”是指本发明化合物的相对无毒的无机和有机酸加成盐和碱加成盐。这些盐可以在化合物的最终分离和纯化过程中原位制备。特别地,酸加成盐可以通过将纯化形式的纯化化合物与有机或无机酸单独反应并分离所得盐来制备。酸加成盐的实例包括由例如s.m.berge等,“pharmaceutical salts”j.pharm.sci,66:p.1
‑
19(1977)所描述的盐。酸加成盐也可以通过将酸形式的纯化化合物与有机或无机碱单独反应并分离所得盐来制备。合适的无机碱加成盐由金属碱制备,包括氢化钠、氢氧化钠、氢氧化钾、氢氧化钙、氢氧化铝、氢氧化锂、氢氧化镁和氢氧化锌。合适的碱氨基加成盐由具有足够碱度以形成稳定盐的胺制备,并且优选包括由于其低毒性和医学用途可接受性而经常用于药物化学中的那些胺。
[0100]
应当理解的是,本发明涵盖了本文提及的特定和优选分组的所有合适组合。
[0101]
根据另一个目的,本发明还涉及制备式(i)的化合物的方法,所述方法包括:
[0102]
‑
络合式(ii)的化合物与金属m的前体的步骤:
[0103]
[化学式6]
[0104][0105]
其中:
[0106]
r1、r2、r3、v、m、r1'、r2'、r3'、v'、m'、z、t、q、p、w、w'、x、x'、x”、x”'、y、y'、y”、y”'、r、u、u'如上所定义,
[0107]
(ii)的化合物为顺式和/或反式异构体及其混合物的形式;和
[0108]
‑
可能地,对获得的产物进行uv照射,以便主要获得顺式异构体。
[0109]
与金属试剂m的络合通常可以在如水的溶剂中、在室温和反应混合物的回流温度之间的温度下(如在40至100℃)进行。通常,溶液的ph值保持在5到7。
[0110]
根据一种实施方式,式(ii)的化合物具有式(iia):
[0111]
[化学式7]
[0112][0113]
其中r1、r2、r3、v、m、r1'、r2'、r3'、v'、m'、p、x、r的定义如式(i)中所定义。
[0114]
根据一种实施方式,式(ii)的化合物具有式(iib):
[0115]
[化学式8]
[0116][0117]
其中p、x、r的定义如式(i)中所定义。
[0118]
根据一种实施方式,式(ii)的化合物具有式(iic):
[0119]
[化学式9]
[0120][0121]
其中x的定义如式(i)中所定义。
[0122]
根据一种实施方式,式(ii)的化合物通过将式(iii)的化合物与式(iv)的化合物偶联获得:
[0123][0124]
其中:
[0125]
r1、r2、r3、v、m、r1'、r2'、r3'、v'、m'、z、t、q、p、w、w'、x、x'、x”、x”'、y、y'、y”、y”'、r、u、u'如上述所定义;和
[0126]
e是直链或支链的c1
‑
c10烷基或烷氧基链,环烷基或芳基,所述环烷基或芳基任选地包含一个或多个选自n、o或s的杂原子,且任选地被一个或多个独立地选自卤素原子和酸酐、羧基、腈、硝基、硫基、氨基、酰胺基、芳基、杂芳基、羟基、酯、羧酸或羧基的取代基中断或取代,条件是e含有亲核取代基,例如羟基、硫醇或伯胺或仲胺官能团,以实现与分子(iii)的偶联;
[0127]
g是直链或支链的c1
‑
c10烷基或烷氧基链,环烷基或芳基,所述环烷基或芳基任选地包含一个或多个选自n、o或s的杂原子,且任选地被一个或多个独立地选自卤素原子和酸酐、羧基、腈、硝基、硫基、氨基(伯或仲)、酰氨基、芳基、杂芳基、羟基、羧酸或羧基的取代基取代,应理解的是,g含有亲电子取代基,例如活化的羧酸官能团或酸酐官能团,以进行与分子(iv)的偶联。
[0128]
偶联反应可以在碱,特别是有机碱如三乙胺的存在下进行。反应可在无水条件下进行。
[0129]
根据一种实施方式,式(iii)的化合物具有式(iiia):
[0130]
[化学式12]
[0131][0132]
其中r1、r2、r3、v、m、r1'、r2'、r3'、v'、m'如上所定义。
[0133]
根据一种实施方式,式(iii)的化合物具有式(iiib):
[0134]
[化学式13]
[0135][0136]
根据一种实施方式,式(iv)的化合物具有式(iva):
[0137]
[化学式14]
[0138][0139]
其中p、x、r如式(i)中所定义。
[0140]
根据一种实施方式,式(iv)的化合物具有式(ivb):
[0141]
[化学式15]
[0142][0143]
其中p、w、w'、x、x'、x”、x”'、y、y'、y”、y”'、r、u、u'如上所定义。
[0144]
根据一种实施方式,式(iv)的化合物具有式(ivc):
[0145]
[化学式16]
[0146][0147]
其中x如式(i)中所定义。
[0148]
式(iii)的化合物通常是市售的。特别地,式(iiib)的化合物是化合物dotaga。
[0149]
式(iv)的化合物可以通过应用和/或修改以下实施例中描述的合成方法来制备。
[0150]
通常,用于本发明的化合物的起始产物和中间体可以通过应用或改进迄今使用的
或文献中描述的已知方法来制备,例如由r.c.larock在综合有机转化(comprehensive organic transformations),vch出版社,1989中所描述的。
[0151]
在反应中,可能有必要保护反应性官能团,例如羟基、氨基、亚氨基、硫基和羧基,当它们在最终产物中需要时,以避免它们不希望地参与反应。可以根据标准做法使用传统保护基;实例参见t.w.green and p.g.m.wuts in"protective groups in organic chemistry",john wiley and sons,1991;j.f.w.mcomie in"protective groups in organic chemistry",plenum press,1973。
[0152]
除非特别说明,对所用溶剂的性质没有特别限制,只要它对反应或所涉及的试剂没有不利影响。合适的溶剂的实例包括:烃,其可以是芳族、脂族或脂环族烃,例如己烷、环己烷、苯、甲苯和二甲苯;酰胺,特别是脂肪酸酰胺,例如二甲基甲酰胺,和醚,例如乙醚和四氢呋喃。
[0153]
反应可以在很宽的温度范围内进行,通常为约0℃至150℃(优选为约室温至约100℃)。反应所需的时间也有很大差异,这取决于许多因素,包括反应温度和试剂的性质。
[0154]
由此制备的化合物可以通过常规方法从反应混合物中回收。例如,可以通过从反应混合物中蒸馏出溶剂或如果必要在从溶液混合物中蒸馏出溶剂后,将剩余物倒入水中,然后用与水不混溶的有机溶剂萃取,并从萃取液中蒸馏出溶剂来回收化合物。此外,如果需要,产物可以通过各种技术进一步纯化,例如重结晶、再沉淀或各种色谱技术,包括柱色谱或制备级薄层色谱。
[0155]
酸加成盐可以与根据本发明的有用的化合物形成,其中存在碱官能团(例如氨基、烷基氨基或二烷基氨基)。优选药学上可接受的,即无毒的酸加成盐。所选择的盐被最佳地选择为与通常的药物溶媒相容并适合肠胃外给药。可用于本发明的化合物的酸加成盐可以通过游离碱与合适的酸的反应,通过应用或修改已知方法来制备。例如,用于本发明的化合物的酸加成盐可以通过将游离碱溶解在水或醇水溶液或含有适当酸的合适溶剂中并通过蒸发溶液或通过反应来分离盐来制备,或通过使有机溶剂中的游离碱和酸反应来制备,在这种情况下,盐直接分离或可通过浓缩溶液获得。可用于本发明的化合物的酸加成盐可以通过应用或修改已知方法由盐再生。例如,可用于本发明的母体化合物可以通过用碱,例如碳酸氢钠水溶液或氨水溶液处理,从它们的酸加成盐再生。
[0156]
根据本发明的有用化合物可以通过应用或修改已知方法从它们的碱加成盐再生。例如,根据本发明的有用的母体化合物可以通过用酸例如盐酸处理而从它们的碱加成盐再生。
[0157]
当根据本发明的有用的化合物含有羧基时,可以形成碱加成盐。可用于制备碱加成盐的碱优选包括当与游离酸组合时产生药学上可接受的盐的那些,即盐的药物剂量中的阳离子对患者是无毒的,使得游离碱固有的有益抑制作用不会被可归因于阳离子的副作用抵消。在本发明范围内,包括那些衍生自碱土金属盐的药学上可接受的盐包括衍生自以下碱的那些:氢化钠、氢氧化钠、氢氧化钾、氢氧化钙、氢氧化铝、氢氧化锂、氢氧化镁、氢氧化锌、氨、乙二胺、n
‑
甲基
‑
葡糖胺、赖氨酸精氨酸、鸟氨酸、胆碱、n,n'
‑
二苄基乙二胺、氯普鲁卡因、二乙醇胺、普鲁卡因、n
‑
苄基苯乙胺、二乙胺、哌嗪、三(羟甲基)
‑
氨基甲烷、四甲基氢氧化铵和类似物。
[0158]
根据本发明的有用化合物可容易地制备或在本发明的方法期间形成为溶剂化物
2695系统(waters)上,使用0.1%水
‑
fa/乙腈梯度体系;0'(95/5)、20'(0/100),流速为0.25ml.min
‑1(注射10μl)进行lcms(液相色谱质谱)分析。质量分析器是tof lct premier(waters)。毛细管电压为2.8kv。锥孔电压为35v。源温度为120℃,去溶剂化温度为280℃。在masslynx上进行数据处理。
[0167]
方法c
[0168]
在与515hplc泵(waters)、pad 2996读数器(waters)以及xbridge c18反相柱(长度:100mm,直径:3.0mm,固定相:3.5μm)偶联的2525二元hplc泵(waters)上,使用0.05%水
‑
tfa/乙腈等度体系,流速为0.75ml.min
‑1(注入10μl)进行hplc分析。检测波长对应于所用流动相中研究的化合物的等吸收点。在masslynx上进行数据处理。
[0169]
ii.总结
[0170]
概括
[0171]
所有试剂均以最高纯度购自sigma
‑
aldrich或alfa aesar,无需进一步纯化即可使用。dotaga酐购自chematech,无需进一步纯化即可使用。用于快速色谱的硅胶(aldrich 717185si 60,40
‑
63μm)购自vwr。rp
‑
18反相快速色谱在combiflash仪器(biotage)上进行。使用涂有60f254硅胶的铝箔进行薄层色谱法(通过254nm紫外灯或茚三酮检测)。1h、
13
c和
19
f nmr谱是在bruker 300mhz或400mhz光谱仪上在环境温度下获得的。化学位移δ以ppm为单位,使用溶剂作为参考。耦合常数j以赫兹为单位测量。耦合分布由缩写d(双峰)、t(三重峰)和m(多重峰)描述。除非另有说明,否则使用配有tof
‑
lct premier质谱仪的飞行时间质量分析仪(waters),在3/7h2o/甲醇混合物中通过电喷雾电离在正模式下进行高分辨率质谱(hrms)实验。分析色谱方法(hplc和lcms)在专门的段落中进行了描述。合成中间体的纯度通过1h nmr或反相hplc确定。最终产物的纯度通过反相hplc测定并确认为>95%。
[0172]
根据以下合成方案合成了以下化合物:
[0173]
[化学式17]
[0174][0175]
其中x=h或f,
[0176]
其中m=cu、ga、y、in、eu、gd、yb或bi,
[0177]
其中n=2或3,
[0178]
[化学式18]
[0179][0180]4‑
羟基
‑
4'
‑
丁氧基偶氮苯的合成(1)
[0181]
将4
‑
丁氧基苯胺(2.00ml,12.0mmol)和亚硝酸钠(0.854g,12.0mmol,1.0当量)溶解在1:1etoh/h2o混合物(24ml)中,并将介质冷却至0℃。在小心加入cc hcl(2.6ml)之前,将冰(12g)加入培养基中。将冷却至0℃的预先制备的苯酚(1.14g,12.0mmol,1.0当量)和naoh(0.960g,24.0mmol,2.0当量)的水溶液(6.3ml)小心地引入到0℃的介质中。将介质在0℃下搅拌20分钟,然后在环境温度(at)下搅拌70分钟。将ph值调节至1(cc hcl)后,在过滤前将介质在环境温度下静置30分钟。用水(4
ⅹ
50ml)洗涤沉淀物,溶解在dcm中,有机相用mgso4干燥,然后浓缩。4
‑
羟基
‑
4'
‑
丁氧基偶氮苯1(2.76g,10.2mmol,85%)被分离为黑色无定形粉末。
[0182]1h nmr(400mhz;cdcl3)δ(ppm):7.87(d;j=8.9hz;2h);7.82(d;j=8.8hz;2h);6.98(d;j=8.9hz;2h);6.91(d;j=8.8hz;2h);4.03(t;j=6.5hz;2h);1.85
‑
1.75(m;2h);1.58
‑
1.45(m;2h);0.99(t;j=7.4hz;3h)。
[0183]
13
c nmr(75mhz;cdcl3)δ(ppm):161.51;158.38;146.89;146.63;124.81;124.56;116.07;114.92;68.22;31.35;19.33;13.96。
[0184]
hrms(m/z):c
16
h
19
n2o2计算为271.1447([m h]
)。实际为271.1440。
[0185]
r
f
=0.36(二氧化硅;dcm;uv)。
[0186]4‑
(n
‑
(叔丁氧基羰基)
‑
乙氧基胺)
‑
4'
‑
丁氧基偶氮苯的合成(2)
[0187]
将4
‑
羟基
‑
4'
‑
丁氧基偶氮苯1(2.00g,7.40mmol)和k2co3(1.53g,11.1mmol,1.5当量)溶解在丙酮(25ml)中。在at和氩气下搅拌30分钟后,将2
‑
(boc
‑
氨基)乙基溴(4.98g,22.2mmol,3.0当量)引入介质中。回流搅拌18小时后,趁热滤出介质,用热丙酮(75ml)洗涤沉淀物。过滤前将滤液在at下静置30分钟,所得沉淀物1用冷丙酮洗涤。过滤前将滤液在0℃下静置3小时。用冷丙酮洗涤所得沉淀物2并加入沉淀物1。将滤液浓缩并通过硅胶快速色谱法(dcm)纯化。4
‑
(n
‑
(叔丁氧基羰基)
‑
乙氧基胺)
‑
4'
‑
丁氧基偶氮苯2(2.27g,5.49mmol,74%)被分离为黄色无定形粉末(沉淀57%,快速色谱17%)。
[0188]1h nmr(400mhz;cdcl3)δ(ppm):7.86(2d;j=8.9hz;4h);6.99(2d;j=8.9hz;4h);4.10(t;j=5.0hz;2h);4.04(t;j=6.5hz;2h);3.57(m;2h);1.87
‑
1.74(m;2h);1.64
‑
1.40(m;11h);1.00(t;j=7.4hz;3h)。
[0189]
13
c nmr(100mhz;cdcl3)δ(ppm):161.46;160.62;156.03;147.51;147.07;124.52;124.48;114.84;114.82;79.79;68.18;67.64;40.27;31.42;28.55;19.38;13.98。
[0190]
hrms(m/z):c
23
h
32
n3o4计算为414.2393([m h]
)。实际为414.2396。
[0191]
r
f
=0.90(二氧化硅;dcm/甲醇98:2;uv)。
[0192]4‑
氨基乙氧基
‑
4'
‑
丁氧基偶氮苯的合成(3)
[0193]
将4
‑
(n
‑
(叔丁氧基羰基)
‑
乙氧基胺)
‑
4'
‑
丁氧基偶氮苯2(1.26g,3.04mmol)溶解在4:1dcm/tfa混合物(63ml)中,将介质在at搅拌1.5小时。浓缩后,加入乙醚(250ml),形成的沉淀物凝固1小时,然后过滤并用乙醚(4
ⅹ
50ml)洗涤。4
‑
氨基乙氧基
‑
4'
‑
丁氧基偶氮苯3(1.25g,2.93mmol,96%,tfa盐)被分离为黄色无定形粉末。
[0194]1h nmr(400mhz;meod)δ(ppm):7.88(d;j=8.9hz;2h);7.85(d;j=8.9hz;2h);7.15(d;j=8.9hz;2h);7.04(d;j=8.9hz;2h);4.32(t;j=4.4hz;2h);4.06(t;j=6.4hz;2h);3.41(t;j=4.4hz;2h);1.86
‑
1.73(m;2h);1.60
‑
1.46(m;2h);1.00(t;j=7.4hz;3h)。
[0195]
13
c nmr(75mhz;meod)δ(ppm):163.03;161.34;148.97;148.10;125.42;125.30;115.99;115.80;69.12;65.59;40.24;32.43;20.28;14.16。
[0196]
hrms(m/z):c
18
h
24
n3o2计算为314.1869([m h]
)。实际为314.1864。
[0197]
hplc(方法a);t
r 17.92分钟(顺式)
‑
20.35分钟(反式)。
[0198]
r
f
=0.70(二氧化硅;dcm/甲醇95:5;uv或茚三酮)。
[0199]4‑
(n
‑
(1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸
‑
10
‑
戊二酰)
‑
乙氧基胺)
‑
4'
‑
丁氧基偶氮苯(azo)的合成
[0200]
将4
‑
氨基乙氧基
‑
4'
‑
丁氧基偶氮苯3(259mg,0.606mmol,tfa盐)溶解在无水dmf(4.3ml)中。引入三乙胺(253μl,1.81mmol,3.0当量)后,将介质在at氩气下搅拌5分钟。然后引入dotaga酸酐(278mg,0.606mmol,1.0当量),并将介质在70℃下在氩气下搅拌21小时。浓缩后,加入乙醚(100ml),形成的沉淀物凝固30分钟,然后过滤并用乙醚(2
ⅹ
50ml)洗涤。通过反相快速色谱法(rp
‑
18,biotage,梯度h2o0.05%fa/ch3cn0.05%fa1:0
‑
2:8)纯化粗产物。4
‑
(n
‑
(1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸
‑
10
‑
戊二酰基)
‑
乙氧基胺)
‑
4'
‑
丁氧基偶氮苯azo(294mg,0.381mmol,63%)通过冷冻干燥以黄色静电粉末的形式分离。
[0201]
hrms(m/z):c
37
h
54
n7o
11
计算为772.3881([m h]
)。实际为772.3883。
[0202]
lcms(方法b);t
r 12.357分钟(顺式)
‑
14.437分钟(反式)。
[0203]
2,6
‑
二氟
‑4‑
羟基
‑
2',6'
‑
二氟
‑
4'
‑
丁氧基偶氮苯的合成(4)
[0204]
将2.6
‑
二氟
‑4‑
丁氧基苯胺(2.20g,10.9mmol)和亚硝酸钠(0.755g,10.9mmol,1.0当量)溶解在1:1etoh/h2o混合物(22ml)中,并将介质冷却至0℃。在小心加入cc hcl(2.37ml)之前,将冰(11g)加入培养基中。将预先制备的3,5
‑
二氟苯酚(1.42g,10.9mmol,1.0当量)和naoh(0.876g,21.9mmol,2.0当量)的水溶液(5.7ml)冷却至0℃,在0℃下小心地将其引入培养基中。将介质在0℃下搅拌20分钟,然后在at下搅拌70分钟。将ph值调节至1(cc hcl)后,在过滤前将介质在at下静置30分钟。沉淀物用水(4
ⅹ
50ml)洗涤,溶解在dcm中,有机相用mgso4干燥,然后浓缩。残余物(黑色粘稠液体)在真空歧管(3.15g)下浓缩,无需进一步纯化即可用于下一步。
[0205]1h nmr(400mhz;dmso)δ(ppm):6.93(d;jh
‑
f=11.4hz;2h);6.64(d;jh
‑
f=11.4hz;2h);4.09(t;j=6.5hz;2h);1.77
‑
1.66(m;2h);1.49
‑
1.38(m;2h);0.94(t;j=7.4hz;3h)。
[0206]
13
c nmr(101mhz;dmso)δ(ppm):161.40(t;j
c
‑
f
=14.8hz);161.11(t;j
c
‑
f
=14.8hz);157.69(dd;j
c
‑
f
=39.1;7.7hz);155.13(dd;j
c
‑
f
=38.2;7.8hz);125.00(t;j
c
‑
f
=10.2hz)124.05(t;j
c
‑
f
=10.3hz);100.26(dd;j
c
‑
f
=22.6;2.0hz);99.68(dd;j
c
‑
f
=24.2;2.1hz);68.74(s);30.31(s);18.55(s);13.58(s)。
[0207]
19
f nmr(376mhz;dmso;解耦)δ(ppm):
‑
118.99;
‑
119.27。
[0208]
19
f nmr(376mhz;dmso)δ(ppm):
‑
118.99(d;j
f
‑
h
=12.1hz);
‑
119.27(d;j
f
‑
h
=12.0hz)。
[0209]
hrms(m/z):c
16
h
15
f4n2o2计算为343.1070([m h]
)。实际为343.1060。
[0210]
r
f
=0.20(二氧化硅;环己烷/乙酸乙酯9:1;uv)。
[0211]
2,6
‑
二氟
‑4‑
(n
‑
(叔丁氧基羰基)
‑
乙氧基胺)
‑
2',6'
‑
二氟
‑
4'
‑
丁氧基偶氮苯的合成(5)
[0212]
将2,6
‑
二氟
‑4‑
羟基
‑
2',6'
‑
二氟
‑
4'
‑
丁氧基偶氮苯4(2.18g粗制,考虑为6.37mmol)和k2co3(1.32g,9.57mmol,1.5当量)溶解在丙酮(22ml)中。在at和氩气下搅拌30分钟后,将2
‑
(boc
‑
氨基)乙基溴(4.29g,19.1mmol,3.0当量)引入介质中。回流搅拌18小时后,将介质浓缩并通过硅胶快速色谱纯化(环己烷/乙酸乙酯,梯度1/0
‑
7/3)。2,6
‑
二氟
‑4‑
(n
‑
(叔丁氧基羰基)
‑
乙氧基胺)
‑
2',6'
‑
二氟
‑
4'
‑
丁氧基偶氮苯5(624mg,1.28mmol,17%分2个步骤)被分离为无定形黄色粉末。
[0213]1h nmr(400mhz;dmso)δ(ppm):7.04(s;1h);6.95(d;jh
‑
f=11.8hz;4h);4.10(m;4h);3.34
‑
3.26(m;2h);1.75
‑
1.67(m;2h);1.49
‑
1.39(m;2h);1.38(s;9h);0.94(t;j=7.4hz;3h)。
[0214]
13
c nmr(101mhz;dmso)δ(ppm):161.53(t;j
c
‑
f
=14.3hz);161.16(t;j
c
‑
f
=14.2hz);157.59(t;j
c
‑
f
=7.9hz);155.64(s);155.03(t;j
c
‑
f
=7.7hz);125.09(t;j
c
‑
f
=8.3hz);124.90(t;j
c
‑
f
=8.3hz);99.90(dd;j
c
‑
f
=11.3;1.7hz);99.67(dd;j
c
‑
f
=11.3;1.7hz);77.86(s);68.80(s);67.87(s);38.71(s);30.29(s);28.17(s);18.53(s);13.57(s)。
[0215]
19
f nmr(376mhz;dmso;解耦)δ(ppm):
‑
118.79;
‑
118.85。
[0216]
19
f nmr(376mhz;dmso)δ(ppm):
‑
118.79(d;j
f
‑
h
=12.0hz);
‑
118.85(d;j
f
‑
h
=11.9hz)。
[0217]
hrms(m/z):c
23
h
28
f4n3o4计算为486.2016([m h]
)。实际为486.2015。
[0218]
r
f
=0.30(二氧化硅;环己烷/乙酸乙酯8:2;uv)。
[0219]
2,6
‑
二氟
‑4‑
氨基乙氧基
‑
2',6'
‑
二氟
‑
4'
‑
丁氧基偶氮苯的合成(6)
[0220]
将2,6
‑
二氟
‑4‑
(n
‑
(叔丁氧基羰基)
‑
乙氧基胺)
‑
2,6
‑
二氟
‑
4'
‑
丁氧基偶氮苯5(879mg,1.81mmol)溶解在4:1dcm/tfa混合物中(37ml),并将介质在at搅拌1.5小时。浓缩后,加入乙醚(200ml),形成的沉淀物1凝结1小时,然后过滤并用乙醚(4
ⅹ
100ml)洗涤。浓缩滤液并在正己烷(200ml)中形成粘性沉淀物2,过滤并用正己烷(3
ⅹ
200ml)洗涤。沉淀物1和2在真空歧管下干燥。2,6
‑
二氟
‑4‑
氨基乙氧基
‑
2',6'
‑
二氟
‑
4'
‑
丁氧基偶氮苯6(858mg,1.72mmol,95%,tfa盐)被分离为无定形黄色粉末(沉淀物1,70%)和黄色油(沉淀2,25%)。
[0221]1h nmr(400mhz;dmso)δ(ppm):7.98(s;3h);7.00(d;j
h
‑
f
=11.3hz;2h);6.96(d;j
h
‑
f
=11.8hz;2h);4.30(t;j=5.0hz;2h);4.11(t;j=6.5hz;2h);3.26(t;j=5.0hz;2h);1.80
‑
1.63(m;2h);1.53
‑
1.33(m;2h);0.94(t;j=7.4hz;3h)。
[0222]
13
c nmr(101mhz;dmso)δ(ppm):161.76(t;j
c
‑
f
=14.4hz);160.27(t;j
c
‑
f
=14.0hz);157.57(dd;j
c
‑
f
=25.8;7.6hz);155.00(dd;j
c
‑
f
=25.9;7.4hz);125.57(s);124.86(s);100.07(dd;j
c
‑
f
=24.7;2.7hz);99.80(dd;j
c
‑
f
=24.7;2.5hz);68.85(s);65.82(s);38.06(s);30.29(s);18.54(s);13.57(s)。
[0223]
19
f nmr(376mhz;dmso;解耦)δ(ppm):
‑
73.46;
‑
118.60;
‑
118.74。
[0224]
19
f nmr(376mhz;dmso)δ(ppm):
‑
73.46(s);
‑
118.60(d;j
f
‑
h
=11.9hz);
‑
118.73(d;j
f
‑
h
=11.8hz)。
[0225]
hrms(m/z):c
18
h
20
f4n3o2计算为386.1492([m h]
)。实际为386.1491。
[0226]
r
f
=0.33(二氧化硅;dcm/甲醇95:5;uv或茚三酮)。
[0227]
2,6
‑
二氟
‑4‑
(n
‑
(1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸
‑
10
‑
戊二酰)
‑
乙氧基胺)
‑
2',6'
‑
二氟
‑
4'
‑
丁氧基偶氮苯(fazo)的合成
[0228]
将2,6
‑
二氟
‑4‑
氨基乙氧基
‑
2',6'
‑
二氟
‑
4'
‑
丁氧基偶氮苯6(267mg,0.535mmol,tfa盐)溶解在无水dmf(3.8ml)中。引入三乙胺(223μl,1.60mmol,3.0当量)后,将介质在氩气下于at下搅拌5分钟。然后引入dotaga酸酐(245mg,0.533mmol,1.0当量)并将介质在70℃下在氩气下搅拌21小时。浓缩后,加入乙醚(100ml),形成的沉淀物凝固30分钟,然后过滤并用乙醚(2
ⅹ
50ml)洗涤。通过反相快速色谱法(rp
‑
18,biotage,梯度h2o 0.05%fa/ch3cn 0.05%fa 1:0
‑
2:8)纯化粗产物。2.6
‑
二氟
‑4‑
(n
‑
(1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸
‑
10
‑
戊二酰)
‑
乙氧基胺)
‑
2,6
‑
二氟
‑
4'
‑
丁氧基偶氮苯fazo(92.1mg,0.109mmol,20%)通过冷冻干燥以黄色静电粉末的形式分离。
[0229]
hrms(m/z):c
37
h
50
f4n7o
11
计算为844.3504([m h]
)。实际为844.3495。
[0230]
lcms(方法b);t
r 14.107分钟(顺式)
‑
14.894分钟(反式)。
[0231]
azo和fazo络合的通用方案
[0232]
azo或fazo和金属试剂溶解在h2o(26mm)中。将ph调至5.5后,将介质在50℃下搅拌17小时,确保ph值保持在5.5
‑
6.0范围内。然后在通过冷冻干燥浓缩介质和反相快速色谱纯化(rp
‑
18,biotage,梯度h2o/ch3cn1:0
‑
0:1)之前将ph值调节至6.5。通过冷冻干燥将最终产
物以静电粉末的形式分离。
[0233]
[表1]
[0234][0235]
*:250mm在6n hcl中。
[0236]
表1.络合反应的实验条件。
[0237]
铜4
‑
(n
‑
(1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸
‑
10
‑
戊二酰)
‑
乙氧基胺)
‑
4'
‑
丁氧基偶氮苯(cuazo)
[0238]
hrms(m/z):c
37
h
51
n7nao
11
cu计算为855.2840([m na]
)。实际为855.2854.
[0239]
lcms(方法b);t
r 13.550分钟(顺式)
‑
15.934分钟(反式)。
[0240]
镓4
‑
(n
‑
(1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸
‑
10
‑
戊二酰)
‑
乙氧基胺)
‑
4'
‑
丁氧基偶氮苯(gaazo)
[0241]
hrms(m/z):c
37
h
51
n7o
11
ga计算为838.2902([m h]
)。实际为838.2906。
[0242]
lcms(方法b);t
r 12.840分钟(顺式)
‑
15.072分钟(反式)。
[0243]
钇4
‑
(n
‑
(1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸
‑
10
‑
戊二酰)
‑
乙氧基胺)
‑
4'
‑
丁氧基偶氮苯(yazo)
[0244]
hrms(m/z):c
37
h
49
n7na2o
11
y计算为902.2344([m
‑
h 2na]
)。实际为902.2349。
[0245]
lcms(方法b);t
r 15.957分钟(顺式)
‑
19.660分钟(反式)。
[0246]
铟4
‑
(n
‑
(1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸
‑
10
‑
戊二酰)
‑
乙氧基胺)
‑
4'
‑
丁氧基偶氮苯(inazo)
[0247]
hrms(m/z):c
37
h
49
n7na2o
11
in计算为928.2324([m
‑
h 2na]
)。实际为928.2319。
[0248]
lcms(方法b);t
r 15.451分钟(顺式)
‑
18.899分钟(反式)。
[0249]
铕4
‑
(n
‑
(1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸
‑
10
‑
戊二酰)
‑
乙氧基胺)
‑
4'
‑
丁氧基偶氮苯(euazo)
[0250]
hrms(m/z)(负离子模式):c
37
h
49
n7o
11
eu计算为920.2702([m
‑
h]
‑
)。实际为920.2694。
[0251]
lcms(方法b);t
r 13.419分钟(顺式)
‑
16.718分钟(反式)。
[0252]
钆4
‑
(n
‑
(1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸
‑
10
‑
戊二酰)
‑
乙氧基胺)
‑
4'
‑
丁氧基偶氮苯(gdazo)
[0253]
hrms(m/z):c
37
h
49
n7na2o
11
gd计算为971.2527([m
‑
h 2na]
)。实际为971.2529。
[0254]
lcms(方法b);t
r 15.956分钟(顺式)
‑
19.633分钟(反式)。
[0255]
钆2,6
‑
二氟
‑4‑
(n
‑
(1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸
‑
10
‑
戊二酰)
‑
乙氧基胺)
‑
2',6'
‑
二氟
‑
4'
‑
丁氧基偶氮苯(gdfazo)
[0256]
hrms(m/z):c
37
h
45
f4n7na2o
11
gd计算为1043.2150([m
‑
h 2na]
)。实际为1043.2158。
[0257]
lcms(方法b);t
r 18.900分钟(顺式)
‑
20.524分钟(反式)。
[0258]
镱4
‑
(n
‑
(1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸
‑
10
‑
戊二酰)
‑
乙氧基胺)
‑
4'
‑
丁氧基偶氮苯(ybazo)
[0259]
hrms(m/z):c
37
h
49
n7na2o
11
yb计算为987.2674([m
‑
h 2na]
)。实际为987.2683。
[0260]
lcms(方法b);t
r 16.112分钟(顺式)
‑
19.968分钟(反式)。
[0261]
铋4
‑
(n
‑
(1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸
‑
10
‑
戊二酰)
‑
乙氧基胺)
‑
4'
‑
丁氧基偶氮苯(biazo)
[0262]
hrms(m/z):c
37
h
49
n7na2o
11
bi计算为1022.3089([m
‑
h 2na]
)。实际为1022.3093。
[0263]
lcms(方法b);t
r 14.235分钟(顺式)
‑
19.055分钟(反式)。
[0264]
iii.电离辐射和细胞实验
[0265]
概括
[0266]
缓冲液培养基如磷酸盐缓冲盐水(pbs)、杜氏改良伊戈尔(dulbecco's modified eagle)培养基
‑
高葡萄糖(dmem)、洛斯维
·
帕克纪念研究所(roswell park memorial institute)培养基(rpmi)、青霉素、链霉素和台盼蓝溶液(0.4%)购自sigma aldrich(法国)。胎牛血清(fbs)购自gibco(法国),碘化丙啶(pi)购自赛默飞世尔科技公司(thermo fisher scientific,法国),盐酸吉西他滨(gem)购自红杉研究产品(sequoia research products,英国)。
[0267]
细胞系
[0268]
人胰腺癌(panc
‑
1)和人急性成淋巴细胞白血病(ccrf
‑
cem)细胞购自atcc(us)并按照供应商的建议进行维持。gem抗性人类急性成淋巴细胞白血病细胞(ccrf
‑
cem arac 8c,hent
‑
1受体未表达)由buddy ullmann博士(俄勒冈健康科学大学(oregon health sciences university))友情提供。简而言之,将panc
‑
1细胞维持在补充有10%(v/v)热灭活fbs的dmem缓冲液中。ccrf
‑
cem和ccrf
‑
cem arac
‑
8c细胞维持在补充有10%(v/v)热灭活fbs的rpmi缓冲液中。所有细胞培养基都补充有青霉素(50u.ml
‑1)和链霉素(0.05mg.ml
‑1)。将细胞维持在37℃和5%co2的潮湿气氛中。在达到第十八代之前使用细胞并在70
‑
80%汇合时收获。
[0269]
电离辐射源
[0270]
uv照射。在配备两个15w管(365nm)的cn
‑
15.lc室(vilber lourmat)中进行uv照射以诱导反式异构体异构化为顺式异构体,其在辐射位置提供0.817mw.cm
‑2的辐射(由cole
‑
parmer vlx
‑
3w微处理器控制的辐射计确定,校准为365nm,vilber lourmat)。
[0271]
x射线照射。使用x射线发生器(xrad 320 dx)进行x射线照射,提供平均能量为80kev(范围0
‑
200kev)的光子,剂量速率约为1gy/min(200kv,20ma)。辐射剂量以灰色表示。
[0272]
伽马射线照射。用铯137源(ibl637)进行伽马辐射,以约1gy/min的剂量速率提供662kev光子。辐射剂量以灰色表示(1gy=1j/l)。
[0273]
电子束照射。使用线性电子加速器(linac,kinetron)以大约4gy/min的剂量速率
以4.5mev的能量传递电子进行电子束照射。辐射剂量以灰色表示。
[0274]
通过吸收分光光度法和hplc对电离辐射诱导的异构化进行量化(图1)
[0275]
将含钆化合物(gdazo或gdfazo)和对照化合物(azo或fazo)(50μm,200μl,pbs)引入两个96孔微孔板(每种化合物10个孔)。将两个微孔板首先在紫外光下照射(365nm,0.817mw.cm
‑2,5分钟)。然后将一个微孔板(板1)保持在黑暗中并用作未照射的对照,而第二块微孔板(板2)则用增加的电离辐射剂量(2gy、3gy、5gy和10gy)进行照射。每次照射后,对未照射(板1)和照射(板2)化合物进行吸光度分光光度法和hplc注射(方法c)分析。使用板1和板2的uv照射之间的26分钟时间延迟,以便在uv照射后同时分析未照射的对照化合物(板1)与电离照射的化合物(板2)。完整的实验在4.6小时内进行。通过三次重复的平均值绘制吸收光谱。每种异构体的相对量通过hplc(在洗脱条件下在等吸收点波长处检测)获得,分子活化(%)由介质中的反式异构体比例的差异确定(3个独立实验)。
[0276]
无电离辐射的共聚焦显微镜(图2a)
[0277]
实验开始前24h将细胞(panc
‑
1)转移至成像板(ibitreat 8孔显微镜玻片,购自德国ibidi),在37℃和5%co2的潮湿环境下维持在培养基中(33000个细胞,200μl/孔)。在实验之前即刻,将细胞培养基替换为不含fbs和酚红的培养基(100μl),然后将ip(5μl,终浓度为1μm)添加到培养基中。添加ip后15分钟,获取了第一组图像。然后将顺式gdazoor、反式gdazo化合物(100μl,终浓度0μm、250μm或500μm)引入细胞培养基中。然后将成像板保持在黑暗中,每5分钟采集一次图像,持续1小时。通过uv照射(365nm,0.817mw.cm
‑2,30min)96孔微孔板中的化合物反式gdazo(105μl,500μm或1mm,顺式gdazo>85%)得到化合物顺式gdazo。利用配有fluotar cs2 10
ⅹ
/0.30秒hc pl物镜的leica tcs sp8门控sted倒置显微镜(leica,德国)观察细胞。该仪器配有白光激光器(激发波长538nm)。在560
‑
650nm的带宽上收集红色荧光发射,并使用相同的光线和pmt
‑
trans检测器获得透射图像。针孔设置为1.0airy(直径73.19μm)。使用leica sp8 las x软件(2.0.1版,leica,德国)获得16位数字图像。
[0278]
存在电离辐射的共聚焦显微镜(图2b
‑
c)
[0279]
在实验开始前24小时,将细胞(panc
‑
1)转移到成像板(lab
‑
tek室载玻片系统,玻璃,8孔,购自thermo fisher scientific,法国),并在37℃和5%co2的潮湿环境下保持在培养基中(10,000个细胞,200μl/孔)。就在实验之前,将细胞培养基替换为pbs(100μl),并将ip(5μl,终浓度1μm)加入培养基中。添加pi后15分钟,将顺式gdazo(100μl,终浓度为0μm、250μm、500μm或850μm)引入细胞培养基中。在这个阶段采集了第一组图像,然后照射成像板(伽马射线,2gy)。然后将成像板保持在37℃(板加温器)的黑暗中,每5分钟采集一次图像,持续30分钟,然后每10分钟采集一次,持续90分钟。除了成像板没有用伽马射线辐射之外,类似的程序用于未辐射的对照实验。顺式gdazo化合物是通过uv照射(365nm,0.817mw.cm
‑2,30分钟)96微孔板中的反式gdazo化合物(105μl、500μm、1mm或1.7mm,顺式gdazo>85%)获得的。使用
ⅹ
10秒物镜、n.a.0.4并配备yokogawa csu
‑
x1头的尼康倒置显微镜(nikon instruments inc.,东京,日本)观察细胞。该仪器配备了激发波长为561nm的激光二极管。在598
‑
672nm的带宽上收集红色荧光发射,并用白色二极管获得透射图像。针孔设置为50μm,放大透镜设置为1.2。图像由e
‑
volves
‑
cmos相机(photometrics)记录。每孔记录四个图像,每个浓度的顺式gdazo使用两个孔。使用imagej软件(带有可调分水岭插件的1.50i版)
对16位数字图像进行分析。辐射前获得的图像中的细胞数量是手动确定的,辐射前后获得的图像中的荧光细胞的数量是使用imagej上编码的脚本自动计算的。细胞通透性是通过伽马辐射之前和之后30分钟荧光细胞比例的差异来确定的(3个独立实验)。
[0280]
电离辐射下的治疗效果(图2d
‑
f)
[0281]
就在实验之前,将细胞(ccrf
‑
cemarac
‑
8c)分散在pbs中并转移到48孔微孔板(tpp细胞培养微孔板,购自thermo fisher scientific,法国)(40,000个细胞,80μl/孔)中。将gem(20μl,终浓度0.1μm)或pbs(20μl)和顺式gdazo化合物(100μl,终浓度0μm、250μm、500μm或850μm)在培养基的伽马辐射之前立即加入(2gy)。将培养基在黑暗和潮湿的气氛中于37℃和5%co2保持1小时。接下来,将培养基(600μl)添加到每个孔中,并通过三个离心循环(300g,5分钟,每次洗涤使用800μl培养基)洗涤细胞。最后将细胞分散在含有或不含gem(终浓度为0μm或0.1μm)的培养基(600μl)中,并在37℃和5%co2的潮湿环境中保持4天。在存在1:1(v/v)台盼蓝(一式三份)的情况下,通过细胞计数确定活细胞的数量。实验独立重复3次。细胞活力表示为处理后的活细胞数量与未经任何处理(无辐射,不存在gem和顺式gdazo)的活细胞数量之比。除了未用伽马射线照射48孔板之外,类似的程序用于未照射的对照实验。除了使用代替顺式gdazo外,类似的程序用于的对照实验(图2f)。通过紫外线照射(365nm,0.817mw.cm
‑2,30min)96孔微孔板中的反式gdazo化合物(105μl,500μm,1mm或1.7mm,顺式gdazo>85%)获得顺式gdazo化合物。
[0282]
这些结果表明:
[0283]
(i)通过使用电离辐射(x射线、伽马射线、电子)激活治疗分子的新构思(图1a
‑
f)。由于这些辐射在活组织中具有非常高的穿透能力,因此这种新的激活构思具有强大的临床应用潜力。当下文献中描述的所有有机分子都需要使用紫外线到近红外线辐射,这种辐射不会深入穿透生物组织(在最佳条件下可以穿透数百微米)。
[0284]
(ii)根据所使用的金属和辐射源,可以通过不同效率的电离辐射来激活mazo化合物(m=cu、ga、y、in、eu、gd、yb、bi)(图1a
‑
c,f)。gdfazo化合物也可以通过电离辐射激活(图1c
‑
e)。这种化合物的特点是足够缓慢的热弛豫,以考虑在体内进行研究。尽管在体外使用的实验条件下,化合物gdfazo的活化相对较低(与化合物gdazo相比,图1c),但很难预测该化合物在体内的分子活化效率。
[0285]
(iii)用于使得能够激活有机单元的金属类型。已经表明,金属的原子序数z必须大于39(钇的原子序数)以允许一致的活化(在5gy下大于30%)。事实上,含有大小大于或等于钇的金属的6种化合物显示出这种活化,而含有较小金属(cu,z=29和ga,z=31)的2种化合物显示出低活化(在5gy下小于10%)(图1f)。
[0286]
(iv)分子活化可以在各种类型的电离辐射(x射线、伽马射线、电子)下进行,具有不同的粒子(光子和电子)和不同的能量(从1kev到4.5mev)(图1c),因此,目前临床使用的一组放射治疗设备应该能有效地产生所描述的分子激活。
[0287]
(v)化合物gdazo能够在低剂量的电离辐射下诱导癌细胞膜的通透性(gdazo在浓度分别为250μm、500μm和850μm,通过2gy的伽马射线时,6.9%(相对于1.2%未辐射的)、8.2%(相对于2.8%未辐射的)和12.4%(相对于7.5%未辐射的))(图2a、b、c)。
[0288]
(vi)在吉西他滨不存在或存在的情况下,化合物gdazo能够在吉西他滨抗性癌细胞系(人急性成淋巴细胞白血病)中诱导毒性(图2d、e),用2gyγ辐射处理后在没有吉西他
滨的情况下,gdazo在浓度分别为250μm、500μm和850μm时,治疗4天后的细胞活力分别降低至23%(相对于79%未辐射的)、18%(相对于42%未辐射的)和3.0%(相对于27%未照射的)。在吉西他滨存在的情况下,gdazo在浓度分别为250μm、500μm和850μm时,治疗4天后的细胞活力分别降低至15%(相对于49%未照射的)、9.1%(相对于30%未照射的)和1.2%(相对于16%未照射的)。该研究表明,该化合物在电离辐射下单独具有活性,并且周围活性物质的存在仅略微增强了该化合物的治疗效果(图2d、e)。
[0289]
(vii)由于商业对照化合物(单独钆螯合物)在电离辐射下没有治疗效果,因此分子上偶氮苯或茋(stillbene)基序的存在是产生治疗效果所必需的(图2f)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。