1.本公开涉及合成紫花前胡素衍生物(,decursin derivative)的新方法。
背景技术:
2.随着人类预期寿命的延长,人们对衰老进程的关注也在积极地提高。然而,目前仍有一些领域尚不清楚,最近的研究聚焦在衰老的遗传或分子机制上,主要针对人类早衰。
3.早衰或hutchinson-gilford早衰综合征(hgps)是一种致命的罕见遗传性疾病,可导致幼儿过早衰老。早衰儿童在婴儿期早期表现正常,但在9-24个月左右开始出现明显的生长迟缓,最终导致身材矮小和体重低下。此外,伴随着面部形状不同、全身动脉粥样硬化、心血管疾病、中风和髋关节脱位,还会出现皮下脂肪层缺失、指甲缺陷、关节硬化和骨骼损伤。这些早衰的儿童患者通常在8-21岁之间死于心脏病,平均预期寿命约为13岁。
4.hgps是一种非常罕见的常染色体显性遗传病,由核纤层蛋白a(lmn a)中g608g的沉默突变引起。该突变导致产生新的切割供体位点并产生早衰蛋白(progerin,prg),该蛋白是选择性切割位点的产物,从该选择性切割位点,核纤层蛋白a的c-端结构域中的50个氨基酸被删除。
5.早衰蛋白的表达诱导细胞形态学改变,如核膜的不规则性或细胞核-细胞质的核纤层蛋白a的减少,早衰蛋白表达的抑制还诱导核转化的减少。因此,早衰蛋白被确定作为hgps的主要因子。
6.因此,在韩国专利公开第10-2018-0019490号中,作为新化合物的紫花前胡素衍生物化合物已被证明在治疗衰老相关疾病以及预防或改善皱纹方面是有效的。然而,由于紫花前胡素衍生物化合物的产量较低,在大规模生产中受到限制。
7.因此,需要研究一种新的合成方法,该方法能够提高所述紫花前胡素衍生物化合物的收率并实现大规模生产。
技术实现要素:
8.技术目标
9.本发明的目的是提供一种合成新型紫花前胡素衍生物的方法,该方法能够提高紫花前胡素衍生物化合物的收率并实现大规模生产。
10.技术方案
11.为实现上述目的,本发明的示例实施方式提供了一种合成紫花前胡素衍生物的方法,该方法包括:(i)通过混合肉桂基溴和作为溶剂的n-甲基-2-吡咯烷酮(nmp),制备溶液;(ii)通过混合紫花前胡醇(,decursinol)、氢化钠(nah)和作为溶剂的四氢呋喃(thf),制备溶液;以及(iii)通过混合(i)和(ii)中制备的所述溶液,获得由化学式1表示的紫花前胡素衍生物。
12.[化学式1]
[0013][0014]
有益效果
[0015]
根据本发明的示例实施方式的合成紫花前胡素衍生物化合物的新方法,可以提高所获得的紫花前胡素衍生物化合物的收率,确保在附加的重结晶过程后的收率为80%或更高,还可以实现大规模生产。
附图说明
[0016]
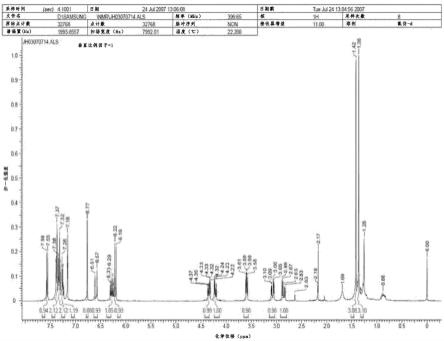
图1是显示根据本公开的示例实施方式合成的紫花前胡素衍生物的nmr结果的图。
具体实施方式
[0017]
下文将详细描述本公开。
[0018]
由肉桂基溴和紫花前胡醇合成紫花前胡素衍生物的过程中,当使用n-甲基-2-吡咯烷酮(nmp)溶剂、四氢呋喃(thf)溶剂和氢化钠(nah)进行合成时,本发明人能够提高所获得的紫花前胡素衍生物化合物的收率,通过确认在附加的重结晶过程后取得80%或更高的高收率的最佳新合成方法,完成了本公开。
[0019]
本公开的示例实施方式提供了一种合成紫花前胡素衍生物的方法,该方法包括:(i)通过混合肉桂基溴和作为溶剂的n-甲基-2-吡咯烷酮(nmp),制备溶液;(ii)通过混合紫花前胡醇、氢化钠(nah)和作为溶剂的四氢呋喃(thf),制备溶液;以及(iii)通过混合(i)和(ii)中制备的所述溶液,获得由化学式1表示的紫花前胡素衍生物。
[0020]
[化学式1]
[0021][0022]
这里,以重量比1:(5-10)含有所述肉桂基溴和nmp,可通过混合肉桂基溴和nmp,然后,在15-25℃下搅拌所得混合物来制备溶液。
[0023]
此外,以重量比(5-10):(50-100):1含有紫花前胡醇、thf和nah,混合紫花前胡醇和thf,并在15-25℃下搅拌所得混合物,然后在-5-5℃下添加nah,由此制备溶液。
[0024]
此外,所述紫花前胡素衍生物的获得包括如下步骤:将(iii)中制备的所述混合溶液的ph值调节至7或更低(第一步骤);从所述第一步骤的所述溶液获得有机层(第二步骤);减压浓缩所述第二步骤获得的所述有机层,获得反应溶液(第三步骤);以及,通过对所述第三步骤中获得的所述反应溶液进行热处理和冷却,然后,通过过滤和干燥,获得所述紫花前胡素衍生物(第四步骤)。
[0025]
通过如上所述的合成紫花前胡素衍生物的方法,可以获得收率为58%的(7s)-( )-8,8-二甲基-7-(3-苯基-烯丙基氧基)-7,8-二氢-6h-吡喃并[3,2-g]色烯-2-酮(slc-d011)化合物,与收率为30-40%的传统合成方法相比,这是获得更高收率的最佳合成方法。
[0026]
在这种情况下,该方法可进一步包括将获得的紫花前胡素衍生物进行重结晶。
[0027]
重结晶可包括如下步骤:通过将丙酮与获得的所述紫花前胡素衍生物混合,制备反应溶液(第一步骤);通过将晶种与所述第一步骤的所述反应溶液混合,制备反应溶液(第二步骤);将所述第二步骤的所述反应溶液与异丙醇混合,将获得的混合物减压浓缩(第三步骤);以及,通过将所述第三步骤中减压浓缩后的溶液进行过滤和干燥,获得所述紫花前胡素衍生物(第四步骤)。
[0028]
此处,晶种是纯度为99.5%以上、单个有关物质为0.10%以下的紫花前胡素衍生物,当晶种用于结晶过程时,有助于结晶。此外,可以去除有关物质,同时易于控制多晶型。
[0029]
通过进一步包括紫花前胡素衍生物的重结晶,确定能够以86%的非常高的收率获得(7s)-( )-8,8-二甲基-7-(3-苯基-烯丙基氧基)-7,8-二氢-6h-吡喃并[3,2-g]色烯-2-酮(slc-d011)化合物。
[0030]
如果反应物含量、温度和混合条件的范围超出合成紫花前胡素衍生物的方法中的范围,则根据本发明示例实施方式的紫花前胡素衍生物化合物的收率显著降低,或者几乎不可能大规模生产,这可能没有成本效益。
[0031]
此外,(7s)-( )-8,8-二甲基-7-(3-苯基-烯丙基氧基)-7,8-二氢-6h-吡喃并[3,2-g]色烯-2-酮(slc-d011)化合物是根据本发明的示例实施例合成的紫花前胡素衍生物,可有效用于治疗衰老相关疾病,例如hutchinson-gilford早衰综合征(hgps)和werner综合征;因为该化合物能够增加皮肤细胞中胶原蛋白的生成,可有效用作预防或改善皱纹的化妆品组合物;该化合物还可用于预防和治疗特应性皮炎的药物组合物和保健功能食品。
[0032]
下文中,将通过示例实施方式更详细地描述本公开。示例实施方式仅用于更详细地描述本发明,对于本公开所属领域的普通技术人员显而易见的是,根据本公开的主旨,本公开的保护范围不受示例实施方式的限制。
[0033]
《比较实施例1》醚形式的( )-紫花前胡素衍生物(slc-d011)的合成
[0034]
[方案1]
[0035][0036]
如方案1所示,在氮气存在下,使用将n,n-二甲基甲酰胺(dmf,10ml)将(s)-( )-紫花前胡醇(slc-b001,2.33g,9.47mmol,1当量)溶解于100ml圆形烧瓶中,然后,将该烧瓶安装在设置为-20℃的低温反应器中。
[0037]
将(e)-肉桂基溴((3-溴丙烯基)-苯,2.8g,14.2mmol,1.5当量)和氢化钠(nah,60%,757mg,18.9mmol,2当量)添加到反应混合物溶液中并搅拌4小时后,添加3ml蒸馏水。10分钟后,将混合物从低温反应器中取出,用200ml二氯甲烷和200ml蒸馏水分离两次。收集
有机层,用硫酸钠脱水,然后过滤。然后,在减压下浓缩滤液。
[0038]
浓缩物经硅胶柱分离(乙酸乙酯:正己烷=从1:10梯度洗脱至1:3),获得1.21g(35.3%)的(7s)-( )-8,8-二甲基-7-(3-苯基-烯丙基氧基)-7,8-二氢-6h-吡喃并[3,2-g]色烯-2-酮(slc-d011);收率35.3%,白色固体,mp:143℃,rf=0.39(2:1正己烷-乙酸乙酯);[α]
25d
117.6(c=1,chcl3);1h nmr(400mhz,cdcl3):δ
h 7.56(1h,d,j=9.6hz,h-4),7.38-7.23(5h,m,h-5’,h-6’,h-7’,h-8’,h-9’),7.15(1h,s,h-5),6.76(1h,s,h-10),6.59(1h,d,j=16.0hz,h-3’),6.30-6.23(1h,m,h-2’),6.20(1h,d,j=9.6hz,h-3),4.34(1h,dd,j=6.0,12.8hz,h-1a’),4.21(1h,dd,j=60,12.4hz,h-1b’),3.59(1h,dd,j=5.2,7.6hz,h-7),3.07(1h,dd,j=4.8,16.0hz,h-6a),2.85(1h,dd,j=7.2,16.4hz,h-6b),1.41(3h,s ch
3-8),1.36(3h,s,ch
3-8);
13
c nmr(100mhz,丙酮-d6)δc161.2(c-2),157.8(c-9a),155.3(c-10a),144.5(c-4),137.9(c-4’),132.9(c-3
′
),130.4(c-5),129.6(c-6’,c-8
′
),128.6(c-7’),127.5(c-2’),127.4(c-5
′
,c-9’),118.3(c-5a),113.7(c-3),113.6(c-4a),104.5(c-10),78.8(c-7),76.4(c-8),70.8(c-1’),27.8(c-6),26.1(ch
3-8),22.2(ch
3-8);esi-ms:m/z=363[m h]
.c
23h22
o4的分析计算值:c,76.22;h,6.12;实测值:c,76.20;h,6.10。
[0039]
《比较实施例2》醚形式的( )-紫花前胡素衍生物(slc-d011)的合成
[0040]
除了使用氢氧化钾代替在《比较实施例1》中的氢化钠(nah)之外,在与《比较实施例1》中相同的条件下进行合成。
[0041]
结果,确定以44%的收率获得了白色固体紫花前胡素衍生物(7s)-( )-8,8-二甲基-7-(3-苯基-烯丙基氧基)-7,8-二氢-6h-吡喃并[3,2-g]色烯-2-酮(slc-d011)。
[0042]1h nmr(400mhz,cdcl3):δ
h 7.56(1h,d,j=9.6hz,h-4),7.38-7.23(5h,m,h-5’,h-6’,h-7’,h-8’,h-9’),7.15(1h,s,h-5),6.76(1h,s,h-10),6.59(1h,d,j=16.0hz,h-3’),6.30-6.23(1h,m,h-2’),6.20(1h,d,j=9.6hz,h-3),4.34(1h,dd,j=6.0,12.8hz,h-1a’),4.21(1h,dd,j=60,12.4hz,h-1b’),3.59(1h,dd,j=5.2,7.6hz,h-7),3.07(1h,dd,j=4.8,16.0hz,h-6a),2.85(1h,dd,j=7.2,16.4hz,h-6b),1.41(3h,s ch
3-8),1.36(3h,s,ch
3-8).
[0043]
《实施例1》新的醚形式的( )-紫花前胡素衍生物(slc-d011)的合成
[0044]
1-1.(e)-肉桂基溴溶液(溶液1)的制备
[0045]
在反应器中,将(e)-肉桂基溴(12.4kg,60.9摩尔,1.24当量)和n-甲基-2-吡咯烷酮(nmp,63.8kg)混合,并在15-25℃下搅拌。
[0046]
1-2.s-紫花前胡醇溶液(溶液2)的制备
[0047]
在反应器中,将s-紫花前胡醇(12.1kg,49.1摩尔,1.0当量)和四氢呋喃(thf,122kg)混合,并在15-25℃下搅拌。缓慢添加氢化钠(nah,1.81kg,75.4摩尔,1.54当量)至少1小时,同时将反应器中的温度保持在-5-5℃。
[0048]
1-3.将所述溶液1和所述溶液2混合
[0049]
将所述溶液1添加到所述溶液2中,同时将内部温度保持在-5-5℃,然后搅拌至少10小时。
[0050]
反应完成后,添加乙酸(6.6kg),检查反应溶液的ph值为7或更低(≤7),并将混合物在-5-5℃下搅拌1-3小时。添加甲基叔丁基醚(mtbe,92kg)和纯化水(124kg),并在20-30
℃下将混合物搅拌20-30分钟。当各层分离时,有机层留在反应器中,水层被丢弃。向有机层中添加纯化水(62kg),搅拌20分钟后分离各层。然后,丢弃水层,再次向有机层中添加纯化水(62kg)。搅拌20分钟分离各层后,丢弃水层。
[0051]
在40℃或更低温度下,向有机层添加甲苯(62kg),然后减压浓缩,直到体积变为25-37l。然后,再添加甲苯(62kg),并在减压下再次浓缩,直到溶液变为25-37l。
[0052]
此后,将反应溶液在80-85℃下加热2小时,在45-50℃下搅拌2-4小时,然后在-5-5℃下搅拌4-8小时后冷却。
[0053]
使用离心过滤器过滤产品,用冷却的甲苯(-5-5℃,27kg)清洗过滤后的产品,然后在35-40℃下干燥,直到残留甲苯变为0.5%或更少。
[0054]
经测定,干燥产物为白色固体(7s)-( )-8,8-二甲基-7-(3-苯基-烯丙基氧基)-7,8-二氢-6h-吡喃并[3,2-g]色烯-2-酮(slc-d011),收率为10.4kg(58%),纯度为99.0%(hplc面积%),含量为98.6%。
[0055]1h nmr(400mhz,cdcl3):δ
h 7.56(1h,d,j=9.6hz,h-4),7.38-7.23(5h,m,h-5’,h-6’,h-7’,h-8’,h-9’),7.15(1h,s,h-5),6.76(1h,s,h-10),6.59(1h,d,j=16.0hz,h-3’),6.30-6.23(1h,m,h-2’),6.20(1h,d,j=9.6hz,h-3),4.34(1h,dd,j=6.0,12.8hz,h-1a’),4.21(1h,dd,j=60,12.4hz,h-1b’),3.59(1h,dd,j=5.2,7.6hz,h-7),3.07(1h,dd,j=4.8,16.0hz,h-6a),2.85(1h,dd,j=7.2,16.4hz,h-6b),1.41(3h,s ch
3-8),1.36(3h,s,ch
3-8)(图1).
[0056]
《实施例2》slc-d011的重结晶
[0057]
将《实施例1》中获得的(7s)-( )-8,8-二甲基-7-(3-苯基-烯丙基氧基)-7,8-二氢-6h-吡喃并[3,2-g]色烯-2-酮(slc-d011,10.4kg,26.7摩尔)添加到反应器中。添加丙酮(68kg),并在50-55℃下搅拌10-40分钟。在保持溶液温度在50-55℃的同时,通过滤芯式过滤器过滤反应溶液。然后,将滤液在50-55℃下搅拌40分钟。
[0058]
用时0.5-1小时,将反应溶液缓慢冷却至40-45℃后,添加100g晶种(纯度99.5%或以上、单个有关物质0.10%或以下的产品),并搅拌1.5-2.5小时。此后,用时0.5-1小时,将反应溶液冷却至20-25℃。
[0059]
在保持20-25℃温度的同时,用时4-8小时,添加异丙醇(127kg)。在25℃或更低温度下对反应溶液进行减压蒸馏,直到溶液体积变为190-210l。然后,用时2-3小时,将反应溶液冷却至0-5℃,并搅拌12-16小时以获得产物。
[0060]
使用过滤干燥器过滤产品,并用异丙醇(1kg
×
2)清洗过滤后的产品两次。将产品置于干燥器中,在20-30℃下干燥20-24小时。经测定,再纯化的(7s)-( )-8,8-二甲基-7-(3-苯基-烯丙基氧基)-7,8-二氢-6h-吡喃并[3,2-g]色烯-2-酮为白色固体,具有以下特征:收率:9.05kg(86%),纯度(hplc):99.9%,手性纯度:100%,含量(hplc):98.3%,lod(干燥失重):0.1%,水含量(卡尔
·
费休):0.1%,dsc:起始149℃、峰值150.3℃,熔点148-149℃。
[0061]1h nmr(400mhz,cdcl3):δ
h 7.56(1h,d,j=9.6hz,h-4),7.38-7.23(5h,m,h-5
′
,h-6’,h-7’,h-8’,h-9’),7.15(1h,s,h-5),6.76(1h,s,h-10),6.59(1h,d,j=16.0hz,h-3’),6.30-6.23(1h,m,h-2’),6.20(1h,d,j=9.6hz,h-3),4.34(1h,dd,j=6.0,12.8hz,h-1a’),4.21(1h,dd,j=60,12.4hz,h-1b’),3.59(1h,dd,j=5.2,7.6hz,h-7),3.07(1h,dd,j=
4.8,16.0hz,h-6a),2.85(1h,dd,j=7.2,16.4hz,h-6b),1.41(3h,s ch
3-8),1.36(3h,s,ch
3-8).
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。