人nr1d1报告基因质粒、稳转细胞株及其构建方法和应用
技术领域
1.本发明涉及生物医药技术领域,特别涉及人nr1d1报告基因质粒、稳转细胞株及其构建方法和应用。
背景技术:
2.核受体rev-erbα(编码基因为nr1d1)是一种转录抑制因子,它通过招募共抑制因子ncor和组蛋白脱乙酰酶3(hdac3)结合到靶基因的rore元件上,行使转录抑制功能。rev-erbα也是分子生物钟的重要组成元件之一,这与其调控核心时钟基因bmal1的表达有关。rev-erbα参与调控细胞增殖、代谢和炎症等过程,在许多疾病(如失眠、代谢性疾病、炎症性疾病、心血管疾病、癌症和退行性疾病)的发生和发展过程中发挥重要作用。例如,rev-erbα能通过直接调控nf-κb表达,抑制nlrp3炎症小体的激活和炎症反应的发生;rev-erbα能调节肝脏和脂肪组织中参与葡萄糖和脂质代谢和转运的关键基因的表达,在调控血糖和脂质代谢及糖尿病和肥胖等代谢性疾病中发挥调控作用。因此,调节nr1d1/rev-erbα的表达或活性是许多疾病的潜在治疗手段。
3.基于rev-erbα蛋白配体结合域(lbd)与小分子的互作研究,目前研究人员已发现多种能直接与rev-erbα蛋白结合并调节其功能的激动剂(如sr9009)和拮抗剂(如sr8278)。sr9009能有效缓解葡聚糖硫酸钠(dss)诱导的结肠炎、脂多糖(lps)诱导的多器官炎症和肝缺血再灌注损伤,且对多种肿瘤细胞的增殖具有抑制作用。sr8278对高同型半胱氨酸血症、急/慢性肾病和急性心肌埂塞等疾病具有一定治疗效果。然而,这些rev-erbα配体由于脱靶效应或药代动力学属性较差等原因,均未进入临床研究。
4.nr1d1是一种节律性基因,其启动子含有能被时钟因子结合的响应元件:e-box(受bmal1/clock调控)、d-box(受dbp/e4bp4调控)或rore/revre(受rors/rev-erbs调控)。此外,肝x受体(lxr)、过氧化物酶体增殖物激活受体γ(pparγ)和糖皮质激素受体(gr)也能调控nr1d1的表达。另一方面,一些药物的作用效果与其调控nr1d1基因的转录表达有关。例如,贝特类药物能通过pparα受体上调nr1d1的表达,而地塞米松则通过gr抑制肝细胞中nr1d1的表达。
5.荧光素酶报告基因系统已被广泛用于研究目的基因的转录调控。然而,基于nr1d1基因启动子的报告系统仍未被建立。并且,目前常用的荧光素酶报告基因测定方法需要裂解细胞后再往裂解液中加入荧光素底物进行发光检测,耗时且花费较高(所使用的商品化试剂盒较为昂贵),在大规模调节剂的筛选中应用受限。因此,构建一种nr1d1报告基因质粒和稳转细胞株,并基于稳转细胞株发展一种高通量方法应用于nr1d1调节剂的筛选,对于发现新的nr1d1调节剂并将其应用于rev-erbα相关疾病的治疗具有重要意义。
技术实现要素:
6.为解决相关问题,本发明的首要目的在于提供人nr1d1报告基因质粒。
7.本发明的另一目的在于提供人nr1d1报告基因稳转细胞株。
8.本发明的再一目的在于提供人nr1d1报告基因稳转细胞株的应用。
9.为了实现上述发明目的,本发明采用以下技术方案:
10.人nr1d1报告基因质粒的制备方法,是将人nr1d1基因启动子序列插入至plv6-bmal-luc质粒(addgene#68833,带有萤火虫荧光素酶序列的plenti6.2载体)中,获得人nr1d1基因启动子驱动的荧光素酶报告质粒;其中,所述的人nr1d1基因启动子序列为人nr1d1基因-1200~ 20bp区间序列,具体如seq id no:1所示。
11.进一步地,所述的人nr1d1基因启动子序列的插入位点为xhoi和bsrgi双酶切位点之间。
12.人nr1d1报告基因质粒,通过上述制备方法得到。
13.人nr1d1报告基因稳转细胞株的制备方法,将上述人nr1d1报告基因质粒经慢病毒包装后感染u-2os细胞,然后采用blasticidin s进行抗性筛选,最后通过有限稀释法获得稳定表达nr1d1-luc的单克隆细胞株。
14.进一步地,所述的感染过程中病毒接种的moi为20~40。
15.进一步地,所述的blasticidin s的使用浓度为3.5~4.5μg/ml;优选为4μg/ml。
16.人nr1d1报告基因稳转细胞株,通过上述制备方法得到。
17.上述人nr1d1报告基因稳转细胞株的应用,用于高通量筛选对nr1d1基因转录活性具有调控作用的药物。
18.进一步地,所述的对nr1d1基因转录活性具有调控作用的药物为nr1d1激动剂或nr1d1抑制剂。
19.进一步地,所述的应用的具体步骤为:将所述的人nr1d1报告基因稳转细胞株接种至培养容器中,加入候选药物作用一定时间后,更换成含有荧光素酶底物的无酚红培养基,采用微孔板发光检测仪进行快速检测,通过发光值表征nr1d1基因转录活性变化,从而筛选出对nr1d1基因转录活性具有调控作用的药物。
20.一种节律细胞模型的制备方法,将上述人nr1d1报告基因稳转细胞株接种至培养容器中,待融合度达100%后更换为节律诱导培养基继续培养,获得萤火虫荧光素酶基因表达呈周期性变化的节律细胞模型;其中,所述的节律诱导培养基的组成如下:含0.29mg/mll-谷氨酰胺的低糖dmem、3.5mg/ml d-葡萄糖、0.35mg/ml碳酸氢钠、100nm地塞米松、10mm羟乙基哌嗪乙硫磺酸(hepes)、10%(v/v)fbs、100单位/ml青霉素、100μg/ml链霉素,ph 7.3。
21.一种节律细胞模型,通过上述制备方法得到。
22.上述节律细胞模型的应用,用于生物节律研究。
23.本发明相对于现有技术具有如下的优点及效果:
24.本发明构建了一种人nr1d1基因启动子驱动的荧光素酶报告质粒和稳转细胞株,可用于快速表征nr1d1转录活性的变化;
25.另外,基于所构建的nr1d1-luc稳转细胞株,发展了一套nr1d1调节剂的高通量筛选方法,并发现了多种对nr1d1转录活性具有调控作用的小分子;
26.此外,nr1d1-luc细胞在地塞米松的诱导下具有明显的节律性,表明其可作为一种新的节律细胞模型应用于生物节律领域相关研究。
附图说明
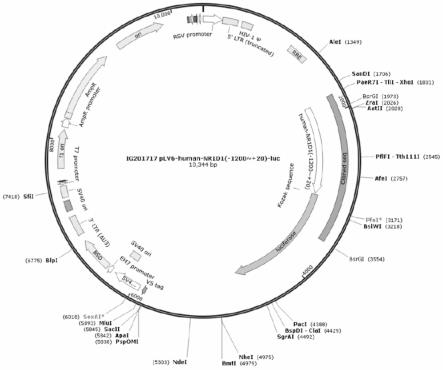
27.图1是nr1d1-luc质粒图谱;
28.图2是nr1d1-luc病毒滴度测定结果图;
29.图3是u-2os细胞加不同浓度blasticidin s的药筛结果图;
30.图4是野生型及转染了nr1d1-luc质粒的u-2os细胞的药筛结果比较图;
31.图5是nr1d1-luc单克隆细胞的形态及pcr鉴定结果图;
32.图6是基于nr1d1-luc细胞的高通量方法测定1760种天然小分子对nr1d1的调控作用测定结果图;
33.图7是高通量方法(intact cells)和传统裂解法(cell lysates)对36种小分子的测定结果对比图;
34.图8是地塞米松诱导的nr1d1-luc细胞节律测定结果图。
具体实施方式
35.下面结合实施例和附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
36.实施例1:nr1d1-luc质粒的构建
37.将表达了bmal-luc基因的plenti6.2质粒(plv6-bmal-luc,addgene#68833)经xhoi和bsrgi酶切后,琼脂糖电泳回收目的片段dna;合成人nr1d1基因的近端启动子(-1200~ 20bp)序列(如seq id no:1所示),并将其克隆至plv6-bmal-luc质粒(addgene#68833)的xhoi和bsrgi酶切位点间替换原载体中bmal1启动子序列(图1);加dna连接酶连接并转化至感受态dh5α细菌后,接种至含氨苄西林的固体培养板培养,次日挑取单克隆细菌于含抗生素的培养基中扩大培养;采用质粒提取试剂盒抽提质粒,经测序验证nr1d1-luc质粒的序列正确。
38.结果显示(图1),nr1d1基因的近端启动子(-1200~ 20bp)成功插入至hnr1d1-luc质粒中,所插入的nr1d1启动子也是驱动萤火虫荧光素酶基因表达的启动子,表明nr1d1-luc质粒构建成功。
39.人nr1d1基因的近端启动子序列(seq id no:1):
40.tggtttcaccgtgttggtcaggctggtctcaaactcctgacctcaggtgatcaacccacctcagcctcacaaagtgctgggattacaggcatgagccaaagcacccggcaatgctggctgtttctaacccctgttcagtatttcacttgtacatctacccaccttcccattcggggtgggcagatgaaactagcaatggacgtctgaccttgggtcggtcacttctcctaagcttcctgttccccactagtaaaaagagggaggcttaagatgatctacatgttcccctctgagtagtaatcttctgtggaattcatattttatcctccagcaccgaggggcaggggtgtcactctgcccccaccccctgcctcacctcttccccattactttaggacctcaaagcactttcactattagttcccctctgttgtcctttttatttcccagacaaagggaaatgactcaccccaaagtcaactggagtgggtggaatggtgtcatacaagcaaacagggagtccctacagacatccctacctctgtgggaactccttcccctggaggtgttctccctaaggcgagtagaagggaaagggggtcacatttcctttccttctctggactttgccctgaagcagagggcagcctaagctcctgactccagggaaatctccctccccggcttctctctctcccggtcaccagtaacctcaggacgaggtcagtcctgcaatcacgtgaagccctcacgtttgcaaggtttgcagaaagggcctcttagctttgatctcccagacagcaaacaagcttgccagtccctccccagaaattcacatgcccctgccatacaggctttctaaacacgccaccctgactcttcagcgcaccccaccccaccccac
tctcagctcctcccaggtcccggcaagcgctttgccaggcagaaaggggaaaggcacgcagtccgcccactttgtcggtggactacaaatcccgacagtcttgtcgttgcgcaggcgcgcaagagctcaacgtgccggctgttggaaaagtgtgtcactggggcacgaggcgctccctgggatcacatggtacctgctccagtgccgcgtgcggcccgggaaccctgggctgctggcgcctgcgcagagccctctgtcccagggaaaggctcgggcaaaaggcggctgagattggcagagtgaaatattactgccg。
41.实施例2:nr1d1-luc质粒的慢病毒包装和病毒滴度测定
42.取两根离心管,管1中加入无血清opti-mem培养液、辅助质粒(pspax2和pmd2g)混合物和nr1d1-luc慢病毒表达质粒,管2中加入无血清opti-mem培养液和lipo2000转染试剂,分别轻轻颠倒混匀并置室温孵育5min;将管1溶液加入至管2中,轻轻颠倒混匀,室温孵育15min,获得dna-lipo2000复合物;hek293t细胞按照4.5
×
106的细胞密度接种至10cm培养皿中,待融合度达到90%后加入dna-lipo2000复合物,转染6h后更换为新鲜培养基继续培养;于转染48和72h后收集细胞培养液,4℃、3000rpm离心15min,去除细胞碎片,回收并合并病毒上清,用0.45μm滤器过滤;根据滤液体积加入对应量的peg6000溶液,充分混匀后放置于4℃冰箱内静置沉淀过夜,4℃、1500g离心30min后弃去上清,加1ml hbss溶解病毒沉淀,缓慢吹打使沉淀分散均匀,得nr1d1-luc病毒溶液,置-80℃保存备用;将含有nr1d1-luc病毒溶液和polybrene(终浓度为5μg/ml)加至u-2os细胞(atcc),感染6h后更换为新鲜培养基,感染48h后收集细胞提取dna,qpcr法检测alb(内参基因)和u5(病毒基因)的拷贝数,计算nr1d1-luc病毒溶液的病毒滴度。
43.结果如图2所示,根据内参基因和病毒基因的标准曲线计算nr1d1-luc病毒感染后u-2os细胞中内参基因和病毒基因的拷贝数分别为6.43e 03和4.59e 03,求得平均每个细胞所含病毒基因数moi值为1.4009,nr1d1-luc病毒的滴度为2.19e 08。
44.实施例3:nr1d1-luc稳转细胞株的构建
45.野生型u-2os细胞按2
×
105个/孔接种于12孔板中,待融合度达60-70%时加入不同浓度的blasticidin s(终浓度为1、2、4、6、8和10μg/ml),之后每天观察细胞的生长情况,选择在6-10天左右细胞被完全筛死的剂量作为后续nr1d1-luc稳转细胞株的最佳药筛浓度;野生型u-2os细胞按3
×
105个/孔接种于6孔板(共两个孔),待融合度达60-70%时更换为含polybrene(终浓度为5μg/ml)的新鲜培养基,其中一孔加入nr1d1-luc病毒(moi=20-40),另一孔作为空白对照;感染48h后更换为含4μg/mlblasticidin s的培养基进行药筛,每两天更换一次含药培养基,至空白细胞完全死亡,然后更换正常的培养基继续培养nr1d1-luc细胞;将nr1d1-luc细胞消化成单细胞悬液,计数,利用有限稀释法稀释至1个/100μl,然后按每孔100μl接种至96孔板中,培养一段时间后置于显微镜下观察,标记单个克隆细胞的孔并扩大培养、冻存细胞和pcr鉴定;采用试剂盒提取野生型u-2os细胞和nr1d1-luc单克隆细胞的基因组dna,取50ng dna进行pcr扩增测定细胞内nr1d1启动子的表达情况,nr1d1启动子的正向引物(nr1d1-f)序列为5'-ctctggactttgccctgaag-3'(seq id no:2),反向引物(nr1d1-r)序列为5'-gctgtctgggagatcaaagc-3'(seq id no:3),pcr反应体系和程序见表1;取15μl pcr产物进行琼脂糖凝胶电泳(200v,25min),使用jy02g型凝胶成像分析仪观察电泳结果,并在波长为302nm紫外灯下观察拍照,用image j软件定量分析电泳条带。
46.表1.pcr扩增体系和程序
[0047][0048]
结果显示,野生型u-2os细胞加不同浓度blasticidin s培养6天后,随着药物浓度的升高细胞出现明显死亡,4μg/ml blasticidin s即可使细胞全部死亡,确定该浓度为最佳药筛浓度(图3)。加4μg/ml blasticidin s作用6天后,空白对照组的u-2os细胞全部死亡,而感染了nr1d1-luc病毒的u-2os细胞生长良好,表明nr1d1-luc质粒被成功递送至细胞内并表达了质粒上含有的blasticidin s脱氨酶基因bsd(图4)。并且,与野生型细胞相比,所构建的3株nr1d1-luc单克隆细胞基因组中均高表达了nr1d1启动子(约4倍),表明成功构建了nr1d1-luc稳转细胞株(图5)。
[0049]
实施例4:基于nr1d1-luc稳转细胞株的高通量筛选方法
[0050]
nr1d1-luc细胞按3
×
103个/孔接种于白色不透光96孔板中,待孔内细胞完全长满(100%融合度)后更换为100μl含1μm目标化合物(1760种天然小分子)或空白溶剂(dmso)的完全dmem培养基,继续培养24h后更换为80μl含0.1mm beetle luciferin的无酚红dmem,37℃培养箱内孵育30min后采用navigator发光检测仪(96通道)快速读取(每孔1s)发光值,分析每种小分子对nr1d1转录活性的调控作用;对于高通量法筛选得到对nr1d1-luc活性有调控作用的化合物(36种),按照上述方法将nr1d1-luc细胞接种于正常透明96孔板内并加药(1μm)处理24h后,弃去培养基,每孔加入30μl裂解液裂解细胞(20min),取10μl细胞裂解液采用dual-双荧光素报告系统测定荧光素酶活性,分析这些小分子对nr1d1转录活性的调控作用。
[0051]
利用所构建的nr1d1-luc稳转细胞株,我们发展了一套高通量筛选方法用于发现新的nr1d1调节剂,并发现了近50种对nr1d1转录活性具有调控作用的天然小分子(图6)。选取36种阳性小分子(13种下调、23种上调)经裂解法验证,结果表明裂解法与高通量法的结果一致(图7)。
[0052]
实施例5:nr1d1-luc细胞的节律研究
[0053]
nr1d1-luc细胞按2
×
105个/皿接种于35mm细胞培养皿中,待融合度达100%后更换为节律监测培养基(ph 7.3):低糖dmem(含0.29mg/ml l-谷氨酰胺)、3.5mg/ml d-葡萄糖、0.35mg/ml碳酸氢钠、100nm地塞米松、10mm hepes、10%(v/v)fbs、100单位/ml青霉素、100μg/ml链霉素和0.1mm beetle luciferin。用胶布将培养皿密封后放入lumicycle32通道生物节律性光度测定仪连续实时监测5天,分析nr1d1-luc细胞的节律特征(如周期)。
[0054]
结果显示(图8),nr1d1-luc细胞具有明显的节律,其周期为23.75
±
0.5h。因此,nr1d1-luc细胞可作为一种新的体外节律模型用于生物节律领域的研究。
[0055]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受所述的实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
