基因组瘢痕测定和相关方法
相关申请的交叉引用
1.本技术要求2019年12月16日提交的标题为“基因组瘢痕测定”(“genomic scarring assays”)的美国临时申请序列号62/948,640的权益,将其通过引用以其整体明确并入本文。
技术领域
2.本公开文本总体上涉及用于检测或预测基因组瘢痕、用于诊断测定领域和用于选择针对人类疾病(例如癌症)的治疗方案的方法。
背景技术:
3.同源重组(hr)是参与修复双链dna断裂(dsb)的主要机制之一。当细胞中的hr途径被破坏时,双链断裂可能无法有效修复(或根本无法修复),从而导致基因组不稳定性(例如,突变、拷贝数改变和结构重排)。由hr修复缺陷导致的基因组不稳定性(也称为“基因组瘢痕”)进而与各种类型的癌症相关。例如,拷贝数改变可能会由于基因的额外拷贝的存在而导致基因的过表达,或者由于杂合性丢失而导致低表达或无表达。在许多类型的癌症中已观察到hr修复缺陷(hrrd)。
4.聚(adp-核糖)聚合酶(parp)已知在各种细胞过程(包括复制、重组、染色质重塑和dna修复)中发挥重要作用。已经发现几种类型的肿瘤(例如,brca1/2突变体)具有缺陷型hr修复途径,并且因此依赖于parp介导的碱基切除修复来存活。鉴于这些发现,parp抑制已经作为一种通过使互补dna修复途径失活来选择性杀死癌细胞的潜在策略出现。然而,鉴于parp抑制剂通常仅对具有hr修复缺陷的癌症有效,因此在施用此疗法之前确定患者是否患有hr修复缺陷型癌症非常重要。目前存在至少两种可商购的基因组瘢痕测定,即myriad genetics,inc.提供的cdx测定和foundation medicine,inc.提供的foundationfocus
tm cdx brca loh测定。然而,这两种测定均需要显著量的测序数据作为先决条件,例如,mychoice测定需要对50,000个单核苷酸多态性(snp)靶标进行测序,并且99%的碱基必须具有平均覆盖率超过500x的100个读段,并且foundationfocus测定需要》99%的外显子覆盖率为》100x的》500x中值覆盖率。这种对大量测序数据的需求增加了成本和处理时间,限制了这些测定的有用性。
技术实现要素:
5.在通用方面,本公开文本提供了用于检测或预测同源重组修复缺陷(“hrrd”)的方法。在一些方面,此类方法可用于为有需要的受试者选择癌症治疗。与如本文所述的已知方法相比,此类方法提供了多种优点。例如,在一些方面,本发明方法需要较少的测序容量,并且因此比已知方法便宜。此外,本发明方法的实施允许使用标准聚合酶链式反应(pcr)设备检测hrrd,并且与多种样品类型(例如,从新鲜冷冻组织或福尔马林固定的石蜡包埋组织中提取的dna,以及无细胞dna)相容。在一些方面,本发明方法可以使用由靶向大约5,000个或
更少的snp的多重pcr测定产生的测序数据来进行。此类方法可以例如作为单管pcr测定来进行,从而节省时间和资源。
6.在一方面,本公开文本涉及一种用于预测hrrd的方法,所述方法包括以下步骤:a)提供获自人类受试者的生物样本,其中所述样本包含基因组dna;b)对所述基因组dna进行多重聚合酶链式反应(pcr)测定以产生扩增产物,其中所述pcr测定被配置为扩增多个扩增子;c)对所述扩增产物的至少一部分进行测序以产生测序结果;以及d)基于所述测序结果,确定所述生物样本的一组参数,其中所述参数组包括:i)区段大小参数,ii)每单位长度的断点计数参数,以及iii)拷贝数参数。
7.在一些方面,所述方法进一步包括:e)基于所确定的参数组,预测所述生物样本是否获自具有hrrd的细胞、组织或肿瘤。在一些方面,所述方法进一步包括:基于所确定的参数组,为提供生物样本的人类受试者选择治疗。在一些方面,所述方法进一步包括:基于所确定的参数组,预测所述人类受试者对包含以下的癌症治疗方案的反应:dna损伤剂、蒽环类、拓扑异构酶i抑制剂、辐射和/或parp抑制剂。在一些方面,所述方法可以进一步包括此段落中所述的步骤的任何组合。在一些方面,所述区段大小参数、所述每单位长度的断点计数参数和所述拷贝数参数可以聚合(例如,使用加法或更复杂的算法)以产生单个度量或得分,然后进行本文所述的预测或选择步骤中的任一个。
8.在一些方面,所述区段大小参数是通过以下方式确定的:鉴定多个区段,其中区段被定义为由至少3个含有杂合多态位置的连续扩增子组成的基因组dna的一部分,所述连续扩增子具有相同的拷贝数;确定所鉴定的多个区段的区段大小分布;以及计算描述所述区段大小的混合组分的后验概率,所述后验概率是通过对开发组的混合建模确定的。在一些方面,所述区段大小参数是通过以下方式确定的:鉴定多个区段,其中区段被定义为由至少3个含有杂合多态位置的连续扩增子组成的基因组dna的一部分,所述连续扩增子具有相同的拷贝数;以及计算所鉴定的多个区段的平均区段大小。在一些方面,所述多个区段包含大小在5-50兆碱基对(mbp)范围内的区段。在一些方面,所述区段各自在1-10、10-20、20-30、30-40、40-50、50-60、60-70、70-80、80-90或90-100mbp的长度范围内。在一些方面,所述区段可以更长(例如,任何长度直至完整的染色体臂的长度)。在一些方面,所述区段各自为至少5、10、15、20、25、30、35、40、45、50、60、70、80、90或100mbp的长度。在一些方面,所述区段各自为小于5、10、15、20、25、30、35、40、45、50、60、70、80、90或100mbp的长度。
9.在一些方面,所述每单位长度的断点计数参数是通过计算每5、6、7、8、9、10、11、12、13、14、15mb的基因组dna的断点数确定的。在一些方面,可以针对基因组dna的一部分计算所述每单位长度的断点计数(例如可以针对存在于所述基因组dna内的一条或多条染色体或染色体臂进行计算)。在一些方面,所述每单位长度的断点计数参数是通过计算描述断点数的混合组分的后验概率确定的,所述后验概率是通过对开发组进行混合建模确定的。
10.在一些方面,所述拷贝数参数是通过计算所述基因组dna的一个或多个区段的拷贝数确定的,其中区段被定义为由至少3个含有杂合多态位置的连续扩增子组成的基因组dna的一部分,所述连续扩增子具有相同的拷贝数。在其他方面,本文公开的任何方法所需的区段可以被定义为这样的基因组dna的一部分,所述基因组dna的一部分由至少1、2、3、4、5、6、7、8、9、10个(或任何其他任意数量的)含有杂合多态性位置的连续扩增子作出,所述连续扩增子具有相同的拷贝数。在一些方面,所述拷贝数参数可以基于所述基因组的多个区
段(例如,至少5、10、15、20、25、30、35、40、45、50、100、150、200、250或500个区段)来计算。在一些方面,一个或多个区段中的每一个的拷贝数可以选自:接近二倍体、接近四倍体、接近六倍体、接近八倍体或接近任何其他倍数。在一些方面,拷贝数可以用数字表示,例如,所述拷贝数参数可以包含所述基因组dna的多个区段的数值平均值或中值拷贝数。在一些方面,所述拷贝数参数是:a)基于所述基因组dna的多个区段,并且通过确定描述所述多个区段的拷贝数的混合组分的后验概率计算,所述后验概率是通过对开发组的混合建模确定的;和/或b)至少部分基于所述多个区段的分类,所述分类基于它们各自的倍性值。
11.在一些方面,所述生物样本获自人类组织、肿瘤或细胞。所述生物样本可以获自健康的人类受试者或获自已经被诊断患有或怀疑患有癌症的人类受试者。
12.在一些方面,所述基因组dna包含整倍体基因组,并且所述pcr测定被配置为扩增至少1,000;2,000;3,000;4,000;5,000;6,000;7,000;或8,000个扩增子。
13.在一些方面,每个扩增子含有至少一个多态性位置,所述至少一个多态性位置具有至少0.1%、0.5%、1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、21%、22%、23%、24%或25%的次要等位基因的平均群体频率。
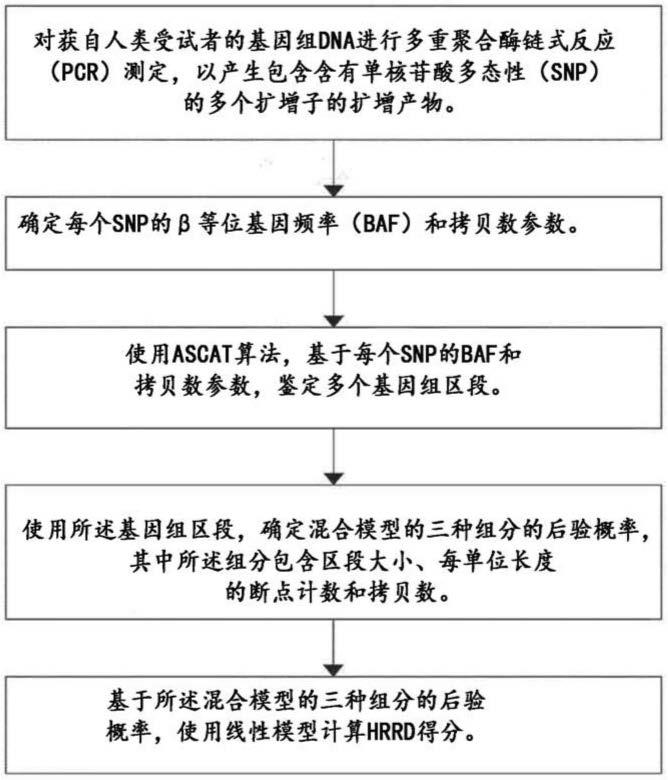
14.在另一个通用方面,本公开文本提供了一种用于检测hrrd的方法,所述方法包括以下步骤:a)对获自人类受试者的基因组dna进行多重聚合酶链式反应(pcr)测定,以产生包含多个含有单核苷酸多态性(snp)的扩增子的扩增产物;b)确定每个所述snp的β等位基因频率(baf)和拷贝数参数;c)使用ascat算法,基于每个所述snp的baf和拷贝数参数,鉴定多个基因组区段;d)使用所述基因组区段,确定混合模型的三种组分的后验概率,其中所述组分包括区段大小、每单位长度的断点计数和拷贝数;以及e)基于所述混合模型的三种组分的后验概率,使用线性模型计算hrrd得分。
15.在一些方面,所述方法进一步包括步骤f)基于所述hrrd得分,预测所述基因组dna是否获自具有hrrd的细胞、组织或肿瘤。在其他方面,步骤f)可以包括基于所述hrrd得分,预测所述人类受试者对包含以下的癌症治疗方案的反应:dna损伤剂、蒽环类、拓扑异构酶i抑制剂、辐射、和/或聚adp-核糖聚合酶(parp)抑制剂。
16.在一些方面,区段大小组分是通过以下方式确定的:鉴定多个区段,其中区段被定义为由至少3个含有杂合多态位置的连续扩增子组成的基因组dna的一部分,所述连续扩增子具有相同的拷贝数;确定所鉴定的多个区段的区段大小分布;以及计算描述所述区段大小的混合组分的后验概率,所述后验概率是通过对开发组的混合建模确定的。
17.在一些方面,所述多个区段包含各自具有以下的区段:a)在5-50兆碱基对(mbp)范围内的大小;b)在1-10、10-20、20-30、30-40或40-50mbp长度范围内的大小;c)至少5、10、15、20、25、30、35、40、45或50mbp的大小。
18.在一些方面,所述基因组dna是从采取自以下的生物样本中提取的:a)健康的人类受试者或b)已经被诊断患有或怀疑患有癌症的人类受试者。所述基因组dna可以包含例如整倍体基因组,并且所述pcr测定被配置为扩增至少1,000;2,000;3,000;4,000;5,000;6,000;7,000;或8,000个扩增子。在一些方面,每个扩增子含有至少一个多态性位置,所述至少一个多态性位置具有至少10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、21%、22%、23%、24%或25%的次要等位基因的平均群体频率。
19.在另一个通用方面,本公开文本提供了一种用于预测hrrd的方法,所述方法包括:a)提供包含一个或多个处理器的电子设备;b)通过所述电子设备接收扩增产物的测序结果,所述扩增产物是使用获自人类受试者的基因组dna通过多重pcr产生的,其中所述测序结果包含多个含有snp的扩增子的序列;c)通过所述电子设备确定每个所述snp的β等位基因频率(baf)和拷贝数参数;d)使用ascat算法,基于每个所述snp的baf和拷贝数参数,通过所述电子设备鉴定多个基因组区段;e)基于所述基因组区段,通过所述电子设备确定混合模型的三种组分的后验概率,其中所述组分包括区段大小、每单位长度的断点计数和拷贝数;以及f)基于所述混合模型的三种组分的后验概率,使用线性模型,通过所述电子设备计算hrrd得分。
20.在另一个通用方面,本公开文本提供了一种用于预测hrrd的系统,所述系统包括:包含一个或多个处理器的电子设备,所述电子设备被配置为:接收扩增产物的测序结果,所述扩增产物是使用获自人类受试者的基因组dna通过多重pcr产生的,其中所述测序结果包含多个含有snp的扩增子的序列;确定每个所述snp的β等位基因频率(baf)和拷贝数参数;使用ascat算法,基于每个所述snp的baf和拷贝数参数,鉴定多个基因组区段;基于所述基因组区段,确定混合模型的三种组分的后验概率,其中所述组分包括区段大小、每单位长度的断点计数和拷贝数;以及基于所述混合模型的三种组分的后验概率,使用线性模型计算hrrd得分。在一些方面,本公开文本提供了一种包含一个或多个处理器的电子设备,所述一个或多个处理器被配置为执行本文所述的任何方法的一个或多个步骤。
21.在另一个通用方面,本公开文本提供了扩增基因组dna的方法,所述方法包括:a)从获自已知患有或怀疑患有癌症的人类受试者的样本中获得基因组dna;以及b)通过使用所述基因组dna进行多重pcr来扩增多个含有单snp的扩增子;其中所述多重pcr是使用被配置为扩增跨所有22条人类体细胞染色体的至少5,000个扩增子的一组pcr引物进行的,其中每个扩增子包含snp。在一些方面,每个扩增子包含100bp的最大长度。在一些方面,平均扩增子密度是1个扩增子/400-600kb体细胞染色体dna。在一些方面,所述多个扩增子包括以下基因的每一种的一个或多个部分:brca1、brca2、brip1、rad51c、rad51d、atm、bard1、chek1、chek2、fanca、fancl、nbn、palb2、rad51b、rad54l、cdk12和tp53。
22.在另一个通用方面,本公开文本提供了产生pcr扩增产物的方法,所述方法包括:a)从获自人类受试者(例如,已知患有或怀疑患有癌症)的样本中获得基因组dna;以及b)通过使用所述基因组dna进行多重pcr扩增多个各自含有单核苷酸多态性(snp)的扩增子,产生所述pcr扩增产物;其中所述多重pcr是使用被配置为扩增跨所有22条人类体细胞染色体的至少5,000个扩增子的一组pcr引物进行的。在一些方面,每个扩增子包含100bp的最大长度。在一些方面,平均扩增子密度是1个扩增子/400-600kb体细胞染色体dna。在一些方面,所述多个扩增子包括以下基因的每一种的一个或多个部分:brca1、brca2、brip1、rad51c、rad51d、atm、bard1、chek1、chek2、fanca、fancl、nbn、palb2、rad51b、rad54l、cdk12和tp53。
23.在另一个通用方面,本公开文本提供了治疗癌症的方法,所述方法包括:a)通过电子设备接收扩增产物的测序结果,所述扩增产物是使用获自在人类受试者中发现的肿瘤的基因组dna通过多重pcr产生的,其中所述测序结果包含多个含有snp的扩增子的序列;b)通过所述电子设备确定每个所述snp的baf和拷贝数参数;c)使用ascat算法,基于每个所述snp的baf和拷贝数参数,通过所述电子设备鉴定多个基因组区段;d)基于所述基因组区段,
通过所述电子设备确定混合模型的三种组分的后验概率,其中所述组分包括区段大小、每单位长度的断点计数和拷贝数;以及e)基于所述混合模型的三种组分的后验概率,使用线性模型,通过所述电子设备计算hrrd得分;以及f)基于所述hrrd,为所述受试者选择和/或施用癌症治疗。在其他方面,本公开文本提供了一种基于本文所述的任何预测方法来治疗癌症的方法。
24.在一些方面,所述癌症治疗是施用dna损伤剂、蒽环类、拓扑异构酶i抑制剂、辐射、和/或parp抑制剂。在一些方面,所选择的癌症治疗是当所述hrrd得分高于(或低于)预选择的阈值时,施用parp抑制剂。
25.在一些方面,根据本公开文本的治疗癌症的方法包括:a)从获自已知患有或怀疑患有癌症的人类受试者的样本中获得基因组dna;以及b)使用所述基因组dna通过进行多重pcr扩增多个含有单核苷酸多态性(snp)的扩增子,其中所述多重pcr是使用被配置为扩增跨所有22条人类体细胞染色体的至少5,000个扩增子的一组pcr引物进行的,其中每个扩增子包含snp;c)基于所述多个扩增子的序列,确定所述人类受试者的hrrd得分;以及d)基于所述hrrd,为所述受试者选择和/或施用癌症治疗。在一些方面,所述癌症是卵巢癌,例如,铂敏感性卵巢癌或铂耐药性卵巢癌。
26.在再另外的方面,本公开文本提供了用于使用多重pcr测定来扩增基因组dna的试剂盒,其中所述试剂盒包含a)pcr反应混合物;b)dna聚合酶;以及c)一组pcr引物,其中所述pcr引物组被配置为扩增跨所有22条人类体细胞染色体的至少5,000个扩增子,其中每个扩增子包含snp和100bp的最大长度。
27.在一些方面,平均扩增子密度是1个扩增子/400-600kb、300-700kb或500-560kb体细胞染色体dna。在一些方面,所述扩增子扩增以下基因的每一种的一个或多个部分:brca1、brca2、brip1、rad51c、rad51d、atm、bard1、chek1、chek2、fanca、fancl、nbn、palb2、rad51b、rad54l、cdk12和tp53。
28.鉴于本公开文本的全部内容,另外的方面对于技术人员将是易于清楚的。
附图说明
29.图1是示出了根据本公开文本的用于预测hrrd的示例性方法的流程图。
30.图2是示出了可以与本文所述的方法一起使用的一组示例性扩增子位置的图,所述扩增子位置是跨人类基因组的22条常染色体作图的。
31.图3是示出了一组示例性样品的图,这些样品是基于它们的基因组瘢痕得分和提供的突变信息分类的。基因组瘢痕得分沿x轴从左到右增加。
32.图4示出了对于用根据本发明方法的基因组瘢痕测定相比于用snp阵列分析的样品,比较拷贝数(归一化值,logr)和β等位基因频率(baf)的一组图。
33.图5示出了将使用本发明方法产生的预测hrrd得分与使用snp阵列确定的基因组瘢痕的已知度量进行比较的一组三个图表。
34.图6是示出了在患有复发性卵巢癌的患者群组中与同源重组修复途径基因突变状态相关的hrrd得分,以及对奥拉帕尼单一疗法的最佳确认反应的图表。
35.图7展示了可以在其上实现所公开的系统和方法的通用计算机系统的例子。
具体实施方式
36.本公开文本提供了用于确定或预测hrrd的各种方法。在一些方面,所述方法可以包括以下步骤:a)提供获自人类受试者的生物样本,其中所述样本包含基因组dna;b)对所述基因组dna进行多重pcr测定以产生扩增产物,其中所述pcr测定被配置为扩增多个扩增子;c)对所述扩增产物的至少一部分进行测序以产生测序结果;以及d)基于所述测序结果,确定所述生物样本的一组参数。在一些方面,所述参数组包括:i)区段大小参数、ii)每单位长度的断点计数参数、和iii)拷贝数参数。在其他方面,所述方法包括对来自获自人类受试者的生物样本的基因组dna进行多重pcr测定以产生扩增产物,其中所述pcr测定被配置为扩增多个扩增子;对所述扩增产物的至少一部分进行测序以产生测序结果;以及基于所述测序结果确定所述生物标本的一组参数。与已知的基因组瘢痕测定相比,本发明方法提供了多个优点,包括减少了成本和处理时间(例如,由于较低的测序要求)、以及提供了可以使用标准pcr设备进行的对hrrd的临床上有用的诊断。
37.在一些方面,所述参数组包括:i)区段大小参数、ii)每单位长度的断点计数参数、和iii)拷贝数参数。在一些方面,这三个参数可以通过添加或使用更复杂的算法来聚合以产生单个得分(例如,代表基因组瘢痕的水平)。在任一情况下,个体参数或基于个体参数的聚合参数可用作用以预测人类受试者对用抗癌疗法治疗的反应性或用以选择抗癌治疗的诊断指标。
38.在一些方面,根据本公开文本的方法可以基于多重pcr测定,所述多重pcr测定包括遍布在22对人类常染色体中多个上的扩增子。图3中示出了映射到人类基因组的一组示例性扩增子。在一些方面,所述扩增子可以靶向特定基因或染色体区域。在其他方面,所述扩增子可以随机遍布在22条常染色体上。在一些方面,所述测定也可以包括能够扩增人类性染色体上的dna的一个或多个扩增子。在一些方面,所述扩增子的大小范围可以为70-90个碱基对(bp)。然而,所述扩增子的大小可以根据需要针对给定实施方式而变化。例如,所述扩增子大小可以是1-50bp、50-100bp、100-150bp、150-200bp、200-250bp、250-300bp、300-350bp、350-400bp、400-450bp、450-500bp、或》500bp。在一些方面,所述扩增子大小可以在通过从任何上述值中选择最小和最大大小而形成的范围内。在一些方面,一些或所有选择的扩增子将含有至少一个多态性位置。在一些方面,扩增子分辨率可以是至少或大约100、150、200、250、300、350、400、450、500、550、600、650、700、750或800kb。在一些方面,可以选择用于多重pcr测定的引物以在单管pcr反应中相容。可以使用本领域已知的任何设备(如通过测序仪器(例如,nextseq系统))对所得的pcr产物进行测序。
39.必须分析测序的输出以确定同源重组(hr)缺陷的水平。在一些方面,分析过程可以由四个步骤组成:1)数据预处理,2)覆盖率归一化和偏倚校正,3)区段化,以及4)确定hrrd得分。然而,应理解,此组织仅仅是非限制性例子。在其他方面,分析过程可以省略这些步骤中的任一个,添加另外的步骤,和/或合并这四个步骤中的一个或多个。
40.数据预处理步骤可以包括基于初始测序结果(例如,一个或多个fastq文件)生成覆盖文件(含有原始读段计数)和snp文件(含有b等位基因频率,“baf”/扩增子)。
41.覆盖率归一化和偏倚校正步骤可以包括a)确定每个扩增子的归一化读段计数(用量商,“dq”)值,并且然后(如果需要)b)产生校正的dq值,所述值说明与样品类型相关的测序偏倚。例如,福尔马林固定的石蜡包埋样品可展示出可通过已知算法校正的测序长度偏
倚。
42.区段化步骤可以包括使用在覆盖率归一化和偏倚校正步骤期间产生的baf值和dq(或校正的dq)值来确定拷贝数区段。可以使用van loo等人“allele-specific copy number analysis of tumors.”pnas 107.39(2010):16910-16915中所述的ascat算法计算区段化,将其通过引用以其整体并入本文。例如,可以对使用本文所述的试剂盒之一产生的多重pcr扩增产物进行测序和分析,以确定每个snp或扩增子的β等位基因频率(baf)和拷贝数参数。baf和拷贝数参数可以用作ascat算法的输入,以鉴定基因组区段。在本文所述的方法的一些方面,区段被定义为由至少3个含有杂合多态位置的连续扩增子组成的基因组dna的一部分,所述连续扩增子具有相同的拷贝数。在其他方面,区段的定义可以基于不同数量的连续扩增子,例如,至少1、2、4、5、6、7、8、9、10(或任何其他任意数量)个连续扩增子。拷贝数区段的大小范围可以例如为大约5-50mbp。
43.hrrd得分的确定可以包括基于在区段化步骤中鉴定的区段确定给定的生物样品的单个整数得分。此过程可以从通过混合建模确定拷贝数区段的几个参数的潜在分布开始,所述参数包括1)区段大小,2)每单位长度(例如,10mbp)的断点计数,和3)拷贝数。这些潜在分布称为组分。可以汇编后验矩阵的和(样品x组分),然后使用非负矩阵分解来降维,允许针对这些参数中的每一个产生值,从而提供关于hrrd的信息。
44.这三个参数的值可以聚合以形成指示hr修复缺陷的水平的单一得分,这进而可以用作用以预测反应性或选择治疗方案的临床诊断指标。图3展示了使用根据本公开文本的方法测定的一组示例性生物样品,根据它们的hrrd得分按顺序排列。在一些方面,此hrrd得分可用于确定是否应将parp抑制剂施用于患有或怀疑患有癌症的人类受试者,以预测对抗癌治疗剂的反应性,或用于本文所述的其他目的。
45.在一些方面,根据本公开文本的基因组瘢痕测定可以开始于对获自人类受试者的基因组dna进行多重pcr测定,以产生包含多个含有snp的扩增子的扩增产物,随后对扩增产物进行测序和分析以确定hrrd得分。可以分析测序结果以确定每个snp的β等位基因频率(baf)和拷贝数参数。这些baf和拷贝数参数可以进而用于使用如loo等人所述的ascat算法将基因组dna划分成多个基因组区段,如上所述。然后可以针对与这些基因组区段相关的一个或多个特征导出拷贝数特征分布,如区段大小、每单位长度(例如,每10mb)的断点计数或拷贝数。可以使用macintyre等人“copy number signatures and mutational processes in ovarian carcinoma,”nature genetics 50.9(2018):1262-1270(本文中,“macintyre”)中所述的方法进行这些拷贝数特征中的任一种的分析和计算,将其全部内容通过引用并入本文。例如,混合建模可用于确定每个组分的潜在分布,如例如macintyre中所述。可以汇编后验矩阵的和(样品x组分),然后使用非负矩阵分解来降维,允许针对这些组分中的每一个产生值。可以将所得的值拟合到广义线性模型(例如,基于具有已知临床结局的群体的hrrd得分产生),以便得出给定样本的聚合hrrd得分。
46.广义线性模型可以以macintyre中所述或本领域中已知的其他方式构建。用户可以根据需要针对给定应用(例如,基于癌症类型/阶段或患者人口统计因素)选择所需的开发组。例如,可以使用获自具有已知临床结局的患者的约250个卵巢癌样品的开发组来构建根据本公开文本的基因组瘢痕测定的广义线性模型。此特定的开发组产生了具有以下公式的广义线性模型:hrrd得分=a (b
×
区段大小) (c
×
每单位长度(10mb)的断点计数) (d
×
拷贝数),其中“a”=77465.2750;“b”=-1.1178;“c”=-515.7440;并且“d”=-1.8780。如上所述,这些组分中的每一个的值可以针对给定样品进行计算,并且将其插入到此广义线性公式中以计算所述样品的hrrd得分。如下文提供的例子中进一步详细描述的,hrrd得分可以针对获自具有已知临床结局的受试者的一组样品进行计算,以便确定用于将样品分类为hrrd阳性或hrrd阴性的适当截止值。可以理解,此截止值可以根据需要针对给定类型的癌症凭经验确定。
47.在其他方面,本公开文本提供了一种治疗癌症的方法,所述方法包括基于本文所述的方法,将癌症治疗施用于已经被诊断具有hrrd的人类受试者。在特定方面,所述方法进一步包括施用dna损伤剂、蒽环类、拓扑异构酶i抑制剂、辐射、和/或聚adp-核糖聚合酶(parp)抑制剂。
48.根据本公开文本的治疗癌症的另外的方法可以包括:a)通过电子设备接收扩增产物的测序结果,所述扩增产物是使用获自在人类受试者中发现的肿瘤的基因组dna通过多重pcr产生的,其中所述测序结果包含多个含有snp的扩增子的序列;b)通过所述电子设备确定每个所述snp的baf和拷贝数参数;c)使用ascat算法,基于每个所述snp的baf和拷贝数参数,通过所述电子设备鉴定多个基因组区段;d)基于所述基因组区段,通过所述电子设备确定混合模型的三种组分的后验概率,其中所述组分包括区段大小、每单位长度的断点计数和拷贝数;e)基于所述混合模型的三种组分的后验概率,使用线性模型,通过所述电子设备计算hrrd得分;以及f)基于所述hrrd得分,为所述受试者选择和/或施用癌症治疗。
49.根据本公开文本的治疗癌症的其他方法可以包括a)从获自已知患有或怀疑患有癌症的人类受试者的样本中获得基因组dna;以及b)使用所述基因组dna通过进行多重pcr扩增多个含有snp的扩增子,其中所述多重pcr是使用被配置为扩增跨所有22条人类体细胞染色体的至少5,000个扩增子的一组pcr引物进行的,其中每个扩增子包含snp;c)基于所述多个扩增子的序列,确定所述人类受试者的hrrd得分;以及d)基于所述hrrd得分,为所述受试者选择和/或施用癌症治疗。
50.在一些方面,所述癌症治疗可以包括施用dna损伤剂、蒽环类、拓扑异构酶i抑制剂、辐射、和/或parp抑制剂(如奥拉帕尼)。如本文所述,用户可以选择hrrd得分截止阈值用于分类样品(例如,肿瘤的样品)是hrrd阳性还是hrrd阴性。此阈值可以基于获自可获得已知临床结局的受试者的样品(例如,肿瘤的样品)的hrrd得分概况eci得分。因此,在一些方面,所选择的癌症治疗是当所述hrrd得分高于或低于预选择的阈值时,施用parp抑制剂。癌症可以是卵巢癌(例如,psoc或proc),或hrrd得分与临床结局相关的任何其他癌症。应理解,人们可以通过分析获自可获得已知临床结局的患者的hrrd得分来确定hrrd得分是否可用作临床结局和/或治疗有效性的诊断指标。
51.在另一个通用方面,本公开文本提供了一种用于预测hrrd的系统,所述系统包括:包含一个或多个处理器的电子设备,所述电子设备被配置为:接收扩增产物的测序结果,所述扩增产物是使用获自人类受试者的基因组dna通过多重pcr产生的,其中所述测序结果包含多个含有snp的扩增子的序列;确定每个所述snp的β等位基因频率(baf)和拷贝数参数;使用ascat算法,基于每个所述snp的baf和拷贝数参数,鉴定多个基因组区段;基于所述基因组区段,确定混合模型的三种组分的后验概率,其中所述组分包括区段大小、每单位长度的断点计数和拷贝数;以及基于所述混合模型的三种组分的后验概率,使用线性模型计算
hrrd得分。所述电子设备可以是具有一个或多个处理器的计算机(例如,台式个人计算机)或基于云的服务器,所述一个或多个处理器被配置为执行用于进行本文所述的任何方法(或其步骤)的指令。例如,此类系统可以被配置为接收多重pcr扩增产物的测序结果,并且执行如本文所述计算hrrd得分所需的所有下游分析。在一些方面,所述软件可以被配置为允许用户选择或修改用于计算hrrd得分的广义线性模型(例如,通过选择与给定类型的癌症相关的得分)。
52.在另一个通用方面,本公开文本提供了扩增基因组dna或产生扩增产物(例如,使用本文所述的试剂盒)的方法。例如,扩增基因组dna的方法可以包括:a)从获自人类受试者(例如,已知患有或怀疑患有癌症)的样本中获得基因组dna;以及b)通过使用所述基因组dna进行多重pcr来扩增多个含有snp的扩增子;其中所述多重pcr是使用被配置为扩增跨所有22条人类体细胞染色体的至少5,000个扩增子的一组pcr引物进行的,其中每个扩增子包含snp。
53.一种产生pcr扩增产物的方法可以类似地包括:a)从获自人类受试者(例如,已知患有或怀疑患有癌症)的样本中获得基因组dna;以及b)通过使用所述基因组dna进行多重pcr扩增多个各自含有单核苷酸多态性(snp)的扩增子,产生pcr扩增产物;其中所述多重pcr是使用被配置为扩增跨所有22条人类体细胞染色体的至少5,000个扩增子的一组pcr引物进行的。
54.在此类方法的一些方面,每个扩增子可以包含100-300bp(例如,100bp)的最大长度,和/或平均扩增子密度可以是1个扩增子/400-600kb体细胞染色体dna。多个扩增子还可以包括与hr修复途径相关的基因的一个或多个部分,所述基因例如brca1、brca2、brip1、rad51c、rad51d、atm、bard1、chek1、chek2、fanca、fancl、nbn、palb2、rad51b、rad54l、cdk12和/或tp53。
55.将通过以下实施例说明根据本公开文本的示例性基因组瘢痕测定的开发、验证和使用。
56.实施例1:根据本公开文本的基因组瘢痕测定的开发
57.在一些方面,本公开文本提供了一种全基因组的、基于多重pcr的瘢痕测定,所述测定利用了考虑杂合性丢失(loh)和拷贝数特征的方法。此类测定可以有利地设计为用于检测基因组瘢痕的基于mastr的通用单重测试。mastr(用于重测序的特定靶标多重扩增)测定能够在有限数量的pcr反应中对目的基因的所有必需编码序列进行多重pcr扩增。mastr测定及当代下一代测序(ngs)技术对dna扩增子的进一步下游汇集和对单独样品的条形码化允许出于研究和诊断两种目的进行简单、高通量和有成本效益的测序。
58.此示例性基因组瘢痕的开发过程开始于选择用于跨所有人类常染色体进行多重pcr扩增的引物,其中分辨率大约为500kb,对应于大约6,000个扩增子。每个扩增子进一步需要含有snp和100bp的最大扩增子长度(用于cfdna相容)。理想情况下,所有引物都应相容以允许单重扩增。
59.为此,使用计算机技术来选择用于此示例性基因组瘢痕测定的一组引物。至此,重复区域(例如,ucsc基因组浏览器中定义的重复区域、重复掩蔽区域和简单重复序列)在人类基因组中被掩蔽。然后,为了强制围绕流行的snp设计扩增子(以允许计算肿瘤组织分数和loh二者),以阶梯方法从1,000个基因组的数据库中提取掩蔽区域之外的snp。snp流行率
是根据全球人口流行率确定的。在选择适用的snp后,从人类参考基因组中提取此snp周围的dna序列,并且设计推定的pcr引物,从而允许扩增含有snp的扩增子(“snp扩增子”)。
60.总之,确定了3批snp扩增子和伴随的推定引物,包含大约2,000,000个snp扩增子。接下来,使用修改的pcr多重化算法执行计算机单重设计,所述修改的pcr多重化算法开始于选择一个初始高流行的snp作为单重第一个扩增子,并且随着snp频率流行率的降低依序添加2,000,000个推定的snp扩增子中的每一个,并且检查所述扩增子与已经在计算机中单重存在的扩增子的相容性。由于从计算方面上对大型单重pcr反应多重化在时间空间中是高度非线性过程,因为找到相容引物对的机会随着处理时间的推移而降低,从而导致按照添加的引物对而言的处理时间更长。总之,需要大约100个计算日才能将2,000,000个推定序列从计算方面上有效地多重化到单管pcr反应中。为了加快这种基因组瘢痕测定的开发,验证并且优化计算机多重化结果的工作并行进行,同时进行这种计算机多重化模拟。
61.在对第一批大约300,000个snp扩增子进行计算机多重化后,订购了3,421个引物对并且等摩尔混合在一起(引物混合),然后进行pcr扩增和ngs。初始pcr扩增条件(缓冲液组成和循环条件)与用于clarigo的条件相同。允许基于ngs的分析来确定哪些引物导致引物二聚体(pd)(随后从引物混合物中去除)形成。通过将来自第二计算机设计批次的1,852个引物对添加到拆分pd的引物混合物中来重复此过程,并且再次扩增、测序和分析形成pd的引物。这是第三次用设计用于填补前两批空位的引物进行的。该第三批引物含有756个引物,并且将其添加到来自第一次和第二次设计的拆分pd的引物混合物中。再次对完成的引物混合物进行测序并且分析形成pd的引物。接下来,从最终的引物混合物中去除所有形成pd的引物,重新制作完整的引物混合物,测序并且分析导致扩增子呈现不足或呈现过度并且显示杂合snp的显著扩增偏倚的引物对。具体地,将覆盖率高于5x平均覆盖率的扩增子、归一化覆盖率低于50x平均覆盖率的扩增子和平均杂合等位基因频率在40%-60%范围之外的扩增子排除并且从所述测定中物理去除。这导致含有分布在所有常染色体上的5,201个扩增子(平均密度为1个扩增子/531kb)的最终基因组瘢痕测定。图2展示了snp扩增子在常染色体上的分布。在计算机上设计并且排序了总共6,029个引物对,其中5,201个(86%)被保留在最终的基因组瘢痕mastr测定中。
62.实施例2:根据本公开文本的基因组瘢痕测定的验证
63.实施例1中开发的基因组瘢痕测定的性能是通过使用先前用snp阵列分析的参考样品测试所述测定来验证的。snp阵列是含有固定的等位基因特异性寡核苷酸探针的微阵列。如图4所展示,每个snp的β等位基因频率(baf)和拷贝数参数(使用log比率值归一化)的图案与snp阵列得到的图案高度相似,但事实是snp阵列的数据点密度是基因组瘢痕测定的160分之一。
64.还测试了基因组瘢痕测定与一组fft(新鲜冷冻组织)、ffpe(福尔马林固定石蜡包埋)和cfdna(无细胞dna)样品的相容性。首先,使用基因组瘢痕测定扩增了62个hgsoc fft样品的组。使用ngs对扩增产物进行测序。对于所有这些样品,tai、lst和hrrd-loh得分可用作参考数据。如上所述计算瘢痕得分,与基于snp阵列数据计算的得分相关。图5示出了可以分类为良好的每个得分的相关性。
65.其次,用基因组瘢痕测定分析了55个高分级浆液性卵巢癌(hgsoc)ffpe样品,其中39个样品与第一项研究的fft样品匹配。此数据证实了可以成功计算源自ffpe的dna样品的
基因组瘢痕得分。
66.第三,使用基因组瘢痕测定分析来自clio研究(nct02822157)的50个cfdna样品和来自复发性肿瘤活检物的匹配hgsoc fft样品。每位患者都可获得来自原发肿瘤的ffpe组织。在包括的154名患者中,103名被随机分配到奥拉帕尼群组(并且接受了这种parp抑制剂),并且51名被分配在化疗群组中。在后一个群组中,32名患者接受了奥拉帕尼的交叉治疗,这意味着135名患者可获得奥拉帕尼反应数据(总体客观反应,主要终点)。对于所有包括的患者,可以获得疗法前和试验期间每月的血浆样品。在135名具有奥拉帕尼反应数据的患者的这一群组中,提取了105个治疗前cfdna样品、105个匹配的种系dna样品和50个源自治疗前活检物的匹配fft dna样品。这是一个高度独特的临床数据集,并且也是同类中最大的数据集之一。我们的结果表明在cfdna样品与匹配的fft肿瘤样品之间的瘢痕得分具有良好的相关性,条件是存在20%的最小肿瘤组织含量。
67.实施例3:使用基因组瘢痕测定来预测患有铂耐药性或铂敏感性卵巢癌的患者对parp抑制剂的反应性
68.clio试验nct 02822154在一组随机分组的患有复发性卵巢癌的患者中评价了奥拉帕尼单一疗法(parp抑制剂)与化学疗法。parp抑制剂治疗被批准作为响应铂敏感性复发性卵巢癌(psoc)的维持。在此研究中,患有psoc的患者被随机分配到两个初始群组中的一组,并且用奥拉帕尼或化学疗法治疗。平行地,第二组患有铂耐药性复发性卵巢癌(proc)的患者被随机分配到奥拉帕尼或化学疗法治疗群组。随后选择两个化学疗法群组的亚组用于奥拉帕尼治疗;这种交叉设计提供了关于化学疗法和奥拉帕尼治疗的组合的另外的见解。
69.源自ffpe的原发性肿瘤dna获自大多数患者,并且用于使用根据本公开文本的基因组瘢痕测定产生每个患者的hrrd得分。在这种情况下,使用基于对来自uz leuven肿瘤库的207例hgsoc病例的分析产生的混合模型确定区段大小、每单位长度的断点计数和拷贝数组分的后验概率。将hrrd得分与随访评价时观察到的患者结局进行比较。如图6所展示,对ffpe样品的基于snp扩增子的基因组瘢痕测定可预测关于psoc群组的患者对奥拉帕尼的反应。例如,图6示出了psoc群组中hrrd得分高的患者更有可能经历完全或部分缓解(分别为cr或pr)。对于此研究,30的截止得分可用于对hrrd阳性与hrrd阴性样品进行分类。应理解,此截止得分可以由基因组瘢痕测定的用户选择。在一些情况下,截止值可基于对具有已知临床结局的癌症患者的hrrd得分的先前经验研究。因此,hrrd截止值可部分基于给定癌症的分期类型、患者的人口统计学或其他因素。
70.实施例4:根据本公开文本的用于基因组瘢痕测定的试剂盒
71.如上所述,本文所述的基因组瘢痕测定可用于分析使用基于mastr的单重测试产生的测序数据。为此,suremastr hrrd scar试剂盒被开发为这样的分子测定,所述分子测定用于半定量评估从来自原发性或转移性癌症的福尔马林固定的石蜡包埋(ffpe)或新鲜冷冻(ff)的肿瘤组织中分离的基因组dna中的肿瘤基因组不稳定性。suremastr hrrd scar试剂盒可用于与illumina ngs系统试剂盒的drmid组合,以允许基于通用pcr掺入使用suremastr hrrd scar试剂盒产生的所有扩增子的分子标识符(mid)或条形码以及下一代测序(ngs)特异性衔接子。suremastr hrrd scar试剂盒和用于一个或多个illumina ngs系统试剂盒的drmid可用作在illumina miseq或nextseq上序列分析的前端扩增测试。所述技术基于靶向重测序,并且依赖于多重pcr扩增和ngs。
72.suremastr hrrd scar试剂盒包括在含有bsa、kcl和mgcl2的tricine缓冲液(ph 8.0)中包含特定寡核苷酸引物和dntp的pcr混合物;taq dna聚合酶和扩增试剂1。用户可以使用所述试剂盒使用单管多重pcr扩增从ffpe或fft样品提取的基因组dna,或cfdna(例如,分离自血浆)。然后可以使用本文所述的方法对所得的扩增产物进行测序和分析,以确定从中提取基因组dna的样品的hrrd得分,和/或以为人类受试者作出诊断或为人类受试者选择治疗(例如,以确定parp抑制剂是否可能有效)。
73.在一些方面,本文所述的方法可以使用通用计算机系统全部或部分地执行。例如,对于给定样品获得的测序结果可以通过实现产生hrrd得分所需的ascat算法和/或其他处理步骤的软件进行分析,如本文所述。
74.图7展示了可以在其上实现所公开的系统和方法的通用计算机系统(其可以是个人计算机或服务器)的例子。如所示,所述计算机系统包括中央处理单元21、系统存储器22和连接各种系统部件(包括与中央处理单元21相关联的存储器)的系统总线23。系统总线23像现有技术中已知的任何总线结构一样实现,进而含有总线存储器或总线存储器控制器、外围总线和能够与任何其他总线架构交互的局部总线。系统存储器包括永久存储器(rom)24和随机存取存储器(ram)25。基本输入/输出系统(bios)26包括确保在个人计算机20的元件之间传输信息的基本程序,如使用rom 24加载操作系统时的那些。
75.进而,个人计算机20包括用于读取和写入数据的硬盘27、用于在可移动磁盘29上读取和写入的磁盘驱动器28以及用于在可移动光盘31上读取和写入的光驱30,如cd-rom、dvd-rom和其他光信息介质。硬盘27、磁盘驱动器28和光驱30分别通过硬盘接口32、磁盘接口33和光驱接口34与系统总线23连接。驱动器和相应的计算机信息介质是独立于电源的模块,用于存储个人计算机20的计算机指令、数据结构、程序模块和其他数据。
76.本公开文本提供了使用硬盘27、可移动磁盘29和可移动光盘31的系统的实现,但是应当理解,可以采用能够以计算机可读形式存储数据的其他类型的计算机信息介质56(固态驱动器、闪存卡、数字磁盘、随机存取存储器(ram)等),所述计算机信息介质经由控制器55与系统总线23连接。
77.计算机20具有文件系统36,其中保存了记录的操作系统35、以及另外的程序应用37、其他程序模块38和程序数据39。用户能够通过使用输入设备(键盘40、鼠标42)将命令和信息输入个人计算机20中。可以使用其他输入设备(未显示):麦克风、操纵杆、游戏控制器、扫描仪等。此类输入设备通常通过串行端口46插入计算机系统20中,进而与系统总线连接,但是也可以以其他方式(例如借助并行端口、游戏端口或通用串行总线(usb))连接。监视器47或其他类型的显示设备也通过接口如视频适配器48与系统总线23连接。除了监视器47外,个人计算机还可以配备有其他外围输出设备(未显示),如扬声器、打印机等。
78.个人计算机20能够使用与一台或多台远程计算机49的网络连接在网络环境中运行。一台远程计算机(或多台计算机)49也是在描述个人计算机20的性质方面具有上述元件(如图7所示)的大部分或全部的个人计算机或服务器。其他设备也可以存在于计算机网络中,如路由器、网络站、对等设备或其他网络节点。
79.网络连接可以形成局域计算机网络(lan)50和广域计算机网络(wan)。此类网络用于公司计算机网络和公司内部网络,并且它们通常可以访问因特网。在lan或wan网络中,个人计算机20通过网络适配器或网络接口51与局域网络50连接。当使用网络时,个人计算机
20可以采用调制解调器54或其他模块来提供与广域计算机网络(如因特网)的通信。调制解调器54是内部或外部设备,通过串行端口46与系统总线23连接。需要注意的是,网络连接只是例子,并且不需要描绘网络的确切配置,即实际上存在通过技术通信模块建立一台计算机与另一台计算机的连接的其他方式。
80.在各个方面,本文所述的系统和方法可以在硬件、软件、固件或其任何组合中实现。如果在软件中实现,所述方法可以作为一个或多个指令或代码存储在非瞬态计算机可读介质上。计算机可读介质包括数据存储器。举例来说而非限制,此类计算机可读介质可以包含ram、rom、eeprom、cd-rom、闪存或其他类型的电、磁或光存储介质,或可用于以指令或数据结构的形式携带或存储所需的程序代码并且可以由通用计算机的处理器访问的任何其他介质。
81.引用本公开文本的原理、方面和实施方案以及其具体实施例的所有本文陈述旨在涵盖其结构和功能等效物二者。另外,此类等效物旨在包括当前已知的等效物和将来开发的等效物,即,所开发的执行相同功能的任何元件,而不管结构如何。因此,本公开文本的范围不旨在受限于本文示出并描述的示例性实施方案。相反,本公开文本的范围和精神由所附权利要求体现。
82.将在本说明书中引用的所有出版物和专利均通过引用并入本文,就好像每个单独的出版物或专利被具体且单独地指出通过引用并入本文,并且通过引用并入本文以公开和描述出版物所引用的相关的方法和/或材料。对任何出版物的引用是针对其在申请日之前的披露内容,并且不应被解释为承认本发明因为先前的发明而不能获得比这种出版物更早的申请日。另外,所提供的出版物的日期可能不同于实际出版日期,实际出版日期可能需要独立确认。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。