雄激素受体n端结构域的抑制剂
1.相关申请
2.本技术要求2019年3月29日提交的序列号为62/826,636的美国临时专利申请的优先权权益。该申请的内容据此通过引用以其整体并入。
3.关于联邦政府赞助研究的声明
4.本发明是根据美国国立卫生研究院授予的授权号ca092131和ca164331在政府资助下完成的。政府对本发明具有一定权利。这项工作得到了美国退伍军人事务部的支持,并且联邦政府对本发明拥有一定的权利。
背景技术:
5.前列腺癌是最常见的癌症,也是西方男性癌症死亡的第二大原因。当癌症局限于局部时,通常可以通过手术或放射疗法来治疗。然而,30%的前列腺癌用这种方法治疗后会复发,并伴有远处转移疾病,一些患者在诊断时已有晚期疾病。晚期疾病通过去势和/或服用抗雄激素治疗,即所谓的雄激素剥夺疗法。去势降低了雄激素的循环水平,降低了雄激素受体(ar)的活性。抗雄激素的施用通过竞争雄激素结合来阻断ar功能,从而降低ar活性。虽然最初有效,但这些治疗很快失败,癌症变为激素难治的,或去势抵抗的。
6.去势抵抗性前列腺癌(crpc)的典型特征是雄激素受体(ar)的持续表达和转录活性。在过去的十年中,临床前模型、涉及患者材料的相关研究和临床研究提供了证据,支持抑制ar代表着有效治疗crpc的可行方法的观点。因此,需要改进的ar抑制剂。
技术实现要素:
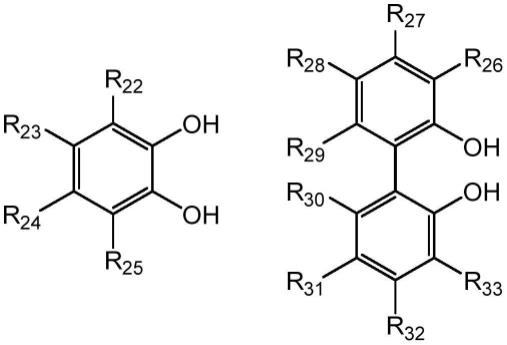
7.在某些方面,本发明提供了具有式i、ii、iii、iv、v、vi、vii或viii的结构的化合物或其药学上可接受的盐:
[0008][0009]
其中:
[0010]
a1是芳基或杂芳基;
[0011]
a2是芳基或杂芳基;
[0012]
r5是h、烷基或卤代;
[0013]
r1是h、烷基、卤代烷基、芳烷基或杂芳烷基;
[0014]
r2是h、烷基或卤代烷基;
[0015]
r3是h、烷基、卤代烷基、芳基或杂芳基;
[0016]r4a
和r
4b
各自独立地为h或烷基,或r
4a
和r
4b
组合形成氧代;
[0017]
是单键或双键;
[0018]
当在式(ii)中为单键时,r
1a
、r
1b
、r
2a
和r
2b
各自独立地是h、烷基或烷氧基;
[0019]
当在式(ii)中为双键时,
[0020]r1a
和r
2a
各自独立地为h、烷基或烷氧基,并且
[0021]r1b
和r
2b
不存在;
[0022]
当在式(vi)中为单键时,r
1a
和r
1b
组合形成ch2;
[0023]
当在式(vi)中为双键时,r
1a
是h或烷基且r
1b
不存在;
[0024]
r6是h、烷基、芳烷基或杂芳烷基;
[0025]
x1和x2各自独立地是nh或o;
[0026]
n是1-4;
[0027]
x是o、nh或s;
[0028]
r7是氨基、炔基、氰基、环烷基、烷基或烯基;
[0029]
z是s或c;
[0030]
当z是s时,r
8a
和r
8b
各自为氧代;
[0031]
当z是c时,
[0032]r8a
和r
8b
各自独立地是h或烷基,或者
[0033]r8a
和r
8b
组合形成氧代,或者
[0034]r8a
和r
8b
组合形成包括z的环丙基环。
[0035]
在某些优选的实施方案中,当a1和a2在式(viii)中均为苯基时,a1和a2中的至少一个被取代。
[0036]
在某些优选实施方案中,式i、ii、iii、iv、v、vi、vii或viii的化合物不是:
[0037]
[0038]
[0039]
[0040]
[0041]
[0042][0043]
式i、ii、iii、iv、v、vi、vii和viii的示例性化合物包括表i中描述的化合物。
[0044]
在某些方面,本发明提供了化合物jn032的固体形式,其特征在于在约21.5
°
、约22.6
°
和约27.3
°
的2θ角处的x射线粉末衍射峰。
[0045]
在某些方面,本发明提供了固体形式,其为化合物jn110的形式i,其特征在于在约17.6
°
、约22.2
°
和约28.8
°
的2θ角处的x射线粉末衍射峰。
[0046]
在某些方面,本发明提供了固体形式,其为化合物jn034的形式i,其特征在于在约8.3
°
、约17.7
°
和约22.4
°
的2θ角处的x射线粉末衍射峰。
[0047]
在某些方面,本发明提供了固体形式,其为化合物jn097的形式i,其特征在于在约20.5
°
、约23.1
°
和约27.0
°
的2θ角处的x射线粉末衍射峰。
[0048]
在某些方面,本发明提供了固体形式,其为化合物jn117的形式i,其特征在于在约7.8
°
、约16.4
°
和约21.5
°
的2θ角处的x射线粉末衍射峰。
[0049]
在某些方面,本发明提供了固体形式,其为化合物jn103的形式i,其特征在于在约6.6
°
、约18.0
°
和约21.6
°
的2θ角处的x射线粉末衍射峰。
[0050]
本发明还涉及主题化合物的药物组合物,以及使用这些化合物或组合物治疗癌症诸如前列腺癌的方法。
附图说明
[0051]
图1是与ar信号传导和治疗靶向相关的细胞过程的示意性描绘。a)雄激素合成的生理调节。lhrh的搏动性分泌诱导垂体前叶分泌促黄体生成素(lh),进而驱动睾丸合成和
分泌睾酮(t),90-95%的雄激素来源于睾丸。lhrh类似物通过持续、不间断地对垂体前叶上的lhrh受体结合来抑制lh分泌。肾上腺是雄激素的次要来源;肾上腺雄激素(例如dhea)在外周组织中转化为t或双氢睾酮(dht)。b)ar工作机制。配体结合后,ar发生二聚化,移位至细胞核,并诱导基因转录。新型ar靶向剂(以红色标示)抑制肿瘤内类固醇生成(例如,阿比特龙,一种17α-羟化酶抑制剂)或用作纯ar拮抗剂(例如mdv3100)。c)全长ar(ar
fl
)、组成型活性arδlbd和可作为高通量筛选测定的基础的y1h系统的示意图。不依赖配体的arδlbd,当在我们遗传改造的药物可渗透的酵母菌株中表达时,会与are的串联拷贝结合,这诱导报告基因的表达。
⊥
抑制;
→
激活;nls:核定位信号。
[0052]
图2.全长ar和缺乏功能性lbd的组成型活性ar剪接变体的一级氨基酸结构的示意图。
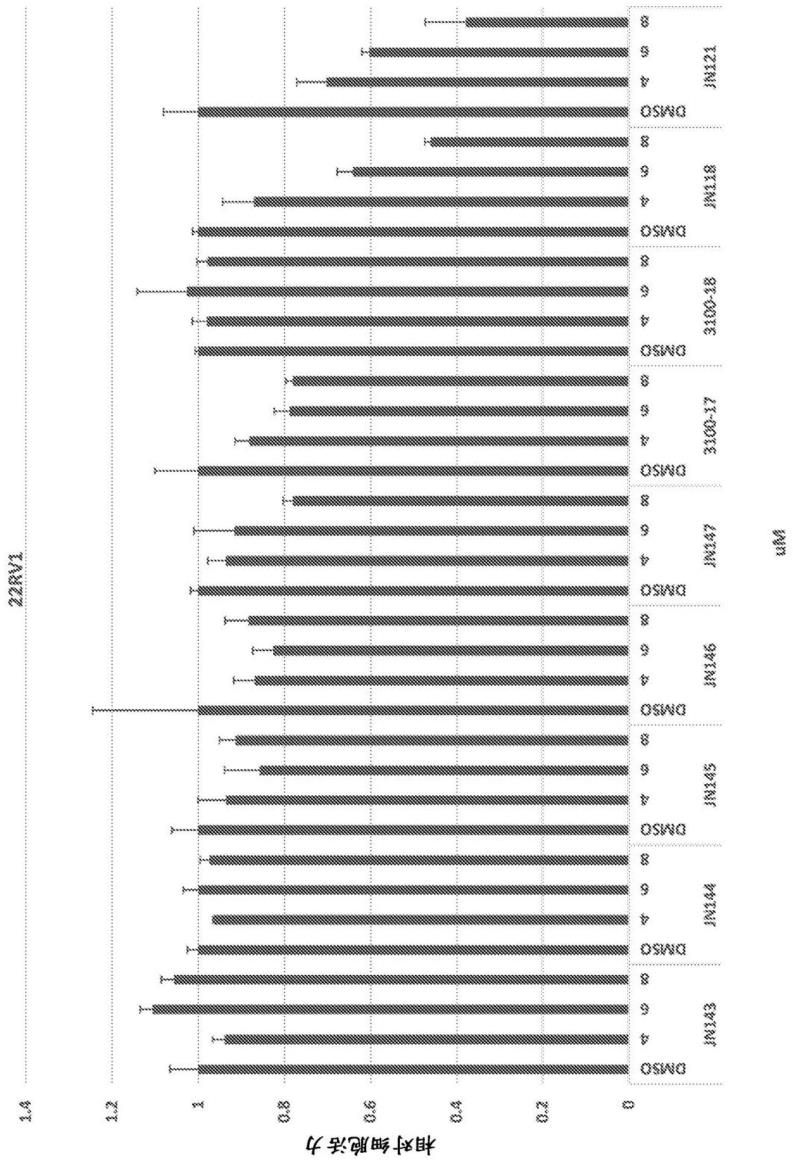
[0053]
图3a至图3q.所选化合物的生长抑制作用。将指定的细胞暴露于指定的化合物6天;通过mtt法测定细胞活力,并使用文献条件测定特定报道分子。将结果以媒介物对照的结果作归一化。以一式四份进行实验;结果为平均值
±
s.d.
[0054]
图3a.22rv1细胞。对于图中的每个浓度,条块从左到右代表针对于jn143、jn144、jn145、jn146、jn147、3100-17、3100-18、jn118和jn121的相对细胞活力。
[0055]
图3b.将22rv1细胞暴露于指定的化合物6天;通过mtt法测定细胞活力。将结果以媒介物对照的结果作归一化。以一式四份进行实验;结果为平均值
±
s.d。对于附图中的每个浓度,条形图从左到右代表针对于jn148、jn149、jn150、jn151、jn152、jn103、jn3100-724、jn3100-18的相对细胞活力。
[0056]
图3c.22rv1细胞(蓝色)、lncap ar细胞(红色)和pc3细胞(绿色)。对于图中的每个浓度,条块从左到右代表针对于jn148、jn149、jn150、jn151、jn152、jn103、jn3100-724、jn3100-18的细胞活力。
[0057]
图3d.22rv1细胞(红色)、lncap ar细胞(蓝色)和pc3细胞(绿色)。对于图中的每个浓度,条块从左到右代表针对于jn152、jn155、jn103和jn154的细胞活力。
[0058]
图3e.22rv1细胞(棕色)、lncap ar细胞(蓝色)和pc3细胞(绿色)。对于图中的每个浓度,条块从左到右代表针对于jn138、jn139、jn140、jn141、jn142、jn103的细胞活力。
[0059]
图3f.lncap ar细胞。对于图中的每个浓度,条块从左到右代表针对于jn143、jn144、jn145、jn146、jn147、3100-17、3100-18、jn118和jn121的细胞活力。
[0060]
图3g.lncap ar细胞。对于图中的每个浓度,条块从左到右代表针对于jn148、jn149、jn150、jn151、jn152、jn103、jn3100-724、jn1300-18的细胞活力。
[0061]
图3h.lncap ar细胞。对于图中的每个浓度,条块从左到右代表针对于jn152、jn153、jn103和jn154的细胞活力。
[0062]
图3i.lncap ar细胞。对于图中的每个浓度,条形图从左到右代表针对于jn152和jn103的mmtv报道基因测定数据。
[0063]
图3j.对于图中的每个浓度,条块从左到右代表针对于jn152和jn103的lncap ar细胞中的mmtv报告基因测定数据(棕色)、pc3细胞中的gal4-ar报道基因测定数据(蓝色)、pc3细胞中的gre报道基因测定数据(黄色)和pc3细胞中的creb报告基因测定数据(绿色)。
[0064]
图3k.lncap ar细胞(棕色)、22rv1细胞(蓝色)和pc3细胞(绿色)。对于图中的每个浓度,条块从左到右代表针对于jn153、jn154、jn155、jn156和jn103的细胞活力。
[0065]
图3l.pc3细胞。对于图中的每个浓度,条形图从左到右代表针对于jn152和jn103的萤光素酶报道基因测定数据。
[0066]
图3m.pc3细胞。对于图中的每个浓度,条形图从左到右代表针对于jn152和jn103的gal4-ar报道基因测定数据。
[0067]
图3n.pc3细胞。对于图中的每个浓度,条形图从左到右代表针对于jn152和jn103的gre报道基因测定数据。
[0068]
图3o.pc3细胞。对于图中的每个浓度,条块从左到右代表针对于jn143、jn144、jn145、jn146、jn147、3100-17、3100-18、jn118和jn121的细胞活力数据。
[0069]
图3p.pc3细胞。对于图中的每个浓度,条块从左到右代表针对于jn148、jn149、jn150、jn151、jn152、jn103、jn3100-724和jn3100-18的细胞活力数据。
[0070]
图3q.pc3细胞。对于图中的每个浓度,条块从左到右代表针对于jn152、jn155、jn103和jn154的细胞活力数据。
[0071]
图4显示了化合物jn032的x射线粉末衍射(xrpd)谱。
[0072]
图5显示了化合物jn110的xrpd谱。
[0073]
图6显示了化合物jn034的xrpd谱。
[0074]
图7显示了化合物jn097的xrpd谱。
[0075]
图8显示了化合物jn117的xrpd谱。
[0076]
图9显示了化合物jn103的xrpd谱。
[0077]
图10显示用jn103(10μm)处理8小时的22rv1和lncap-ar细胞的基因集组表达分析。显示了ar转录程序的负富集分数(nes)。
[0078]
图11a显示了jn103对lncap-ar细胞的选择性降解。
[0079]
图11b显示了jn103对lncap-95细胞的选择性降解。
[0080]
图11c显示了jn103对被改造成为异位表达arδ567的hek-293细胞的选择性降解。
[0081]
图11d显示了jn103对pc3细胞的选择性降解。
[0082]
图11e显示了jn103对t47d乳腺癌细胞的选择性降解。
[0083]
图12显示了用jn103处理的du145、pc3lncap-ar(全长ar)、22rv1(全长和剪接变体ar)和vcap细胞的集落形成测定。
[0084]
图13显示了jn103在mtt测定中对20种非前列腺癌细胞系的生长抑制作用。
具体实施方式
[0085]
在某些方面,本公开提供了具有式i、ii、iii、iv、v、vi、vii或viii的结构的化合物及其药学上可接受的盐:
[0086][0087]
其中:
[0088]
a1是芳基或杂芳基;
[0089]
a2是芳基或杂芳基;
[0090]
r5是h、烷基或卤代;
[0091]
r1是h、烷基、卤代烷基、芳烷基或杂芳烷基;
[0092]
r2是h、烷基或卤代烷基;
[0093]
r3是h、烷基、卤代烷基、芳基或杂芳基;
[0094]r4a
和r
4b
各自独立地为h或烷基,或r
4a
和r
4b
组合形成氧代;
[0095]
是单键或双键;
[0096]
当在式(ii)中为单键时,r
1a
、r
1b
、r
2a
和r
2b
各自独立地是h、烷基或烷氧基;
[0097]
当在式(ii)中为双键时,
[0098]r1a
和r
2a
各自独立地为h、烷基或烷氧基,并且
[0099]r1b
和r
2b
不存在;
[0100]
当在式(vi)中为单键时,r
1a
和r
1b
组合形成ch2;
[0101]
当在式(vi)中为双键时,r
1a
是h或烷基且r
1b
不存在;
[0102]
r6是h、烷基、芳烷基或杂芳烷基;
[0103]
x1和x2各自独立地是nh或o;
[0104]
n是1-4;
[0105]
x是o、nh或s;
[0106]
r7是氨基、炔基、氰基、环烷基、烷基或烯基;
[0107]
z是s或c;
[0108]
当z是s时,r
8a
和r
8b
各自为氧代;
[0109]
当z是c时,
[0110]r8a
和r
8b
各自独立地是h或烷基,或者
[0111]r8a
和r
8b
组合形成氧代,或者
[0112]r8a
和r
8b
组合形成包括z的环丙基环。
[0113]
在某些实施方案中,本公开提供式viii的化合物,其中当a1和a2均为苯基时,a1和a2中的至少一个被取代。
[0114]
在某些实施方案中,本公开提供了具有式(ia)、(ib)、(iia)、(iib)、(iic)、(va)、(vb)、(via)、(vib)、(viia)、(viib)或(viic)的结构的化合物:
[0115]
[0116]
[0117][0118]
在某些实施方案中,化合物由式i(诸如式ia或式ib)表示。在某些实施方案中,化合物由式ii(诸如式iia或式iib)表示。在某些实施方案中,化合物由式iii表示。在某些实施方案中,化合物由式iv表示。在某些实施方案中,化合物由式v(诸如式va或式vb)表示。在某些实施方案中,化合物由式vi(诸如式via或式vib)表示。在某些实施方案中,化合物由式vii(诸如式viia、viib或viic)表示。在某些实施方案中,化合物由式viii表示。
[0119]
在本文所述式的某些优选实施方案中,a1和a2互为顺式。
[0120]
在某些实施方案中,a2是未取代的芳基或被一个或多个r
11
取代的芳基,其中每个r
11
独立地选自卤代、烷基、卤代烷基、羟基、氰基、烷氧基、炔基或叠氮基。在某些此类实施方案中,a2是氯苯基。
[0121]
在某些实施方案中,a2是未取代的杂芳基或被一个或多个r
11
取代的杂芳基,其中每个r
11
独立地选自卤代、烷基、卤代烷基、羟基、氰基、烷氧基、炔基或叠氮基。在某些此类实施方案中,a2是被三氟甲基取代的吡啶基(例如吡啶-3-基),诸如5-三氟甲基吡啶-3-基。
[0122]
在某些实施方案中,a1是苯基。
[0123]
在某些实施方案中,a1是未取代的。
[0124]
在某些实施方案中,a1是未取代的或被至少一个r
12
取代的芳基,其中每个r
12
独立地选自卤代、烷基、卤代烷基、羟基、氰基、烷氧基、炔基或叠氮基。在某些此类实施方案中,a1被至少一个r
12
取代。
[0125]
在某些实施方案中,其中r5是h或烷基。在某些此类实施方案中,r5是h。
[0126]
在某些实施方案中,r1是h或甲基。
[0127]
在某些实施方案中,r2是h。
[0128]
在某些实施方案中,r3是h、卤代烷基或芳基。
[0129]
在某些实施方案中,r
4a
和r
4b
各自是h。在某些其它实施方案中,r
4a
和r
4b
组合形成氧代。
[0130]
在式iii的某些实施方案中,r6是芳基。在某些实施方案中,r6是苄基。
[0131]
在式iv的某些实施方案中,r3是h、卤代烷基或芳基,诸如h、三氟甲基或苯基。在某些其它实施方案中,r1是h、甲基或苄基。在式iv的某些实施方案中,诸如当r3是h、卤代烷基或芳基时,r1和r2互为反式。
[0132]
在某些实施方案中,本公开提供了选自以下的化合物:
[0133]
[0134][0135]
在某些实施方案中,本公开提供了选自以下的化合物:
[0136][0137]
在某些方面,本公开提供了本文公开的化合物的固体形式。
[0138]
在某些实施方案中,本公开提供了化合物jn032的形式i,其特征在于在约21.5
°
、约22.6
°
和约27.3
°
的2θ角处的x射线粉末衍射峰。在某些实施方案中,jn032的形式i的特征还在于在约16.5
°
、约20.5
°
和约28.2
°
的2θ角处的x射线粉末衍射峰。jn032的形式i的特征也在于基本上如图4所示的x射线粉末衍射图。
[0139]
在某些实施方案中,本公开提供了化合物jn110的形式i,其特征在于在约17.6
°
、约22.2
°
和约28.8
°
的2θ角处的x射线粉末衍射峰。在某些实施方案中,jn110的形式i的特征还在于在约10.2
°
、约15.0
°
和约21.3
°
的2θ角处的x射线粉末衍射峰。jn110的形式i的特征也在于基本上如图5所示的x射线粉末衍射图。
[0140]
在某些实施方案中,本公开提供了化合物jn034的形式i,其特征在于在约8.3
°
、约17.7
°
和约22.4
°
的2θ角处的x射线粉末衍射峰。在某些实施方案中,jn034的形式i的特征还在于在约9.7
°
、约14.4
°
和约25.0
°
的2θ角处的x射线粉末衍射峰。jn034的形式i的特征也在于基本上如图6所示的x射线粉末衍射图。
[0141]
在某些实施方案中,本公开提供了化合物jn097的形式i,其特征在于在约20.5
°
、约23.1
°
和约27.0
°
的2θ角处的x射线粉末衍射峰。在某些实施方案中,jn097的形式i的特征还在于在约12.1
°
、约18.7
°
和约22.1
°
的2θ角处的x射线粉末衍射峰。jn097的形式i的特征也在于基本上如图7所示的x射线粉末衍射图。
[0142]
在某些实施方案中,本公开提供了化合物jn117的形式i,其特征在于在约7.8
°
、约16.4
°
和约21.5
°
的2θ角处的x射线粉末衍射峰。在某些实施方案中,jn117的形式i的特征还在于在约18.5
°
、约19.1
°
和约20.1
°
的2θ角处的x射线粉末衍射峰。jn117的形式i的特征也在于基本上如图8所示的x射线粉末衍射图。
[0143]
在某些实施方案中,本公开提供了化合物jn103的形式i,其特征在于在约6.6
°
、约18.0
°
和约21.6
°
的2θ角处的x射线粉末衍射峰。在某些实施方案中,jn103的形式i的特征还在于在约23.7
°
、约25.1
°
和约28.1
°
的2θ角处的x射线粉末衍射峰。jn103的形式i的特征也在于基本上如图9所示的x射线粉末衍射图。
[0144]
在某些方面,本公开提供了包含本文公开的化合物之一(诸如本文公开的固体形式)和药学上可接受的赋形剂的药物组合物。
[0145]
在某些方面,本公开提供了使用本文公开的化合物(例如本文公开的固体形式)的方法。在某些实施方案中,所述方法用于抑制雄激素受体,并且包括使雄激素受体与本文公开的化合物或组合物接触。在某些实施方案中,所述方法用于诱导细胞中雄激素受体的降解,其包括使雄激素受体与本文公开的化合物或组合物接触。
[0146]
在某些实施方案中,本公开提供了用于治疗患有癌症的哺乳动物的方法,其包括施用本文公开的化合物或组合物。在某些实施方案中,癌症是前列腺癌,例如去势抵抗性前列腺癌。癌症可以是转移性的或非转移性的。在某些优选实施方案中,癌症对抗雄激素疗法(诸如利用恩扎鲁胺、比卡鲁胺、阿比特龙、氟他胺、尼鲁米特、达洛鲁胺或阿帕鲁胺(apalutamide)的治疗)有抗性。在进一步的实施方案中,癌症对利用恩扎鲁胺、比卡鲁胺、阿比特龙(例如醋酸阿比特龙)、氟他胺或尼鲁米特的治疗具有抗性。在某些此类实施方案中,癌症可能对利用醋酸阿比特龙和泼尼松或醋酸阿比特龙和泼尼松龙的联合治疗具有抗性。
[0147]
在某些方面,本公开提供了本文所述的化合物:本文所述的化合物可用作例如癌症治疗剂,特别是用作ar抑制剂和降解剂。在某些方面,本公开提供了使用本文所述的化合物治疗增殖性疾病诸如前列腺癌的方法、抑制ar的方法和提高ar降解速率的方法。
[0148]
在某些实施方案中,本发明的化合物是本文所述化合物的前药。例如,其中母体化合物中的羟基以酯或碳酸酯的形式存在,或者母体化合物中存在的羧酸以酯的形式存在。在某些此类实施方案中,前药在体内被代谢成活性母体化合物(例如,酯水解为相应的羟基或羧酸)。
[0149]
在某些实施方案中,本发明的化合物可以是外消旋的。在某些实施方案中,本发明的化合物可富集在一个对映异构体中。例如,本发明的化合物可具有大于30%ee、40%ee、50%ee、60%ee、70%ee、80%ee、90%ee或甚至95%或更大的ee。在某些实施方案中,本发明的化合物可具有不止一个立体中心。在某些此类实施方案中,本发明的化合物可富集在一个或多个非对映异构体中。例如,本发明的化合物可具有大于30%de、40%de、50%de、60%de、70%de、80%de、90%de或甚至95%或更大de。
[0150]
在某些实施方案中,本发明提供了包含式i、ii、iii、iv、v、vi、vii或viii的化合物的药物组合物。在某些实施方案中,药物组合物还包含药学上可接受的赋形剂。
[0151]
在某些实施方案中,药物组合物可用于治疗或预防本文所述的疾患或疾病。
[0152]
在某些实施方案中,本发明涉及用式i化合物进行治疗的方法。在某些实施方案中,可富集治疗制剂以主要提供化合物的一种对映异构体或异构体。对映异构体富集的混合物可包含例如至少60mol%,或更优选至少75mol%、90mol%、95mol%或甚至99mol%的一种对映异构体。在某些实施方案中,富集在一个对映异构体中的化合物基本上不含另一个对映异构体,其中基本上不含是指与例如组合物或化合物混合物中的另一个对映异构体的量相比,所述物质占小于10%、或小于5%、或小于4%、或小于3%、或小于2%、或小于1%。例如,如果组合物或化合物混合物包含98克的第一对映异构体和2克的第二对映异构体,那么可以说其含有98mol%的第一对映异构体和仅2%的第二对映异构体。
[0153]
在某些实施方案中,可富集治疗制剂以主要提供化合物的一个非对映异构体。非对映异构体富集的混合物可包含例如至少60mol%,或更优选至少75mol%、90mol%、95mol%或甚至99mol%的一个对映异构体。
[0154]
在某些实施方案中,本发明提供适用于人患者的药物制剂,其包含任何上述化合物和一种或多种药学上可接受的赋形剂。
[0155]
任何上述结构的化合物可用于制造用于治疗本文公开的任何疾病或疾患的药物。
[0156]
在某些方面,本公开的化合物用于抑制雄激素受体。
[0157]
在某些方面,本公开的化合物用于在表达雄激素受体的细胞中诱导雄激素受体的降解。
[0158]
在某些方面,本公开的化合物用于治疗患有癌症的哺乳动物。在某些实施方案中,癌症是前列腺癌。在某些实施方案中,癌症是去势抵抗性前列腺癌。在某些实施方案中,癌症是转移性的。在某些实施方案中,癌症是非转移性的。
[0159]
在上述方面的某些实施方案中,癌症对抗雄激素疗法具有抗性。在某些实施方案中,癌症对利用恩扎鲁胺、比卡鲁胺、阿比特龙、氟他胺或尼鲁米特的治疗具有抗性。在某些实施方案中,癌症对利用醋酸阿比特龙的治疗具有抗性。在某些实施方案中,癌症对利用醋酸阿比特龙和泼尼松龙的联合治疗具有抗性。
[0160]
在某些方面,本公开提供了抑制雄激素受体的方法,其包括使雄激素受体与本公开的化合物或组合物接触。
[0161]
在某些方面,本公开提供了诱导雄激素受体降解的方法,其包括使雄激素受体与本公开的化合物或组合物接触。
[0162]
在某些方面,本公开提供了治疗患有癌症的哺乳动物的方法,其包括施用本公开的化合物或组合物。在某些实施方案中,癌症是前列腺癌。在某些实施方案中,癌症是去势抵抗性前列腺癌。在某些实施方案中,癌症是转移性的。在某些实施方案中,癌症是非转移性的。
[0163]
在上述方面的某些实施方案中,癌症对抗雄激素疗法具有抗性。在某些实施方案中,癌症对利用恩扎鲁胺、比卡鲁胺、阿比特龙、氟他胺或尼鲁米特的治疗具有抗性。在某些实施方案中,癌症对利用醋酸阿比特龙的治疗具有抗性。在某些实施方案中,癌症对利用醋酸阿比特龙和泼尼松龙的联合治疗具有抗性。
[0164]
讨论
[0165]
本公开描述了以新型方式抑制ar的化合物。在哺乳动物细胞系统中,式i、ii、iii、iv、v、vi、vii或viii的化合物抑制配体诱导的和组成型ar转录活性,并增强ar降解。
[0166]
本文公开的化合物靶向ar n-端tad。这些化合物可用于治疗其生长由ar或其剪接变体驱动的疾病。前列腺癌是这样的疾病的一个实例。这些化合物比现有的、批准的靶向ar的化合物具有竞争优势,因为现有化合物靶向ar的lbd,而本文公开的化合物对于抗缺乏功能性lbd的全长和组成型活性ar变体具有活性。本文公开的化合物靶向ar n-端并抑制缺乏功能性lbd的组成型活性ar变体的活性(更多细节参见下文第6节)。这些ar变体已被证明赋予对当前批准的ar靶向剂的抗性。另外,这些化合物诱导包括ar剪接变体在内的ar降解,这不是已获得监管批准的任何ar靶向剂的已知机制。这些ar变体已被证明赋予对当前ar靶向剂的抗性。
[0167]
组合物和施用方式
[0168]
本发明的化合物可以以游离碱、盐(优选药学上可接受的盐)、溶剂化物、水合物、前药、异构体或其混合物的形式用于治疗本文所述的疾患。所有形式都在本公开的范围内。可以形成酸加成盐并提供更方便的使用形式;实际上,盐形式的使用本质上等同于碱形式的使用。可用于制备酸加成盐的酸优选包括当与游离碱组合时产生药学上可接受的盐的那些,即其阴离子在该盐的药学剂量中对主题生物无毒的盐,从而使游离碱固有的有益特性不会因阴离子引起的副作用而受损。尽管碱性化合物的药学上可接受的盐是优选的,但所有酸加成盐都可用作游离碱形式的来源,即使期望特定的盐本身只作为中间产物,例如,当形成盐仅用于纯化和鉴定的目的时,或当其在通过离子交换方法制备药学上可接受的盐中用作中间体时。
[0169]
本公开范围内的药学上可接受的盐包括衍生自以下酸的盐;诸如盐酸、硫酸、磷酸和氨基磺酸等无机酸;诸如乙酸、柠檬酸、乳酸、酒石酸、丙二酸、甲磺酸、乙磺酸、苯磺酸、对甲苯磺酸、环己基氨基磺酸、奎尼酸等有机酸。
[0170]
本发明的化合物可以配制成药物组合物,并以适合所选施用途径的各种形式向需要治疗的受试者,例如哺乳动物(诸如人患者)施用,所述施用途径是例如口服、鼻内、腹膜内或肠胃外途径(例如,通过静脉内、腹膜内、皮下、肌内、经上皮、经鼻、肺内、鞘内、直肠或局部途径)。肠胃外施用可以通过在选定的时间段内连续输注进行。
[0171]
根据本公开的方法,如本领域技术人员将理解的,根据所选择的施用途径,所述化合物可以以多种形式向患者施用。含有本公开的化合物的组合物可以通过制备可向受试者施用的药学上可接受的组合物的已知方法来制备,从而将有效量的活性物质与药学上可接受的赋形剂组合成混合物。合适的媒介物描述于例如remington's pharmaceutical sciences(remington's pharmaceutical sciences,mack publishing company,easton,pa.,usa 1985)中。在此基础上,所述组合物包括但不限于与一种或多种药学上可接受的载体或稀释剂结合的物质的溶液,并包含在具有合适的ph值和与生理液体等渗的缓冲溶液中。
[0172]
包含本公开的化合物的组合物还可含有佐剂诸如防腐剂、润湿剂、乳化剂和分散剂。可以通过包含各种抗细菌剂和抗真菌剂诸如对羟基苯甲酸酯、氯丁醇、苯酚、山梨酸等确保对微生物作用的预防。还可合乎需要的是在组合物中包含等渗剂,诸如糖、氯化钠等。另外,可通过包含延迟吸收的剂如单硬脂酸铝和明胶来实现可注射药物形式的延长吸收。
[0173]
本领域技术人员会知道如何制备合适的制剂。用于选择和制备合适制剂的常规程序和成分描述于例如remington's pharmaceutical sciences(1990-第18版)和1999年出
版的the united states pharmacopeia:the national formulary(usp 24nf19)中。
[0174]
因此,本发明的化合物可与药学上可接受的媒介物(诸如惰性稀释剂或可吸收食用载体)组合来全身性施用,例如经口施用;或者通过吸入或吹入全身性施用。它们可封闭在硬质或软质外壳明胶胶囊中,可压制成片剂,或可直接与患者的膳食的食物合并。对于经口治疗性施药,化合物可与一种或多种赋形剂组合且以可摄取片剂、经颊片剂、锭剂、胶囊、酏剂、混悬液、糖浆、粉片等形式使用。化合物可以与细的惰性粉末载体结合并由受试者吸入或吹入。此类组合物和制剂应包含至少0.1%的式i、ii、iii、iv、v、vi、vii或viii的化合物。组合物和制剂的百分比当然可以变化且可宜在给定单位剂型的重量的约2%至约60%之间。化合物在所述治疗适用组合物中的量是使得将获得有效剂量水平的量。
[0175]
在本公开的某些实施方案中,适用于口服施用的包含本公开的化合物的组合物包括:胶囊、扁囊剂、丸剂、片剂、锭剂(使用经调味的基质,通常为蔗糖和阿拉伯胶或西黄蓍胶)、粉剂、颗粒剂或作为水性液体或非水性液体中的溶液或混悬剂、或作为水包油或油包水乳剂、或作为酏剂或糖浆剂、或作为软锭剂(pastille)(使用惰性基质,诸如明胶和甘油,或蔗糖和阿拉伯胶)等,每种均含有预定量的作为活性成分的本公开化合物。
[0176]
在用于口服施用的固体剂型(胶囊、片剂、糖锭、丸剂、糖锭剂、粉末、颗粒等)中,可将一种或多种包含本公开的化合物的组合物与一种或两种药学上可接受的载体(如柠檬酸钠或磷酸二钙)和/或以下中的任何一项进行混合:(1)填充剂或增充剂,诸如淀粉、乳糖、蔗糖、葡萄糖、甘露醇和/或硅酸;(2)粘合剂,诸如,例如,羧甲基纤维素、藻酸盐、明胶、聚乙烯吡咯烷酮、蔗糖、黄芪胶、玉米淀粉和/或阿拉伯胶;(3)湿润剂,诸如甘油;(4)崩解剂,诸如琼脂、碳酸钙、马铃薯或木薯淀粉、藻酸、某些硅酸盐、以及碳酸钠;(5)溶液延迟剂,诸如石蜡;(6)吸收加速剂,如季铵化合物;(7)润湿剂,诸如,例如,鲸蜡醇和单硬脂酸甘油酯;(8)吸收剂,诸如高岭土和膨润土;(9)润滑剂,诸如滑石、硬脂酸钙、硬脂酸镁、固体聚乙二醇、月桂基硫酸钠及其混合物;以及(10)着色剂。在胶囊、片剂以及丸剂的情况下,所述药物组合物还可以包含缓冲剂。在使用赋形剂(诸如乳糖(lactose)或乳糖(milk sugar))以及高分子量聚乙二醇等的软质和硬质填充的明胶胶囊中还可采用相似类型的固体组合物作为填充剂。各种其它物质可作为包衣存在或可存在以另外改进固体单位剂型的实物形式。举例来说,片剂、丸剂或胶囊可用明胶、蜡、虫胶或糖等包覆。糖浆或酏剂可含有活性化合物、作为甜味剂的蔗糖或果糖、作为防腐剂的对羟基苯甲酸甲酯和对羟基苯甲酸丙酯、染料和调味剂,如樱桃或橙调味剂。用于制备任何单位剂型的任何物质都应为药学上可接受的且在所用量下实质上无毒。此外,化合物可并入持续释放制剂和装置中。例如,可将化合物掺入缓释胶囊、缓释片剂和缓释丸剂中。
[0177]
用于口服施用的液体剂型包括药学上可接受的乳剂、微型乳剂、溶液、混悬剂、糖浆剂以及酏剂。除所述本公开的化合物之外,所述液体剂型可以包含本领域常用的惰性稀释剂,诸如,水或其他溶剂、增溶剂以及乳化剂,诸如乙醇(乙醇(ethanol))、异丙醇、碳酸乙酯、乙酸乙酯、苯甲醇、苯甲酸苄酯、丙二醇、1,3-丁二醇、油(具体地说,棉籽油、花生油、玉米油、胚芽油、橄榄油、蓖麻油以及芝麻油)、甘油、四氢呋喃醇、聚乙二醇以及脱水山梨醇的脂肪酸酯及其混合物。除了惰性稀释剂,所述口服组合物还可包含助剂,诸如润湿剂、乳化剂和助悬剂、甜味剂、矫味剂、着色剂、芳香剂和防腐剂。
[0178]
除活性化合物、盐和/或其前药以外,混悬剂还可含有助悬剂,诸如乙氧基化异硬
脂醇、聚氧乙烯山梨醇和脱水山梨糖醇酯、微晶纤维素、偏氢氧化铝、膨润土、琼脂和黄蓍胶及其混合物。
[0179]
在某些实施方案中,适合于肠胃外施用的药物组合物可包含一种或多种活性化合物与一种或多种药学上可接受的无菌等渗水性溶液或非水性溶液、分散液、混悬剂或乳剂,或可在临使用前复原成无菌可注射溶液或分散液的无菌散剂的组合,所述组合可以含有抗氧化剂、缓冲剂、抑菌剂、使制剂与预期受体的血液等渗的溶质、或助悬剂或增稠剂。本公开药物组合物中可以采用的适合的水性和非水性载体的实例包括水、乙醇、多元醇(诸如甘油、丙二醇、聚乙二醇等)、及其适合混合物、植物油(诸如橄榄油)、以及可注射的有机酯(诸如油酸乙酯)。适当流动性可以例如通过使用包衣材料(诸如卵磷脂)、在分散液的情况下通过维持所需粒度以及通过使用表面活性剂加以维持。
[0180]
化合物可通过输注或注射加以静脉内或腹膜内施用。可于水中制备化合物或它们的盐的溶液,任选与无毒界表面活性剂混合。也可于甘油、液体聚乙二醇、三乙酸甘油酯及其混合物中以及于油中制备分散液。在普通的储存和使用条件下,这些制剂可含有防腐剂以防止微生物的生长。
[0181]
适于注射或输注的医药剂型可包括无菌水溶液或分散液或包含适合于临时制备无菌可注射或可输注溶液或分散液,任选囊封于脂质体中的化合物的无菌粉末。在所有情况下,最终剂型在制造和储存条件下都应为无菌的、流动的和稳定的。液体载体或媒介物可为溶剂或液体分散介质,其包括例如水、乙醇、多元醇(例如甘油、丙二醇、液体聚乙二醇等)、植物油或无毒甘油酯及其适合混合物。适当流动性可例如通过形成脂质体、在分散液的情况下通过维持所需粒度或通过使用表面活性剂加以维持。防止微生物作用可通过各种抗细菌剂和抗真菌剂(例如对羟基苯甲酸酯、氯丁醇、苯酚、山梨酸、硫柳汞(thimerosal)等)来达成。在许多情况下,将为优选的是包括等张剂,例如糖、缓冲剂或氯化钠。延长可注射组合物的吸收可通过在组合物中使用延迟吸收的试剂,例如单硬脂酸铝和明胶来达成。
[0182]
无菌可注射溶液是通过将化合物以所需量在必要时与以上列举的各种其它成分一起并入适当溶剂中,随后进行过滤灭菌来制备。在用于制备无菌可注射溶液的无菌粉末的情况下,优选制备方法是真空干燥和冷冻干燥技术,其产生活性成分外加存在于先前无菌过滤溶液中的任何其它所需成分的粉末。
[0183]
对于表面施用,化合物可以纯净形式加以施用。然而,将通常合乎需要的是与皮肤病学上可接受的可为固体或液体的载体组合,以组合物或制剂形式向皮肤施用它们。
[0184]
适用固体载体包括微细固体,如滑石、粘土、微晶纤维素、二氧化硅、氧化铝等。其他固体载体包括无毒聚合物纳米颗粒或微粒。适用液体载体包括化合物可任选借助于无毒表面活性剂在有效水平下溶解或分散于其中的水、醇或二醇或水-醇/二醇掺合物。如芳香剂及其它抗微生物剂的佐剂可被添加来使给定用途的性质最优化。所得液体组合物可自吸收垫施用,用于浸渍绷带和其它敷料,或使用泵型喷雾器或气雾剂喷雾器喷雾于受影响区域上。
[0185]
如合成聚合物、脂肪酸、脂肪酸盐和酯、脂肪醇、改性纤维素或改性矿物质的增稠剂也可与液体载体一起用于形成用于直接向使用者的皮肤施用的可涂敷糊剂、凝胶剂、软膏剂、皂剂等。
[0186]
可用于向皮肤递送化合物的适用皮肤学组合物的实例为本领域所知;例如参见
jacquet等(美国专利第4,608,392号)、geria(美国专利第4,992,478号)、smith等(美国专利第4,559,157号)和wortzman(美国专利第4,820,508号),所述专利全部据此通过引用并入。
[0187]
式i、ii、iii、iv、v、vi、vii或viii的化合物的适用剂量可通过比较它们的体外活性与在动物模型中的体内活性来确定。用于将小鼠和其它动物中的有效剂量外推至人的方法为本领域所知;例如参见美国专利第4,938,949号,其据此通过引用并入。
[0188]
例如,化合物在液体组合物诸如洗剂中的浓度可为约0.1-25重量%,或约0.5-10重量%。半固体或固体组合物诸如凝胶或粉末的浓度可为约0.1-5重量%,或约0.5-2.5重量%。
[0189]
为在治疗中使用所需的化合物的量将不仅随所选特定盐而变化,而且也随施药途径、所治疗病状的性质以及患者的年龄和状况而变化,且将最终由主治医师或临床医师裁量。
[0190]
本发明剂的有效剂量和施用途径是常规的。剂的确切量(有效剂量)将因受试者而异,取决于例如受试者的物种、年龄、体重和一般或临床状况、所治疗的任何病症的严重性或机制、所使用的特定剂或媒介物、施用方法和时间安排等。治疗有效剂量可通过本领域技术人员已知的常规程序凭经验确定。参见,例如,the pharmacological basis of therapeutics,goodman and gilman,编辑,macmillan publishing co.,new york。例如,可在细胞培养测定或合适的动物模型中初步估计有效剂量。动物模型也可用于确定合适的浓度范围和施用途径。这样的信息可用于确定对于在人中施用的有效剂量和途径。治疗剂量也可以通过类比可比治疗剂的剂量来选择。
[0191]
具体施用方式和剂量方案将由主治临床医生考虑病例的具体情况(例如,受试者、疾病、所涉及的疾病状态以及治疗是否是预防性的)来选择。治疗可包括在几天到几个月甚至几年的时间内服用一种或多种化合物的每日一次或多日一次的剂量。
[0192]
然而,一般而言,合适的剂量范围为约0.001至约100mg/kg,例如,约0.01至约100mg/kg体重/天,诸如高于约0.1mg/千克,或在约1至约10mg/千克接受者体重/天的范围内。例如,合适的剂量可以是约1mg/kg体重/天、10mg/kg体重/天或50mg/kg体重/天。
[0193]
式i、ii、iii、iv、v、vi、vii或viii的化合物以单位剂型方便地施用;例如,每单位剂型含有0.05至10000mg、0.5至10000mg、5至1000mg或约100mg的活性成分。
[0194]
可施用化合物以达到例如约0.5至约75μm、约1至50μm、约2至约30μm或约5至约25μm的峰值血浆浓度。示例性的理想血浆浓度包括至少或不超过0.25μm、0.5μm、1μm、5μm、10μm、25μm、50μm、75μm、100μm或200μm。例如,血浆水平可为约1至100微摩尔或约10至约25微摩尔。例如,这可通过静脉内注射0.05至5%的化合物溶液(任选地在盐水中),或作为含有约1-100mg化合物的大丸剂口服施用。可通过连续输注维持理想的血液水平以提供每小时每千克体重约0.00005-5mg,例如至少或不超过0.00005mg/kg/hr、0.0005mg/kg/hr、0.005mg/kg/hr、0.05mg/kg/hr、0.5mg/kg/hr或5mg/kg/hr。或者,此类水平可通过包含约0.0002-20mg/kg体重,例如,至少或不超过0.0002mg化合物/kg体重、0.002mg化合物/kg体重、0.02mg化合物/kg体重、0.2mg化合物/kg体重、2mg化合物/kg体重、20mg化合物/kg体重或50mg化合物/kg体重的间歇性输注而获得。
[0195]
化合物可宜以单次剂量形式提供或以在适当间隔下施用的分次剂量形式,例如以
每天两次、三次、四次或四次以上亚剂量形式提供。亚剂量自身可进一步例如分成许多次个别宽松间隔施药;如自吹入器进行多次吸入。
[0196]
本公开的化合物和/或组合物的剂量可根据许多因素而变化,所述因素是诸如化合物的药效学特性、施用方式、接受者的年龄、健康和体重、症状的性质和程度、治疗的频率和同时治疗的类型(如果有的话)以及化合物在待治疗的受试者中的清除率。本领域技术人员可以根据上述因素确定合适的剂量。本公开的化合物最初可以以合适的剂量施用,所述剂量可以根据临床反应按需要进行调整。为了从用于治疗大鼠的年龄依赖性认知障碍的剂量中计算人的等效剂量(hed),可采用公式hed(mg/kg)=大鼠剂量(mg/kg)x0.16(参见estimating the safe starting dose in clinical trials for therapeutics in adult healthy volunteers,2002年12月,center for biologics evaluation and research)。例如,通过使用该公式,大鼠中10mg/kg的剂量相当于人中的1.6mg/kg。此转换基于更通用的公式hed=以mg/kg表示的动物剂量x(以kg表示的动物体重/以kg表示的人体重)0.33。类似地,为了从用于大鼠治疗的剂量中计算hed,可采用公式hed(mg/kg)=小鼠剂量(mg/kg)x 0.08(参见estimating the safe starting dose in clinical trials for therapeutics in adult healthy volunteers,2002年12月,center for biologics evaluation and research)。
[0197]
本公开的化合物和/或组合物可以单独或与其它治疗剂联合,或与其它类型的治疗组合用于治疗细胞增殖性病症诸如前列腺癌。例如,在一些实施方案中,本公开的化合物和组合物可用于治疗crpc或用于治疗对抗雄激素疗法诸如恩扎鲁胺、比卡鲁胺、阿比特龙、氟他胺或尼鲁米特具有抗性的癌症。例如,根据本公开的方法,这些其它治疗上有用的剂可以按照本公开的方法,在单一制剂中施用,与本公开的化合物同时或依次施用。
[0198]
许多上面鉴定的化合物对激素难治性前列腺癌细胞表现出几乎没有或没有激动活性。由于这些化合物是强ar抑制剂,因此它们不仅可用于治疗前列腺癌,而且还可用于治疗其它ar相关疾病或疾患,诸如良性前列腺增生、脱发和痤疮。由于ar属于核受体家族,因此这些化合物可作为靶向其它核受体(诸如雌激素受体和过氧化物酶体增殖子激活受体)的药物合成的支架。因此,它们可被进一步开发用于核受体在其中发挥作用的其它疾病,诸如乳腺癌、卵巢癌、糖尿病、心脏病和代谢相关疾病。
[0199]
晶型
[0200]
在某些方面,本发明提供了本文所述化合物的固体形式。在某些优选的实施方案中,固体形式是晶型。本文所述化合物的晶型可用于促进化合物的纯化(例如,通过重结晶)并且/或者调节/改善化合物的物理化学性质,包括但不限于固态性质(例如,结晶度、吸湿性、熔点或水合作用)、药物性质(例如,溶解度/溶解速率、稳定性或相容性)以及结晶特性(例如,纯度、产率或形态学)。
[0201]
在某些方面,本发明提供了化合物jn032的固体形式,其特征
在于在约21.5
°
、约22.6
°
和约27.3
°
的2θ角处的x射线粉末衍射(xrpd)峰。在某些优选实施方案中,化合物jn032的固体形式的特征在于基本上如图4所示的xrpd衍射图。
[0202]
在某些方面,本发明提供了固体形式,其为化合物jn110的形式i,其特征在于在约17.6
°
、约22.2
°
和约28.8
°
的2θ角处的xrpd峰。在某些优选实施方案中,化合物jn110的固体形式的特征在于基本上如图5所示的xrpd图。
[0203]
在某些方面,本发明提供了固体形式,其为化合物jn034的形式i,其特征在于在约8.3
°
、约17.7
°
和约22.4
°
的2θ角处的xrpd峰。在某些优选实施方案中,化合物jn034的固体形式的特征在于基本上如图6所示的xrpd图。
[0204]
在某些方面,本发明提供了固体形式,其为化合物jn097的形式i,其特征在于在约20.5
°
、约23.1
°
和约27.0
°
的2θ角处的xrpd峰。在某些优选实施方案中,化合物jn097的固体形式的特征在于基本上如图7所示的xrpd图。
[0205]
在某些方面,本发明提供了固体形式,其为化合物jn117的形式i,其特征在于在约7.8
°
、约16.4
°
和约21.5
°
的2θ角处的xrpd峰。在某些优选实施方案中,化合物jn117的固体形式的特征在于基本上如图8所示的xrpd图。
[0206]
在某些方面,本发明提供了固体形式,其为化合物jn103的形式i,其特征在于在约6.6
°
、约18.0
°
和约21.6
°
的2θ角处的xrpd峰。在某些优选实施方
案中,化合物jn103的固体形式的特征在于基本上如图9所示的xrpd图。
[0207]
图4-9中每个峰的相对强度以及两个θ值在某些条件下可能会发生变化或偏移,尽管晶型是相同的。本领域普通技术人员应该能够通过比较它们的xrpd数据容易地确定给定的晶型是否与图4-9之一中描述的晶型相同。如本文所用,如果一个数据集中的一个或多个峰在另一数据集中的对应峰的
±
0.2
°
2θ内,则xrpd数据集“基本上如在”另一xrpd数据集中所示。
[0208]
如本文所用,术语“约”被定义为接近如本领域普通技术人员所理解的。在一个非限制性实施方案中,当用于提及化合物、试剂或溶剂的量或体积时,术语“约”被定义为在10%以内,优选在5%以内,更优选在1%以内,并且最优选地在0.5%以内。在另一个非限制性实施方案中,当参考xrpd峰使用时,如果峰在所述值的
±
0.2
°
2θ内,则峰处于“大约”所述值处。
[0209]
在某些实施方案中,晶型基本上是纯的。如本文中所用,术语“基本上纯的”,当用于指给定的晶形时,是指纯度至少约为90%的晶型。这意味着晶型不含超过约10%的任何其它形式的化合物。甚至更优选地,术语“基本上纯的”是指纯度至少约为95%的化合物的晶型。这意味着所述化合物的晶型不含超过约5%的任何其它形式的化合物。甚至更优选地,术语“基本上纯的”是指纯度至少约为97%的化合物的晶型。这意味着所述化合物的晶型不含超过约3%的任何其它形式的化合物。
[0210]
定义
[0211]
除非本文另外定义,否则本技术中使用的科学和技术术语应具有本领域普通技术人员通常理解的含义。通常,本文所述的与化学、细胞和组织培养、分子生物学、细胞和癌症生物学、神经生物学、神经化学、病毒学、免疫学、微生物学、药理学、遗传学以及蛋白质和核酸化学结合使用的术语和技术是本领域熟知和常用的那些。
[0212]
除非另外指示,否则本公开的方法和技术一般是根据本领域中熟知并且如本说明书通篇引用和论述的各种一般性和更特定参考文献中所述的常规方法来执行。参见例如,“principles of neural science”,mcgraw-hill medical,new york,n.y.(2000);motulsky,“intuitive biostatistics”,oxford university press,inc.(1995);lodish等人,“molecular cell biology,第4版”,w.h.freeman&co.,new york(2000);griffiths等人,“introduction to genetic analysis,第7版”,w.h.freeman&co.,n.y.(1999);和gilbert等人,“developmental biology,第6版”,sinauer associates,inc.,sunderland,ma(2000)。
[0213]
本文使用的化学术语根据本领域中的常规用法来使用,如由“the mcgraw-hill dictionary of chemical terms”,parker s.编,mcgraw-hill,san francisco,c.a.(1985)所示例的。
[0214]
以上所有内容以及本技术中提及的任何其他公布、专利和公布的专利申请均以引用的方式明确并入本文。当发生冲突时,以本说明书(包括其特定定义)为准。
[0215]
本文使用的术语“剂”表示化合物(诸如有机或无机化合物、化合物的混合物)、生物大分子(诸如核酸、抗体,包括其部分以及人源化、嵌合和人抗体以及单克隆抗体、蛋白质或其部分,例如肽、脂质、碳水化合物)或由生物材料诸如细菌、植物、真菌或动物(特别是哺乳动物)细胞或组织制成的提取物。剂包括例如结构已知的剂和结构未知的剂。此类剂抑制
ar或促进ar降解的能力可以使它们适合作为本公开的方法和组合物中的“治疗剂”。
[0216]“患者”、“受试者”或“个体”可互换使用并且是指人或非人动物。这些术语包括哺乳动物,诸如人类、灵长类动物、家畜动物(包括牛、猪等)、伴侣动物(例如,犬、猫科动物等)以及啮齿动物(例如,小鼠和大鼠)。
[0217]“治疗”病症或患者是指采取措施以获得有益的或所需的结果,包括临床结果。如本文所用以及本领域中很好地理解的,“治疗”是用于获得有益的或所需的结果(包括临床结果)的方式。有益的或所需的临床结果可包括但不限于一种或多种症状或病症的减缓或改善、疾病程度的减小、疾病状态的稳定(即不恶化)、疾病扩散的预防、疾病进程的延缓或减慢、疾病状态的改善或缓和以及缓解(无论是部分缓解或是全部缓解),无论是可检测的或是不可检测的。“治疗”还可以意指与未接受治疗时期望的存活相比延长存活。
[0218]
术语“预防”是本领域公认的,并且当相对于病状诸如局部复发(例如,疼痛)、疾病诸如癌症、征候诸如心力衰竭或任何其他医学病状使用时,是在本领域中所熟知的,并且包括施用组合物,相对于不接受所述组合物的受试者,所述组合物减少医学病状的症状的频率,或延缓其发病。因此,癌症的预防例如包括,例如以统计上和/或临床上显著的量,减少接受预防性治疗的患者群体相对于未治疗对照群体的可检测癌性生长的数量,和/或延缓治疗群体相对于未治疗对照群体的可检测癌性生长的出现。
[0219]
向受试者“施用”物质、化合物或剂或者对物质、化合物或剂的“施用”可使用本领域的技术人员已知的各种方法中的一种来实施。例如,化合物或剂可通过以下方式施用:静脉内、动脉内、真皮内、肌内、腹膜内、皮下、经眼部、舌下、口服(通过摄取)、鼻内(通过吸入)、脊柱内、脑内以及透皮(通过吸收,例如,通过皮肤管)。化合物或剂还可适当地通过可再充电或可生物降解聚合物装置或其他提供对化合物或剂的延长的、缓慢的或受控的释放的装置(例如贴剂和泵剂)或制剂来引入。施用还可以例如进行一次、多次和/或在一个或多个延长时间段内进行。
[0220]
向受试者施用物质、化合物或剂的适当方法还将取决于例如受试者的年龄和/或身体状况以及化合物或剂的化学和生物特性(例如,溶解性、可消化性、生物可用性、稳定性以及毒性)。在一些实施方案中,化合物或剂例如通过摄取向受试者口服施用。在一些实施方案中,口服施用化合物或剂在延长释放或缓慢释放的制剂中,或使用用于此缓慢或延长释放的装置来施用。
[0221]
如本文所用,短语“联合施用”是指两种或更多种不同治疗剂的任何形式的施用,使得当先前施用的治疗剂在体内仍然有效时施用第二剂(例如,两种剂在患者中同时有效,其可以包括两种剂的协同作用)。例如,不同的治疗化合物可以在相同的制剂中或在单独的制剂中同时地或依序地施用。因此,接受这种治疗的个体可以受益于不同治疗剂的组合作用。
[0222]
药物或剂的“治疗有效量”或“治疗有效剂量”是药物或剂当向受试者施用时将具有预期的治疗性作用的量。完全的治疗性作用并不一定通过施用一次剂量而出现,而可能仅在施用一系列剂量之后才出现。因此,可以一次或多次施用来施用治疗有效量。受试者所需的精确有效量将取决于例如受试者的身材、健康和年龄,以及受治疗的病状(诸如癌症或mds)的性质和程度。技术人员可以易于通过常规实验确定给定情况的有效量。
[0223]
本文所用术语“任选的”或“任选地”意指随后描述的事件或情况可以发生或可以
不发生,且该描述包括所述事件或情况发生的情况以及所述事件或情况不发生的情况。例如,“任选地取代的烷基”是指烷基可以是被取代的以及其中烷基是未取代的。
[0224]
应当理解,本发明的化合物上的取代基和取代模式可由本领域普通技术人员选择用来产生化学稳定的化合物,其可通过本领域已知的技术以及下面所示的那些方法从可容易获得的起始材料开始容易地合成。如果取代基本身被不止一个基团取代,应理解这些多个基团可存在于同一个碳上或不同的碳上,只要产生稳定的结构即可。
[0225]
如本文所用,术语“任选地取代的”是指用指定的取代基的基团替换给定结构中的1至6个氢基团,所述取代基包括但不限于:羟基、羟基烷基、烷氧基、卤素、烷基、硝基、硅烷基、酰基、酰氧基、芳基、环烷基、杂环基、氨基、氨基烷基、氰基、卤代烷基、卤代烷氧基、-oco-ch
2-o-烷基、-op(o)(o-烷基)2或
‑‑
ch
2-op(o)(o-烷基)2。优选地,“任选地取代的”是指用上述取代基替换给定结构中的1至4个氢基团。更优选地,1至3个氢基被如上所述的取代基取代。应当理解,所述取代基可被进一步取代。
[0226]
术语“酰基”是本领域公认的,并且是指由通式烃基c(o)-,优选烷基c(o)-表示的基团。
[0227]
术语“酰基氨基”是本领域公认的,并且是指被酰基取代的氨基,并且可以例如由式烃基c(o)nh-表示。
[0228]
术语“酰基氧基”是本领域公认的,并且是指由通式烃基c(o)o-,优选烷基c(o)o-表示的基团。
[0229]
术语“烷氧基”是指连接有氧的烷基。代表性烷氧基包括甲氧基、乙氧基、丙氧基、叔丁氧基等。
[0230]
术语“烷氧基烷基”是指被烷氧基取代的烷基,并且可以由通式烷基-o-烷基表示。
[0231]
术语“烷基”是指饱和脂族基团,包括直链烷基、支链烷基、环烷基(脂环族)基团、烷基取代的环烷基和环烷基取代的烷基。在优选的实施方案中,直链或支链烷基在其主链中具有30个或更少的碳原子(例如,对于直链为c
1-30
,对于支链为c
3-30
),并且更优选为20个或更少。
[0232]
此外,在整个说明书、实施例和权利要求书中使用的术语“烷基”旨在包括未取代的和取代的烷基,后者是指在烃主链的一个或多个碳原子上具有替换氢的取代基的烷基部分,包括卤代烷基,诸如三氟甲基和2,2,2-三氟乙基等。
[0233]
当与诸如酰基、酰氧基、烷基、烯基、炔基或烷氧基的化学部分结合使用时,术语“c
x-y”或“c
x-c
y”意指包括在链中含有x至y个碳的基团。c0烷基表示氢,其中基团在末端位置,如果在内部则为键。例如,c
1-6
烷基在链中含有1至6个碳原子。
[0234]
如本文所用,术语“烷基氨基”是指被至少一个烷基取代的氨基。
[0235]
如本文所用,术语“烷硫基”是指被烷基取代的硫醇基,并且可以由通式烷基s-表示。
[0236]
如本文所用,术语“酰胺”是指基团
[0237][0238]
其中r9和r
10
各自独立地表示氢或烃基,或者r9和r
10
与它们所连接的n原子一起形
成在环结构中具有4至8个原子的杂环。
[0239]
术语“胺”和“氨基”是本领域公认的,并且是指未取代的和取代的胺及其盐,例如可以由下式表示的部分
[0240][0241]
其中r9、r
10
和r
10’各自独立地表示氢或烃基,或者r9和r
10
与它们所连接的n原子一起形成在环结构中具有4至8个原子的杂环。
[0242]
如本文所用,术语“氨基烷基”是指被氨基取代的烷基。
[0243]
如本文所用,术语“芳烷基”是指被芳基取代的烷基。
[0244]
如本文所用,术语“芳基”包括取代或未取代的单环芳族基团,其中环的每个原子为碳。优选地,所述环是5至7元环,更优选6元环。术语“芳基”还包括具有两个或更多个环的多环系统,其中两个或更多个碳是两个相邻环共有的,其中至少一个环是芳族的,例如,其他环可以是环烷基、环烯基、环炔基、芳基、杂芳基和/或杂环基。芳基包括苯、萘、菲、苯酚、苯胺等。
[0245]
术语“氨基甲酸酯”是本领域公认的,并且是指以下基团
[0246][0247]
其中r9和r
10
独立地表示氢或烃基。
[0248]
如本文所用,术语“碳环基烷基”是指被碳环基团取代的烷基。
[0249]
如本文所用,术语“碳环”、“碳环基”和“碳环的”是指其中环的每个原子为碳的非芳族饱和或不饱和环。优选地,碳环含有3至10个原子,更优选5至7个原子。
[0250]
如本文所用,术语“碳环基烷基”是指被碳环基团取代的烷基。
[0251]
术语“碳酸酯”是本领域公认的,并且是指基团-oco
2-。
[0252]
如本文所用,术语“羧基”是指由式-co2h表示的基团。
[0253]
如本文所用,术语“酯”是指基团-c(o)or9,其中r9表示烃基。
[0254]
如本文所用,术语“醚”是指通过氧连接至另一个烃基的烃基。因此,烃基的醚取代基可以是烃基-o-。醚可以是对称的或不对称的。醚的实例包括但不限于杂环-o-杂环和芳基-o-杂环。醚包括“烷氧基烷基”基团,其可以由通式烷基-o-烷基表示。
[0255]
如本文所用,术语“卤基”和“卤素”意指卤素,并且包括氯、氟、溴和碘。
[0256]
如本文所用,术语“杂芳烷基(hetaralkyl)”和“杂芳烷基(heteroaralkyl)”是指被杂芳基取代的烷基。
[0257]
术语“杂芳基(heteroaryl)”和“杂芳基(hetaryl)”包括取代或未取代的芳族单环结构,优选5至7元环,更优选5至6元环,其环结构包含至少一个杂原子,优选一至四个杂原子,更优选一个或两个杂原子。术语“杂芳基(heteroaryl)”和“杂芳基(hetaryl)”还包括具有两个或更多个环的多环系统,其中两个或更多个碳是两个相邻环共有的,其中至少一个环是杂芳族的,例如,其他环可以是环烷基、环烯基、环炔基、芳基、杂芳基和/或杂环基。杂
芳基包括例如吡咯、呋喃、噻吩、咪唑、噁唑、噻唑、吡唑、吡啶、吡嗪、哒嗪和嘧啶等。
[0258]
如本文所用,术语“杂原子”意指除碳或氢以外的任何元素的原子。优选的杂原子是氮、氧和硫。
[0259]
如本文所述的术语“杂环基烷基”是指被杂环基团取代的烷基。
[0260]
术语“杂环基”、“杂环”和“杂环的”是指取代或未取代的非芳族环结构,优选3至10元环,更优选3至7元环,其环结构包含至少一个杂原子,优选一至四个杂原子,更优选一个或两个杂原子。术语“杂环基”和“杂环的”还包括具有两个或更多个环的多环系统,其中两个或更多个碳是两个相邻环共有的,其中至少一个环是杂环的,例如,其他环可以是环烷基、环烯基、环炔基、芳基、杂芳基和/或杂环基。杂环基包括例如哌啶、哌嗪、吡咯烷、吗啉、内酯、内酰胺等。
[0261]
如本文所用,术语“烃基”是指通过不具有=o或=s取代基的碳原子键合的基团,并且通常具有至少一个碳-氢键和主要为碳的主链,但是可以任选地包含杂原子。因此,出于本技术的目的,诸如甲基、乙氧基乙基、2-吡啶基和甚至三氟甲基的基团被认为是烃基,但是诸如乙酰基(其在连接碳上具有=o取代基)和乙氧基(其通过氧而不是碳连接)的取代基不是。烃基包括但不限于芳基、杂芳基、碳环、杂环、烷基、烯基、炔基及其组合。
[0262]
如本文所用,术语“羟烷基”是指被羟基取代的烷基。
[0263]
当与化学部分诸如酰基、酰氧基、烷基、烯基、炔基或烷氧基结合使用时,术语“低级”意指包括其中取代基中有十个或更少原子,优选六个或更少原子的基团。例如,“低级烷基”是指含有十个或更少,优选六个或更少的碳原子的烷基。在某些实施方案中,本文所定义的酰基、酰氧基、烷基、烯基、炔基或烷氧基取代基分别是低级酰基、低级酰氧基、低级烷基、低级烯基、低级炔基或低级烷氧基,无论它们是单独出现还是与其他取代基组合出现,诸如在叙述羟烷基和芳烷基中(在这种情况下,例如,当计算烷基取代基中的碳原子时,不计算芳基内的原子)。
[0264]
术语“多环基”、“多环”和“多环的”是指两个或更多个环(例如,环烷基、环烯基、环炔基、芳基、杂芳基和/或杂环基),其中两个或更多个原子是两个相邻环共用的,例如,环是“稠合环”。多环的每个环可以是取代或未取代的。在某些实施方案中,多环的每个环在环中含有3至10个原子,优选5至7个原子。
[0265]
术语“硫酸酯”是本领域公认的,并且是指基团-oso3h或其药学上可接受的盐。
[0266]
术语“磺酰胺”是本领域公认的,并且是指由以下通式表示的基团
[0267][0268]
其中r9和r
10
独立地表示氢或烃基。
[0269]
术语“亚砜”是本领域公认的,并且是指基团-s(o)-。
[0270]
术语“磺酸酯”是本领域公认的,并指基团so3h或其药学上可接受的盐。
[0271]
术语“砜”是本领域公认的,并且是指基团-s(o)
2-。
[0272]
术语“取代的”是指在主链的一个或多个碳上具有替换氢的取代基的部分。应理解,“取代”或“被
……
取代”包括隐含的条件,即这种取代是根据取代原子和取代基的允许
化合价,并且所述取代产生稳定的化合物,例如,其不会自发地进行诸如通过重排、环化、消除等的转化。如本文所用,术语“取代的”考虑包括有机化合物的所有可允许的取代基。在广义方面,可允许的取代基包括有机化合物的非环状的和环状的、支链的和非支链的、碳环的和杂环的、芳族的和非芳族的取代基。可允许的取代基可以是一个或多个取代基并且对于适当的有机化合物而言是相同或不同的。出于本发明的目的,杂原子诸如氮可以具有氢取代基和/或本文所述的有机化合物的满足杂原子的化合价的任何可允许的取代基。取代基可以包括本文所述的任何取代基,例如卤素、羟基、羰基(诸如羧基、烷氧基羰基、甲酰基或酰基)、硫代羰基(诸如硫代酸酯、硫代乙酸酯或硫代甲酸酯)、烷氧基、磷酰基、磷酸根、膦酸根、次膦酸根、氨基、酰胺基、脒、亚胺、氰基、硝基、叠氮基、巯基、烷硫基、硫酸根、磺酸根、磺酰胺基、亚磺酰胺基、磺酰基、杂环基、芳烷基或芳族或杂芳族部分。本领域技术人员将理解,如果合适,在烃链上取代的部分本身可以被取代。
[0273]
如本文所用,术语“硫代烷基”是指被硫醇基团取代的烷基。
[0274]
如本文所用,术语“硫酯”是指基团-c(o)sr9或-sc(o)r9[0275]
其中r9表示烃基。
[0276]
如本文所用,术语“硫醚”等同于醚,其中氧被硫替换。
[0277]
术语“脲”是本领域公认的并且可以由以下通式表示
[0278][0279]
其中r9和r
10
独立地表示氢或烃基。
[0280]
如本文所用,术语“调节”包括抑制或压制功能或活性(诸如细胞增殖)以及增强功能或活性。
[0281]
短语“药学上可接受的”是本领域公认的。在某些实施方案中,所述术语包括在合理医学判断范围内,适用于与人类和动物的组织相接触而没有过量毒性、刺激、过敏反应或其他问题或并发症、与合理的利益/风险比相称的组合物、赋形剂、助剂、聚合物以及其他材料和/或剂型。
[0282]“药学上可接受的盐”在本文中用于是指适用于治疗患者或与患者的治疗相容的酸加成盐或碱加成盐。
[0283]
如本文所用,术语“药学上可接受的酸加成盐”意指由式i、ii、iii、iv、v、vi、vii或viii表示的任何碱化合物的任何无毒的有机或无机盐。形成合适的盐的例示性无机酸包括盐酸、氢溴酸、硫酸和磷酸,以及金属盐诸如正磷酸一氢钠和硫酸氢钾。形成合适的盐的例示性有机酸包括一元羧酸、二元羧酸和三元羧酸,诸如乙醇酸、乳酸、丙酮酸、丙二酸、琥珀酸、戊二酸、富马酸、苹果酸、酒石酸、柠檬酸、抗坏血酸、马来酸、苯甲酸、苯乙酸、肉桂酸和水杨酸以及磺酸,诸如对甲苯磺酸和甲磺酸。可以形成一元酸盐或二元酸盐,并且此类盐可以水合形式、溶剂化形式或基本上无水形式存在。通常,式ii、ii、iii、iv、v、vi、vii或viii的化合物的酸加成盐更易溶于水和各种亲水性有机溶剂,并且与它们的游离碱形式相比,通常表现出更高的熔点。合适的盐的选择是本领域技术人员已知的。可以使用其他非药学上可接受的盐,例如草酸盐,例如,用于分离供实验室使用的式i、ii、iii、iv、v、vi、vii或viii的化合物,或用于随后转化为药学上可接受的酸加成盐。
[0284]
如本文所用的术语“药学上可接受的碱加成盐”意指由式i、ii、iii、iv、v、vi、vii或viii表示的任何酸化合物或其任何中间体的任何无毒的有机或无机碱加成盐。形成合适的盐的例示性无机碱包括氢氧化锂、氢氧化钠、氢氧化钾、氢氧化钙、氢氧化镁或氢氧化钡。形成合适的盐的例示性有机碱包括脂族、脂环族或芳族有机胺,诸如甲胺、三甲胺和甲基吡啶或氨。适当盐的选择将对本领域的技术人员是已知的。
[0285]
可用于本公开的方法和组合物的许多化合物在其结构中具有至少一个立构中心。这个立构中心可以r或s构型存在,所述r和s符号根据pure appli.chem.(1976),45,11-30中所描述的规则来使用。本公开考虑所有立体异构形式,诸如化合物、盐、前药或其混合物的对映异构体和非对映异构体形式(包括立体异构体的所有可能的混合物)。参见例如wo 01/062726。
[0286]
此外,某些含有烯基的化合物可以作为z(同侧)或e(异侧)异构体存在。在每种情况下,本公开包括混合物和单独的个别异构体两者。
[0287]
一些化合物也可以作为互变异构形式存在。尽管未在本文所述的式中明确指出,但此类形式意图包括在本公开的范围内。
[0288]“前药”或“药学上可接受的前药”是指一种化合物,其在施用之后在宿主中被代谢,例如水解或氧化以形成本公开的化合物(例如,式i、ii、iii、iv、v、vi、vii或viii的化合物)。前药的典型实例包括在活性化合物的官能部分上具有生物不稳定或可裂解(保护)基团的化合物。前药包括可以被氧化、还原、胺化、脱胺化、羟基化、脱羟基化、水解、脱水解、烷基化、脱烷基化、酰化、脱酰化、磷酸化或脱磷酸化以产生活性化合物的化合物。在美国专利6,875,751、7,585,851和7,964,580中公开了使用酯或氨基磷酸酯(phosphoramidate)作为生物不稳定或可裂解(保护)基团的前药的实例,其公开内容以引用的方式并入本文。本公开的前药被代谢以产生式i、ii、iii、iv、v、vi、vii或viii的化合物。本公开在其范围内包括本文所述化合物的前药。例如,在“design of prodrugs”ed.h.bundgaard,elsevier,1985中描述了合适前药的选择和制备的常规程序。
[0289]
如本文所用的术语“药学上可接受的载体”意指可用于配制用于医学或治疗性用途的药物的药学上可接受的材料、组合物或媒介物,诸如液体或固体助滤剂、稀释剂、赋形剂、溶剂或包封材料。
[0290]
如本文所用,术语“溶解度的对数”、“logs”或“logs”在本领域中用于定量化合物的水溶性。化合物的水溶性显著地影响其吸收和分布特性。低溶解度通常伴随着不良吸收。logs值是以摩尔/升为单位测量的溶解度的单位剥离对数(以10为底数)。
[0291]
讨论
[0292]
前列腺腺癌(pca)是美国男性中诊断出的最常见的非皮肤实体瘤,并代表男性癌症相关死亡的第二大原因,仅次于肺癌。pca最初是依赖于雄激素的(ad),并且雄激素剥夺疗法(adt)(其通过手术去势或化学去势(以促黄体激素释放激素(lhrh)类似物的形式(图1a))递送)导致ad pca细胞的细胞凋亡和生长停滞并在几乎所有患者中诱导临床反应。不幸的是,去势抵抗性前列腺癌(crpc)不可避免地发展,不仅代表疾病的终末期(其中中位生存期约为12-15个月),而且与严重的发病率相关。直到最近,化学治疗剂多西他赛还是唯一延长中位总生存期的crpc的全身性疗法,尽管只延长了两到三个月。2010年,另一种细胞毒性化学治疗剂卡巴他赛基于3个月的生存期改善也获得了监管批准,用于多西他赛耐药患
者,细胞疫苗provenge也是如此,所述细胞疫苗在一个高度选择的亚组的体能状态优良的患者中延长了四个月的生存期。因此,尽管有这些适度的、渐进的进步,但仍需要基于对去势抵抗性背后生物学的理解的新型治疗方法来更实质性地改善crpc患者的结果。
[0293]
大量实验和临床证据已经证实,ar活性的恢复是绝大多数crpc患者的治疗抵抗的基础。尽管ar具有非基因引向效应(non-genotropic effect),但ar转录活性的重新激活代表了去势抵抗性所必需和充足的主要生化驱动力。细胞适应,包括1)ar基因扩增,2)肿瘤内类固醇生成,3)允许配体混杂的功能获得性ar基因突变,4)ar的体细胞嵌合,5)ar转录共激活剂的表达增加,6)以及由生长因子、细胞因子和ar磷酸化介导的真正不依赖于配体的ar激活,是相互非排斥的机制,尽管为雄激素的去势血清水平,但仍然驱动ar转录活性。在最近对200多名crpc患者进行的综合基因组分析中,几乎在所有crpc病例中都鉴定了ar信号传导轴的激活突变。
[0294]
基于这些观察,通过新型方法靶向ar信号传导轴的药物,包括纯ar拮抗剂(例如恩扎鲁胺)和旨在抑制肿瘤内类固醇生成的cyp17抑制剂(例如醋酸阿比特龙),已经在临床上取得了进展(图1b)。醋酸阿比特龙和恩扎鲁胺都已被批准用于治疗转移性crpc(mcrpc)。然而,约三分之一的患者出现对这些剂的原发性抗性,而其余患者在不同持续时间的初始反应期后发展了继发性抗性,表现为疾病的进展。
[0295]
证明醋酸阿比特龙和恩扎鲁胺在化学疗法初期和化学疗法后患者中的临床成功的3期研究,证实了ar作为去势抵抗的驱动因素的病理生理学相关性。阿比特龙与恩扎鲁胺之间的交叉抗性是常态,如当这些剂中的一种在另一种剂进展后使用时,反应率低所证明的。自从这些第二代内分泌疗法的临床实施以来,临床前模型以及mcrpc患者队列的测序研究已经证明了在后阿比特龙/后恩扎鲁胺mcrpc中正在进行的ar表达和信号传导。事实上,ar是最频繁突变的基因,在这种情况下,依赖于ar的转录程序被重新激活。因此,在新开发的crpc和后阿比特龙/后恩扎鲁胺crpc中,ar均代表去势抵抗性生长的关键驱动力。
[0296]
缺乏功能性lbd的ar的组成型活性变异体最近被证明在前列腺癌标本中表达,其中在mcrpc标本中的频率不断增加。这些组成型活性变体赋予对醋酸阿比特龙和恩扎鲁胺的抗性;事实上,这些变体预计不会对任何直接或间接靶向lbd的现有药物产生反应。鉴于对阿比特龙和恩扎鲁胺的原发性或继发性抗性的不可避免的发展,以及在去势抵抗性状态的自然和治疗史中ar的病理生理学相关性,开发新型ar靶向剂以改善转移性crpc患者的临床结果的需求尚未得到满足。
[0297]
临床上用于治疗pca的所有现有内分泌疗法,包括但不限于阿比特龙和恩扎鲁胺,都直接或间接靶向ar的c端配体结合结构域(lbd)。ar的c端lbd代表了开发中的新ar靶向剂以及长期使用的那些靶向剂(包括促黄体生成素释放激素(lhrh)类似物(例如亮丙瑞林,一种“化学去势”)和部分ar拮抗剂(例如比卡鲁胺))的直接或间接分子靶标(图1c)。ar的另外的主要结构域,包括位于中心的dna结合结构域(dbd)和n端反式激活结构域(tad),尚未被直接靶向和利用以实现治疗益处。这些结构域是ar转录活动所需要的,但迄今为止,还没有靶向这两个结构域中的任一个的药物成功达到监管批准的程度。位于中心的dbd与核类固醇受体家族的其它成员(例如糖皮质激素受体[gr]、孕酮受体[pr])共享显著的同源性,而位于n端的ar tad与该家族的其他成员的n端的ar tad共享最低的同源性,因此可被选择性靶向。
[0298]
ar tad是内在无序的蛋白质,不适合结晶。因此,其结构还未得到解析,并且通过扩展,ar tad不适合于基于结构的药物设计。靶向tad的概念的原理证明支持来自于其中tad诱饵分子抑制依赖于ar的生长的研究。
[0299]
对靶向tad的概念的原理证明支持来自一个小组最近的研究,所述研究鉴定了tad诱饵分子以及选择性靶向ar tad的海洋海绵(marine sponge)提取物。重要的是,这种被称为epi-001的海洋海绵提取物,通过与tad的af1区的相互作用抑制了crpc的生长。epi-001不是通过高通量筛选鉴定的,很可能已作为工业化合物被海洋海绵体内吸收。其它化合物已被证明对组成型活性ar剪接变体具有抑制作用。galeterone与ar lbd结合,但据报道可诱导ar剪接变体降解。galeterone进入了临床试验,但由于无效,最近在中期分析中中止了一项3期研究。氯硝柳胺(一种抗真菌剂)也抑制ar剪接变体,并已进入早期临床试验。其他ar tad抑制剂包括在国际公布第wo 2018/136792号中描述的那些,所述国际公布通过引用完全并入本文。
[0300]
已经制备本文公开的化合物并测试了其抗ar活性,如表1所列:
[0301]
表1
[0302]
[0303][0304]
本文公开的化合物被认为是直接靶向tad的ar降解剂。通过靶向ar及其剪接变体,这些化合物有望克服依赖于ar的去势抵抗性,而不管潜在的一种或多种分子机制如何,包括但不限于缺乏功能性c端lbd的组成型活性arsv的表达。
[0305]
在某些方面,本公开包括本公开的化合物和药学上可接受的赋形剂。
[0306]
实施例
[0307]
现已大体上描述本发明,参考以下实施例将更容易理解本发明,这些实施例被包括仅出于说明本发明的某些方面和实施方案的目的并且不意图限制本发明。
[0308]
化学
[0309]
一般材料和方法
[0310]
除非另有说明,否则所有溶剂和试剂均购自商业来源,无需进一步纯化即可使用。用于反应的二氯甲烷(氢化钙)、乙醚(钠)和四氢呋喃(钠)通过在指定的干燥剂上蒸馏来干燥。所有反应均在干燥氩气的惰性气氛下进行,并通过薄层色谱法(tlc)在预涂覆的emd硅胶60 f
254 tlc铝板上进行监测,并用uv灯进行显现。快速柱色谱在siliaflash p60(silicycle inc.)硅胶(40-63μm,孔径)上进行。在玻璃背衬的20
×
20cm(1500μm厚)制备型tlc板(analtech,z513040)上进行制备级薄层色谱。在ucla mic magnetic resonance
实验室的bruker av500仪器上获得nmr谱。使用mestrenova nmr软件(mestrelab research s.l.,11.0.2版)分析nmr数据。化学位移(δ)以ppm表示,并用于1h nmr(chcl
3 7.26ppm,dmso-d
6 2.50ppm)和
13
c nmr(cdcl
3 77.16ppm,dmso-d
6 39.52ppm)的内部参照。在配有id-cube离子源和vapur界面(ionsense)的thermo pure plus msd(thermo scientific)上收集的dart-ms谱。离子源和msd均由excalibur 3.0版控制。使用二氯甲烷或氯仿作为溶剂,将分析物点在openspot采样卡(ionsense)上。电离是使用he等离子体完成的,没有使用额外的电离剂。在b-545熔点仪上记录熔点。在2.0
×
50mm waters corp.1.5μm c
18
分析hplc柱上进行分析hplc。在5分钟内使用从5%至95%的含有0.2%hcooh的mecn/水的流动相线性梯度。流速为0.4ml/min,峰通过lct-premier esi-tof质谱仪以正离子模式检测。
[0311]
合成
[0312][0313]
方案1:n-甲基丙烯酰基丙烯酰胺jn103的合成。
[0314]
(e)-3-(4-氯苯基)-2-(4-氟苯基)丙烯酸(1)
[0315]
向烧瓶中的4-氟苯乙酸(15.0g,95.4mmol,1.0当量)和4-氯苯甲醛(13.61g,95.4mmol,1.0当量)中加入乙酸酐和三乙胺的混合物(v/v 1:1,每份37.5ml)。将所得悬浮液在120℃下搅拌6h。然后将其冷却至23℃,并在搅拌的条件下加入75ml浓hcl和225ml水。然后将烧瓶在23℃下放置过夜,过滤所得沉淀并用水洗涤。将该粗产物从乙醇/水中重结晶(在23℃放置过夜以完成沉淀),得到呈浅棕色固体的丙烯酸1(15.50g,56.0mmol,59%)。1h nmr(500mhz,dmso-d6)δ12.84(br s,1h),7.76(s,1h),7.30(d,j=8.6hz,2h),7.22-7.19(m,4h),7.07(d,j=8.6hz,2h);
13
c nmr(126mhz,dmso-d6)δ168.02,161.67(d,j=244.4hz),138.07,133.64,133.30,133.04,131.76,131.68(d,j=8.2hz),128.45,128.14,128.08,115.54(d,j=21.3hz)。
[0316]
(e)-3-(4-氯苯基)-2-(4-氟苯基)-n-甲基丙烯酰基丙烯酰胺(2,jn103)
[0317]
将丙烯酸1(5.0g,18.1mmol,1.0当量)悬浮在二氯甲烷(75ml)中,并将烧瓶冷却至0℃。向其中加入草酰氯(1.87ml,21.7mmol,1.2当量),然后加入无水dmf(0.50ml,缓慢加入),并将溶液在0℃搅拌4h。然后真空除去挥发物,得到呈棕色蜡状固体的粗酰氯。
[0318]
在干冰-丙酮浴中冷却的独立烧瓶中,将n-buli(7.20ml的2.40m己烷溶液,17.2mmol,0.95当量)加入甲基丙烯酰胺(1.49g,17.2mmol,0.95当量)于四氢呋喃(100ml)中的悬浮液中,并在23℃下继续搅拌4h。然后将上述合成的酰氯作为四氢呋喃(25ml)溶液缓慢加入烧瓶中。将所得混合物在23℃搅拌过夜,然后在etoac(200ml)和饱和nh4cl/水(160:40ml)之间分配。分离有机层,依次用饱和nahco3/水(75:75ml)和盐水(100ml)洗涤。
然后将其经无水mgso4干燥,过滤,并真空浓缩。通过硅胶柱色谱纯化,使用0-20%etoac/己烷的流动相梯度,随后使用15-20%的含有2%三乙胺添加剂的etoac/己烷的梯度来纯化粗残余物。然后通过在二氯甲烷/己烷中重结晶来进一步纯化分离的淡黄色固体,得到呈白色固体的n-甲基丙烯酰基丙烯酰胺2(jn103)(953.6mg,2.8mmol,15%)。熔点146.2-146.9℃;1h nmr(500mhz,dmso-d6)δ10.56(br s,1h),7.38(s,1h),7.32(d,j=8.6hz,2h),7.28-7.20(m,4h),7.08(d,j=8.6hz,2h),5.83(s,1h),5.63(q,j=1.5hz,1h),1.84(t,j=1.2hz,3h);
13
c nmr(126mhz,dmso-d6)δ168.86,167.99,161.87(d,j=245.4hz),139.20,136.05,134.77,133.44,133.35,131.75(d,j=8.4hz),131.50,131.44(d,j=3.4hz),128.45,123.05,115.79(d,j=21.5hz),18.09;c
19h16
clfno2[m h]
的hrms m/z计算值为344.08481,实测值为344.08296;分析hplc tr=4.26min。
[0319]
(z)-3-(4-氯苯基)-2-(4-氟苯基)-n-甲基丙烯酰基丙烯酰胺(jn117)
[0320]
z-异构体(jn117)可分离自上面色谱分离的相同反应,得到jn103。类白色固体。1h nmr(500mhz,cdcl3)δ8.33(br s,1h),7.48(dd,j=8.9,5.2hz,2h),7.35-7.29(m,4h),7.08(t,j=8.7hz,2h),6.88(s,1h),5.48(q,j=1.6hz,1h),5.46(q,j=1.0hz,1h),1.83(dd,j=1.6,0.9hz,3h).
13
c nmr(126mhz,cdcl3)δ170.12,165.12,163.07(d,j=248.6hz),139.29,137.28,134.50,133.91,132.42(d,j=3.4hz),129.69,129.07,128.62,128.61(d,j=8.1hz),123.07,115.96(d,j=21.7hz),18.22;c
19h16
clfno2[m h]
的hrms m/z计算值为344.08481,实测值为344.08448。
[0321][0322]
方案2;甲基丙烯酰胺jn138的合成。
[0323]
(e)-3-(4-氯苯基)-2-苯基丙-2-烯-1-醇(4)
[0324]
在0℃下向丙烯酸3(5.1g,19.7mmol,1.0当量)于乙醚(60ml)中的溶液中分小份加入氢化铝锂(1.58g,39.4mmol,2.0当量)。将所得溶液在23℃下搅拌1.5h,然后通过缓慢加入水(8ml)进行淬灭。向该烧瓶中加入乙醚(50ml)、15%naoh溶液(水溶液,50ml)和水(50ml),并将溶液在室温搅拌15min。然后将其通过硅藻土塞过滤,并将硅藻土用乙醚洗涤。在滤液中分离各层,将水层进一步用乙醚(50ml
×
2)萃取。将合并的有机层用盐水(150ml)洗涤,经无水mgso4干燥,过滤并在真空中除去挥发物,得到呈黄色油状物的α-羟基烯烃4(4.81g,19.7mmol,定量)。1h nmr(500mhz,cdcl3)δ7.37-7.30(m,3h),7.20(dd,j=7.9,1.7hz,2h),7.08(d,j=8.5hz,2h),6.91(d,j=8.6hz,2h),6.64(d,j=1.5hz,1h),4.46(d,j=1.5hz,2h);
13
c nmr(126mhz,cdcl3)δ142.37,138.24,135.06,132.59,130.55,129.08,128.77,128.29,127.93,125.21,68.43。
[0325]
(e)-1-氯-4-(3-氯-2-苯基丙-1-烯-1-基)苯(5)
[0326]
在0℃下,向α-羟基烯烃4(255.3mg,1.04mmol,1.0当量)和三乙胺(0.43ml,3.1mmol,3.0当量)于二氯甲烷(8ml)中的溶液中加入对-甲苯磺酰氯(242.8mg,1.25mmol,1.2当量)和催化性4-二甲氨基吡啶(12.8mg,0.10mmol,0.10当量)。在所得溶液于23℃下搅拌过夜后,将反应混合物用etoac(40ml)稀释并用水(20ml
×
2)和盐水(20ml)洗涤。将所得有机层经无水mgso4干燥,过滤,并真空浓缩。使用0-3%etoac/己烷的流动相梯度通过硅胶柱色谱法纯化粗蜡状残余物,得到呈无色油状物的α-氯代烯烃5(249.4mg,0.95mmol,91%)。1h nmr(500mhz,cdcl3)δ7.41-7.30(m,3h),7.23(dd,j=7.6,2.0hz,2h),7.09(d,j=8.6hz,2h),6.90(d,j=8.6hz,2h),6.74(s,1h),4.43(d,j=1.0hz,2h);
13
c nmr(126mhz,cdcl3)δ138.61,137.83,134.40,133.30,130.68,129.84,129.03,128.88,128.38,128.21,51.39。
[0327]
(e)-1-(3-叠氮基-2-苯基丙-1-烯-1-基)-4-氯苯(6)
[0328]
将α-氯代烯烃5(144.0mg,0.55mmol,1.0当量)溶解在3ml dmso中。向其中加入叠氮化钠(106.7mg,1.6mmol,3.0当量)于水(1ml)中的溶液,并将所得悬浮液在23℃下搅拌过夜。然后将反应混合物用水(10ml)稀释并用乙醚(8ml
×
3)萃取。将合并的有机层用水(10ml)和盐水(10ml)洗涤,经无水mgso4干燥,过滤并真空浓缩,得到呈淡黄色油状物的α-叠氮基烯烃6(129.1mg,0.48mmol,87%)。1h nmr(500mhz,cdcl3)δ7.39-7.32(m,3h),7.21(dd,j=7.7,1.8hz,2h),7.09(d,j=8.6hz,2h),6.92(d,j=8.5hz,2h),6.63(s,1h),4.15(d,j=1.2hz,2h);
13
c nmr(126mhz,cdcl3)δ138.14,137.21,134.45,133.15,130.69,129.16,128.81,128.63,128.39,128.22,59.05。
[0329]
(e)-n-(3-(4-氯苯基)-2-苯基烯丙基)甲基丙烯酰胺(7,jn138)
[0330]
在23℃下,向四氢呋喃/水(分别为3ml和0.6ml)中的α-叠氮基烯烃6(118.7mg,0.44mmol,1.0当量)中加入三苯基膦(256.5mg,0.97mmol,2.2当量),并将所得溶液搅拌过夜。然后将反应混合物在etoac与水(各自10ml)之间分配。将水层用etoac(3ml
×
2)进一步萃取。将合并的有机层用盐水(10ml)洗涤,经无水mgso4干燥,过滤并真空浓缩。将由此获得的粗α-氨基烯烃溶解在四氢呋喃(3ml)中并将溶液冷却至0℃。向其中加入三乙胺(0.12ml,0.88mmol,2.0当量)和甲基丙烯酰基氯(40μl,0.44mmol,1.0当量)。在23℃下搅拌该混合物1小时后,用乙醚(8ml)稀释内容物,并用0.1n hcl(水溶液,5ml)、水(2ml)和饱和nahco3(5ml)洗涤。然后将有机层经无水mgso4干燥,过滤,并真空浓缩。使用70:30:2己烷/etoac/三乙胺的流动相,通过硅胶制备型tlc纯化粗残余物,得到呈白色固体的甲基丙烯酰胺7(jn138)(74.1mg,0.24mmol,54%)。1h nmr(500mhz,cdcl3)δ7.38-7.28(m,3h),7.18(d,j=7.1hz,2h),7.06(d,j=8.2hz,2h),6.88(d,j=8.2hz,2h),6.54(s,1h),5.98-5.83(br m,1h),5.55(s,1h),5.27(s,1h),4.32(d,j=5.9hz,2h),1.90(s,3h);
13
c nmr(126mhz,cdcl3)δ168.36,140.14,139.35,138.36,134.93,132.65,130.58,129.09,128.70,128.26,127.98,126.53,119.57,47.24,18.76。
[0331][0332]
方案3:2h-吡咯-2-酮衍生物jn140。
[0333]
(e)-1-(3-(4-氯苯基)-2-苯基丙烯酰基)-3-乙基-4-甲基-1,5-二氢-2h-吡咯-2-酮(9,jn140)
[0334]
将丙烯酸3(1.0g,3.87mmol,1.0当量)悬浮在二氯甲烷(16ml)中,并将烧瓶冷却至0℃。向其中加入草酰氯(0.40ml,4.6mmol,1.2当量),然后加入无水dmf(2滴),并将溶液在0℃搅拌3h。然后真空除去挥发物,得到呈棕色蜡状固体的粗酰氯8,将其溶解在10ml无水四氢呋喃中制成约0.39m的8的溶液。
[0335]
在-78℃下向无水四氢呋喃(6ml)中的3-乙基-4-甲基-1,5-二氢-2h-吡咯-2-酮(140.5mg,1.10mmol,1.0当量)中添加n-buli(0.45ml的2.46m己烷溶液,1.10mmol,1.0当量),并将溶液再搅拌30分钟。然后加入2.82ml(1.10mmol,1.0当量)的上述酰氯(8)溶液。在-78℃下再搅拌1小时后,将反应混合物在etoac(10ml)与饱和nh4cl/水(8:2ml)之间分配。将有机层用饱和nahco3(10ml)洗涤,经无水mgso4干燥,过滤并真空浓缩。使用0-20%etoac/己烷的流动相梯度,通过用2%三乙胺/己烷缓冲的硅胶柱色谱纯化所得的粗物质,得到呈淡黄色蜡状物的2h-吡咯-2-酮9(jn140)(61.2mg,0.17mmol,15%)。1h nmr(500mhz,cdcl3)δ7.40-7.35(m,2h),7.32-7.27(m,3h),7.12(d,j=8.6hz,2h),7.04(d,j=8.5hz,2h),6.77(s,1h),4.26(q,j=1.0hz,2h),2.22(q,j=7.6hz,2h),2.04(t,j=1.0hz,3h),1.00(t,j=7.6hz,3h);
13
c nmr(126mhz,cdcl3)δ169.60,169.36,151.00,137.86,134.72,134.21,133.92,133.81,131.18,131.07,129.79,128.56,128.43,128.24,52.11,16.81,13.63,12.92;c
22h21
clno2[m h]
的hrms m/z计算值为366.12553,实测值为366.12318。
[0336][0337]
方案4:四氢吡啶基衍生物jn142的合成。
[0338]
n'-(1-苄基哌啶-4-亚基)-4-甲基苯磺酰肼(10)
[0339]
在23℃下,向乙醇(25ml)中的甲苯基磺酰肼(2.26g,11.7mmol,1.1当量)中加入1-苄基哌啶-4-酮(2.0ml,10.7mmol,1.0当量),并将溶液搅拌3.5h。将所得固体过滤,用乙醇洗涤,真空干燥,得到呈白色固体的酰肼衍生物10(2.53g,7.1mmol,66%)。1h nmr(500mhz,dmso-d6)δ10.20(s,1h),7.71(d,j=8.3hz,2h),7.38(d,j=8.0hz,2h),7.35-7.27(m,4h),
7.27-7.21(m,1h),3.48(s,2h),2.45-2.31(m,9h),2.17(t,j=5.8hz,2h);
13
c nmr(126mhz,dmso-d6)δ159.26,143.04,138.27,136.32,129.37,128.70,128.19,127.50,126.95,61.20,53.03,51.77,33.94,27.49,21.01。
[0340]
(e)-3-(4-氯苯基)-2-苯基丙烯醛(11)
[0341]
分三部分向溶解在二氯甲烷(90ml)中的α-羟基烯烃4(4.57g,18.7mmol,1.0当量)的冷却溶液(冰水浴)中加入戴斯-马丁高碘剂(dess-martin periodinane)(8.80g,20.5mmol,1.1当量)。将所得悬浮液在4℃下搅拌2.5h。然后向烧瓶中加入20ml饱和nahco3水溶液并搅拌5min。然后将烧瓶内容物在另外的二氯甲烷(60ml)与饱和nahco3(水溶液,80ml)之间进行分配。将有机层取出,依次用饱和nahco3(水溶液,50ml
×
3)和盐水(50ml)洗涤。然后将其经无水mgso4干燥,过滤,并真空浓缩。使用3-10%etoac/己烷的流动相梯度,通过硅胶柱色谱法纯化残余物,得到呈淡黄色固体的烯醛11(2.79g,11.5mmol,61%)。1h nmr(500mhz,cdcl3)δ9.77(s,1h),7.44-7.38(m,3h),7.34(s,1h),7.20(d,j=8.7hz,2h),7.19-7.16(m,2h),7.13(d,j=8.7hz,2h);
13
c nmr(126mhz,cdcl3)δ193.77,148.51,142.27,136.35,133.08,132.62,131.99,129.37,129.14,128.97,128.68。
[0342]
(e)-1-(1-苄基1,2,3,6-四氢吡啶-4-基)-3-(4-氯苯基)-2-苯基丙-2-烯-1-醇(12)
[0343]
将四甲基乙二胺(0.21ml,1.4mmol,5.0当量)添加到酰肼10(100.0mg,0.28mmol,1.0当量)于己烷(3ml)中的冷却(-78℃)溶液中,并将该溶液搅拌10分钟。向其中加入n-buli(0.57ml的2.46m己烷溶液,1.4mmol,5.0当量),然后将所述溶液在-78℃下搅拌15min,在23℃下搅拌2.5h。将所得溶液在冰水浴中冷却,并一次性加入烯醛11(135.9mg,0.56mmol,2.0当量)。然后使反应升温至23℃并搅拌过夜,之后将反应混合物冷却(冰水浴)并通过加入水(2ml)进行淬灭。然后将烧瓶内容物在乙醚(10ml)与水(10ml)之间进行分配。将有机层用盐水(10ml)洗涤,经无水mgso4干燥,过滤并真空浓缩。使用75:25:2己烷/etoac/三乙胺的流动相,通过硅胶制备型tlc纯化粗混合物,得到呈黄色蜡状残余物的醇12(43.1mg,0.10mmol,37%)。1h nmr(500mhz,cdcl3)δ7.35-7.27(m,7h),7.16-7.09(m,3h),7.05(d,j=8.6hz,2h),6.86(d,j=8.6hz,2h),6.72-6.68(m,1h),5.58-5.40(m,1h),4.82(s,1h),3.55(s,2h),3.02-2.83(m,2h),2.64-2.57(m,1h),2.56-2.49(m,1h),2.28-2.13(m,2h);
13
c nmr(126mhz,cdcl3)δ143.04,138.25,135.91,135.15,132.51,130.61,129.30,129.25,128.70,128.36,128.20,127.64,127.22,126.25,122.67,一个重叠的sp2峰,79.81,62.57,52.57,49.81,25.26。
[0344]
(e)-1-(1-苄基-1,2,3,6-四氢吡啶-4-基)-3-(4-氯苯基)-2-苯基丙-2-烯-1-酮(13,jn142)
[0345]
向醇12(40.1mg,96.4μmol,1.0当量)于二氯甲烷(3ml)中的冷却溶液(冰水浴)中加入戴斯-马丁高碘剂(51.6mg,160μmol,1.2当量)。将所得混合物在4℃下搅拌25min。然后将烧瓶内容物在另外的二氯甲烷(5ml)与饱和nahco3(水溶液,5ml)之间进行分配。将水层用另外的二氯甲烷(3ml)进行进一步萃取。将合并的有机层经无水mgso4干燥,过滤并真空浓缩。使用80:20:2己烷/etoac/三乙胺的流动相,通过硅胶制备型tlc纯化粗混合物,得到呈黄色蜡状残余物的四氢吡啶基衍生物13(jn142)。1h nmr(500mhz,cdcl3)δ7.38-7.27(m,8h),7.22-7.17(m,2h),7.13(d,j=8.6hz,2h),6.98(d,j=8.5hz,2h),6.93(s,1h),6.78
(tt,j=3.5,1.5hz,1h),3.63(s,2h),3.23-3.17(m,2h),2.64(t,j=5.7hz,2h),2.55-2.47(m,2h);
13
c nmr(126mhz,cdcl3)δ197.20,141.23,140.35,137.86,137.12,136.40,134.41,134.31,133.60,131.30,129.30,129.26,128.99,128.58,128.50,128.16,127.43,62.67,53.12,49.49,24.99;c
27h25
clno[m h]
的hrms m/z计算值为414.16192,实测值为414.16044。
[0346][0347]
方案5:丙烯酰胺jn144、噁唑衍生物jn148和二氢噁唑衍生物jn149的合成。
[0348]
(e)-3-(4-氯苯基)-2-(2,4-二氟苯基)-n-(丙-2-炔-1-基)丙烯酰胺(15,jn144)
[0349]
在0℃下,向三乙胺(0.87ml,6.24mmol,3.0当量)和炔丙胺(0.41ml,6.24mmol,3.0当量)于四氢呋喃(8ml)中的溶液中加入酰氯14(4.0ml 0.52m溶液,2.08mmol,1.0当量)。将所得溶液在0℃下搅拌1h,然后在23℃下搅拌1h。然后将烧瓶内容物在etoac(30ml)与饱和nh4cl/水(24:6ml)之间进行分配。有机层用水(20ml)和饱和nahco3(20ml)洗涤,经无水mgso4干燥,过滤并真空浓缩。使用0-30%etoac/己烷的流动相梯度,通过用2%三乙胺/己烷缓冲的硅胶柱色谱纯化粗残余物,得到呈白色固体的丙烯酰胺15(jn144)(502.1mg,1.51mmol,73%)。1h nmr(500mhz,cdcl3)δ7.90(s,1h),7.23-7.13(m,3h),7.02-6.91(m,4h),5.59(br t,j=5.5hz,1h),4.13(dd,j=5.4,2.6hz,2h),2.21(t,j=2.6hz,1h);
13
c nmr(126mhz,cdcl3)δ165.71,163.69(dd,j=253.1,12.2hz),160.38(dd,j=251.4,11.6hz),139.33,135.28,133.02,132.85(dd,j=9.6,4.2hz),131.04,128.89,127.32,118.88(dd,j=16.6,3.9hz),113.06(dd,j=21.1,3.9hz),105.58(t,j=25.4hz),79.30,71.91,30.07。
[0350]
(e)-2-(2-(4-氯苯基)-1-(2,4-二氟苯基)乙烯基)-5-甲基噁唑(16,jn148)
[0351]
在23℃下,将氯化铁(iii)(24.3mg,0.15mmol,0.5当量)加入丙烯酰胺15(100.0mg,0.30mmol,1.0当量)于1,2-二氯乙烷(1.5ml)中的溶液中。将所得混合物在80℃下加热3h,然后冷却至23℃。然后将烧瓶内容物在二氯甲烷(5ml)与水(5ml)之间进行分配。将水层用另外的二氯甲烷(2ml
×
2)进行进一步萃取。将合并的有机层用盐水(5ml)洗涤,经无水mgso4干燥,过滤并真空浓缩。使用75:25:2己烷/etoac/三乙胺的流动相通过硅胶制备型tlc纯化粗混合物,得到呈淡黄色固体的噁唑衍生物16(jn148)(54.3mg,0.16mmol,55%)。1h nmr(500mhz,cdcl3)δ7.63(s,1h),7.23(td,j=8.3,6.4hz,1h),7.17(d,j=8.6hz,2h),7.00(d,j=8.4hz,2h),6.96-6.88(m,2h),6.79(q,j=1.2hz,1h),2.36(d,j=
1.2hz,3h);
13
c nmr(126mhz,cdcl3)δ163.35(dd,j=250.9,12.1hz),161.12,160.46(dd,j=250.8,12.3hz),149.38,134.39,133.81,132.78(dd,j=9.4,4.7hz),132.67,130.65,128.80,124.97,122.86,119.57(dd,j=16.4,4.2hz),112.25(dd,j=21.3,3.8hz),104.96(t,j=25.5hz),11.27;c
18h13
clf2no[m h]
的hrms m/z计算值为332.06482,实测值为332.06348。
[0352]
(e)-2-(2-(4-氯苯基)-1-(2,4-二氟苯基)乙烯基)-5-亚甲基-4,5-二氢噁唑(17,jn149)
[0353]
将二碘锌(95.7mg,0.30mmol,1.0当量)加入丙烯酰胺15(100.0mg,0.30mmol,1.0当量)于二氯甲烷(1.5ml)中的溶液中,并将所得混合物于23℃下搅拌3h。然后将烧瓶内容物在二氯甲烷(5ml)与水(5ml)之间进行分配。将水层用另外的二氯甲烷(2ml
×
2)进行进一步萃取。将合并的有机层用盐水(5ml)洗涤,经无水mgso4干燥,过滤并真空浓缩。使用80:20:2己烷/etoac/三乙胺的流动相通过硅胶制备型tlc纯化粗混合物,得到呈类白色固体的二氢噁唑衍生物17(jn149)(59.3mg,0.18mmol,60%)。1h nmr(500mhz,cdcl3)δ7.64(s,1h),7.23-7.16(m,3h),7.00(d,j=8.3hz,2h),6.96-6.85(m,2h),4.79(q,j=3.0hz,1h),4.63-4.55(m,2h),4.33(q,j=2.7hz,1h);
13
c nmr(126mhz,cdcl3)δ164.29,163.34(dd,j=251.2,12.0hz),160.25(dd,j=251.1,12.6hz),158.74,138.31,135.27,133.14,132.62(dd,j=9.5,4.7hz),131.01,128.90,122.45,119.29(dd,j=16.5,4.1hz),112.21(dd,j=21.2,3.9hz),104.93(t,j=25.5hz),83.81,58.45;c
18h13
clf2no[m h]
的hrms m/z计算值为332.06482,实测值为332.06337。
[0354][0355]
方案6:n-氨磺酰基丙烯酰胺衍生物jn145的合成。
[0356]
(e)-3-(4-氯苯基)-2-(2,4-二氟苯基)-n-氨磺酰基丙烯酰胺(18,jn145)
[0357]
在0℃下,向搅拌的三乙胺(0.87ml,6.24mmol,3.0当量)和硫酸二酰胺(833.0mg,8.32mmol,4.0当量)于四氢呋喃(8ml)中的溶液中加入酰氯14(4.0ml的0.52m溶液,2.08mmol,1.0当量)。使反应在0℃下进行1h,然后在23℃下进行1h。然后将烧瓶内容物在etoac(5ml)与饱和nh4cl/水(4:1ml)之间进行分配。将有机层分离,并用饱和nahco3(5ml)洗涤,经无水mgso4干燥,过滤并真空浓缩。使用60:40:2etoac/己烷/三乙胺的流动相,通过硅胶制备型tlc纯化粗残余物,得到呈白色固体的n-氨磺酰基丙烯酰胺18(jn145)(213.3mg,0.57mmol,28%)。1h nmr(500mhz,cdcl3)δ7.89(s,1h),7.22(td,j=8.3,6.3hz,1h),7.17(d,j=8.6hz,2h),7.02-6.91(m,4h),5.79(br s,1h),5.43(br s,1h);3c nmr(126mhz,cdcl3)δ167.94,163.61(dd,j=252.8,11.8hz),160.29(dd,j=250.8,11.5hz),139.59,135.38,132.97,132.70(dd,j=9.5,4.3hz),131.10,128.91,127.30,119.54(dd,j=16.9,4.0hz),112.95(dd,j=21.4,3.8hz),105.49(t,j=25.3hz)。
[0358][0359]
方案7:环丙烷甲酰胺衍生物jn147的合成。
[0360]
(e)-n-(3-(4-氯苯基)-2-(2,4-二氟苯基)丙烯酰基)环丙烷甲酰胺(19,jn147)
[0361]
在-78℃下,向环丙烷甲酰胺(25.2mg,0.29mmol,0.90当量)于四氢呋喃(3ml)的溶液中加入n-buli(0.12ml的2.40m己烷溶液,0.29mmol,0.90当量),并将溶液在-78℃下继续再搅拌45min。然后将酰氯14作为四氢呋喃中的溶液(0.62ml 0.52m溶液,0.32mmol,1.0当量)缓慢加入烧瓶中。将所得混合物在-78℃下再搅拌1.5h,并通过加入0.2ml饱和nh4cl溶液终止反应。使反应混合物升温至23℃后,将其在etoac(5ml)与饱和nh4cl/水(4:1ml)之间进行分配。分离有机层,依次用饱和nh4cl/水(4:1ml)和饱和nahco3(5ml)洗涤。然后将其经无水mgso4干燥,过滤,并真空浓缩。使用75:25:2己烷/etoac/三乙胺的流动相,通过硅胶制备型tlc纯化粗残余物,得到呈类白色固体的环丙烷甲酰胺衍生物19(jn147)(17.1mg,47.3μmol,16%)。1h nmr(500mhz,cdcl3)δ7.94(s,1h),7.80(br s,1h),7.23-7.16(m,3h),7.05-6.93(m,4h),3.05(tt,j=7.9,4.6hz,1h),1.15(dt,j=4.8,3.3hz,2h),1.04(dt,j=8.3,3.4hz,2h);
13
c nmr(126mhz,cdcl3)δ176.74,164.80,164.05(dd,j=253.5,11.5hz),160.46(dd,j=251.7,11.9hz),141.86,136.16,132.84(dd,j=9.7,4.0hz),132.47,131.38,129.10,127.69,117.94(dd,j=16.7,3.9hz),113.44(dd,j=21.5,3.7hz),105.88(t,j=25.3hz),14.62,11.39。
[0362][0363]
方案8:甲基丙烯酰胺衍生物jn156的合成。
[0364]
(e)-3-(4-氯苯基)-2-(2,4-二氟苯基)-n-(3-甲基丙烯酰胺基丙基)丙烯酰胺(20,jn156)
[0365]
将三乙胺(0.31ml,1.6mmol,5.0当量)和n-(3-氨基丙基)甲基丙烯酰胺(90.3mg,0.48mmol,1.5当量)于四氢呋喃(3ml)中的溶液在冰水浴中冷却,然后加入酰氯14(0.62ml0.52m溶液,0.32mmol,1.0当量)。使反应在23℃下进行3h,然后将内容物在etoac(5ml)与饱和nh4cl/水(4:1ml)之间进行分配。分离有机层,依次用饱和nh4cl/水(4:1ml)和饱和nahco3(5ml)洗涤。然后将其经无水mgso4干燥,过滤,并真空浓缩。使用70:30:2etoac/己烷/三乙胺的流动相,通过硅胶制备型tlc纯化粗残余物,得到呈类白色固体的甲基丙烯酰胺20(jn156)(59.3mg,0.14mmol,44%)。1h nmr(500mhz,dmso-d6)δ7.90(br t,j=5.8hz,1h),7.75(br t,j=5.9hz,1h),7.57(s,1h),7.35-7.29(m,3h),7.23(td,j=8.5,6.6hz,
1h),7.13(td,j=8.5,2.6hz,1h),7.04(d,j=8.7hz,2h),5.65-5.60(m,1h),5.31(p,j=1.6hz,1h),3.15(q,j=6.8hz,2h),3.10(q,j=6.8hz,2h),1.84(t,j=1.2hz,3h),1.60(p,j=6.9hz,2h);
13
c nmr(dmso-d6)δ167.43,166.07,162.53(dd,j=248.6,13.6hz),159.77(dd,j=247.7,13.0hz),140.00,135.26,133.68,133.24,133.05(dd,j=9.7,4.6hz),130.86,130.30,128.55,119.69(dd,j=16.6,4.0hz),118.85,112.37(dd,j=21.6,3.4hz),104.78(t,j=26.0hz),37.07,36.40,29.21,18.62。
[0366][0367]
方案9:环戊烯酮23的合成。
[0368]
3-(4-氯苯基)-5-亚甲基-2-苯基环戊-2-烯-1-酮(23)
[0369]
将烯酮21(1.0g,3.9mmol,1.0当量)、多聚甲醛(0.72g,23.4mmol,6.0当量)和n-苄基甲基胺盐酸盐(1.36g,8.6mmol,2.2当量)溶解在甲苯(8ml)中,并在回流下加热1h。然后在搅拌下通过添加1ml 10%na2co3(水溶液)来淬灭反应。然后将溶液在et2o(30ml)与10%na2co3(水溶液,30ml)之间进行分配。将层分离,并且将水层用et2o(10ml
×
2)进行进一步萃取。将合并的有机层用盐水(30ml)洗涤,经无水mgso4干燥,过滤并真空浓缩。使用3:100至15:100ml et2o/已烷的流动相梯度,通过用1%于已烷中的三乙胺缓冲的硅胶柱色谱纯化残余物。使用15%etoac/已烷的流动相,通过硅胶制备型tlc进一步纯化含有23的级分,得到呈类白色固体的环戊烯酮23(10.4mg,37.0μmol,0.9%)。r
f 0.16(10%et2o/hexanes).1h nmr(500mhz,cdcl3)δ7.39-7.32(m,3h),7.31-7.22(m,7h),6.31-6.26(m,1h),5.61-5.57(m,1h),3.97-3.21(m,2h);
13
c nmr(126mhz,cdcl3)δ194.08,160.47,141.99,141.33,136.20,133.66,132.27,129.79,129.51,128.96,128.77,128.36,117.62,35.20;c
18h14
clo[m h]
的hrms m/z计算值为281.07277,实测值为281.07161。
[0370][0371]
(z)-3-(4-氯苯基)-2-苯基丙烯腈(z-24)
[0372]
在23℃下,向苄基腈化物(10.0ml,84.9mmol,1.0当量)和4-氯苯甲醛(12.1g,84.9mmol,1.0当量)于无水乙醇中的混合物中加入新鲜制备的乙醇钠的乙醇溶液(100ml的1.27m溶液,127.0mmol,1.5当量)。将所得混合物在回流下加热1.5h,然后逐渐冷却至0℃。将所得沉淀物过滤,用冰冷的无水乙醇洗涤,真空干燥,得到呈白色固体的丙烯腈z-24(11.9g,49.6mmol,58%)。1h nmr(400mhz,cdcl3)δ7.83(d,j=8.5hz,2h),7.70-7.65(m,2h),7.50-7.40(m,6h);
13
c nmr(101mhz,cdcl3)δ140.76,136.57,134.28,132.29,130.60,129.56,129.38,129.26,126.13,117.87,112.43。
[0373][0374]
1-(4-氯苯基)-4-甲基-2-苯基戊-1,4-二烯-3-醇(26;jn034和jn033)
[0375]
向丙烯腈z-24(2.0g,8.3mmol,1.0当量)于甲苯中的冷却(-78℃)溶液中加入1.0m dibal-h(10.0ml,10.0mmol,1.2当量)溶液。将所得悬浮液在-78℃下搅拌1h。使反应升温至0℃并通过在0℃下加入5ml 5%h2so4(水溶液)进行猝灭。向其中再添加5%h2so4(水溶液,45ml)和et2o(50ml),并将混合物在0℃下剧烈搅拌30min。将层分离后,将水层用et2o(50ml
×
2)进行萃取。将合并的有机层用盐水(75ml)洗涤,经无水mgso4干燥,过滤并真空浓缩。将由此获得的粗烯醛25(2:1,e:z)不经进一步纯化而用于下一步骤。
[0376]
将上述粗烯醛25(8.3mmol,1.0当量)于thf(40ml)中的溶液冷却至0℃。向其中加入异丙烯基溴化镁溶液(18.3ml的0.50m thf溶液,9.1mmol,1.1当量),并将反应在0℃下搅拌1h。向该混合物中加入饱和nh4cl(水溶液,5ml),并将反应内容物在饱和nh4cl(水溶液,50ml)、水(50ml)和dcm(100ml)之间进行分配。将水层用dcm(100ml
×
2)进行进一步萃取。将合并的有机层用盐水(150ml)洗涤,经无水mgso4干燥,过滤并真空浓缩。使用0%至10%etoac/已烷梯度,通过硅胶柱色谱纯化粗物质,得到呈淡黄色油状物的醇z-26(485.0mg,1.7mmol,21%)和e-26(364.4mg,1.3mmol,15%)。
[0377]
z-26:1h nmrδ7.57-7.53(m,2h),7.36-7.34(m,4h),7.34-7.29(m,3h),6.87(s,1h),5.26(d,j=5.6hz,1h),5.10(s,1h),4.95(q,j=1.6hz,1h),1.89(d,j=5.6hz,1h),1.63(d,j=1.4hz,3h);
13
c nmrδ145.63,142.52,139.65,135.44,133.31,131.82,130.31,128.74,128.33,128.21,127.77,111.38,73.15,20.10;c
18h16
cl[m-oh]
的hrms m/z计算值为267.09350,实测值为267.09213。
[0378]
e-26:1h nmr(500mhz,cdcl3)δ7.33-7.28(m,3h),7.15-7.11(m,2h),7.06(d,j=8.6hz,2h),6.87(d,j=8.7hz,2h),6.71(s,1h),4.91(s,2h),4.89(d,j=4.8hz,1h),1.86(d,j=4.4hz,1h),1.78(s,3h);
13
c nmr(126mhz,cdcl3)δ144.51,142.89,137.97,135.11,132.63,130.65,129.24,128.77,128.25,127.77,126.62,113.26,80.62,18.43;c
18h16
cl[m-oh]
的hrms m/z计算值为267.09350,实测值为267.09195。
[0379][0380]
(z)-1-(4-氯苯基)-4-甲基-2-苯基戊-1,4-二烯-3-酮(z-27)
[0381]
将醇z-26(450.9mg,1.58mmol,1.0当量)于dcm(10ml)中的溶液在冰水浴中冷却。向其中加入戴斯-马丁高碘剂(738.7mg,1.74mmol,1.1当量)并将反应在0℃下搅拌20min。向该混合物中加入饱和nahco3(水溶液,3ml)并将混合物搅拌5min。然后将内容物在dcm(40ml)与饱和nahco3(水溶液,50ml)之间进行分配,并将层分离。将水层用另外的dcm(20ml
×
2)进行萃取。将合并的有机层经无水mgso4干燥,过滤并真空浓缩。使用0至3%的etoac/已烷流动相梯度,通过硅胶柱色谱纯化粗物质,得到呈淡黄色蜡状物的二烯酮z-27(258.3mg,0.91mmol,58%)。1h nmrδ7.42-7.29(m,5h),7.26(d,j=8.5hz,2h),7.18(d,j=8.5hz,2h),7.02(s,1h),5.99(s,1h),5.81(s,1h),1.94(s,3h);
13
c nmrδ201.53,144.39,141.85,138.18,134.58,133.96,130.30,129.95,128.99,128.86,128.49,128.38,126.31,16.96;c
18h16
clo[m h]
的hrms m/z计算值为283.08842,实测值为283.08642。
[0382][0383]
(e)-1-(4-氯苯基)-4-甲基-2-苯基戊-1,4-二烯-3-酮(e-27)
[0384]
使用与上面针对z-27所概述的相同的程序,得到呈白色固体的异构体e-27(54%)。1h nmrδ7.37-7.31(m,3h),7.21-7.17(m,2h),7.14(d,j=8.6hz,2h),7.11(s,1h),6.99(d,j=8.3hz,2h),5.84(p,j=1.0hz,1h),5.81(p,j=1.5hz,1h),2.00(dd,j=1.5,0.9hz,3h);
13
c nmrδ199.00,144.31,141.27,136.37,136.27,134.63,133.50,131.51,129.40,129.01,128.63,128.20,126.40,18.76;c
18h16
clo[m h]
的hrms m/z计算值为283.08842,实测值为283.08634。
[0385][0386]
(z)-1-(4-氯苯基)-4-(甲基-d)-2-苯基戊-1,4-二烯-3-酮(jn025-d,h:d 0.84:1混合物)
[0387]
将烯酮jn110(77.0mg,0.30mmol,1.0当量)、多聚甲醛(30.3mg,0.98mmol,3.3当量)和n-甲基-1-苯基甲烷-d
2-胺盐酸盐2(100.0mg,0.63mmol,2.1当量)溶解在二甲基甲酰胺(1ml)中并在125℃下加热3h。然后在真空中除去挥发物并且将剩余的内容物在et2o(7ml)与10%na2co3(水溶液,7ml)之间进行分配。将层分离,并且将水层用et2o(5ml
×
2)进行进一步萃取。将合并的有机层用盐水(5ml)洗涤,经无水mgso4干燥,过滤并真空浓缩。使用3:100至15:100ml的et2o/已烷流动相梯度,通过用1%于己烷中的三乙胺缓冲的硅胶柱色谱纯化残余物,得到呈黄色蜡状物的jn025-d(h:d 0.84:1混合物)(28.4mg,0.10mmol,33%)。1h nmr(500mhz,cdcl3)δ7.43-7.31(m,5h),7.26(d,j=8.5hz,2h),7.18(d,j=8.4hz,2h),7.02(s,1h),5.99(t,j=0.9hz,1h),5.83-5.80(m,1h),1.95(s,1.34h,-ch3),1.94-1.92(br m,1.07h,-ch2d);2h nmr(77mhz,cdcl3)δ1.94(t,j=2.2hz);
13
c nmr(126mhz,cdcl3)δ201.53,201.51,144.39,144.36,141.85,138.18,134.58,133.96,130.30,129.95,128.99,128.86,128.49,128.38,126.31,16.97(ch3),16.72(t,j=19.7hz,-ch2d);c
18h15
dclo[m h]
的hrms m/z计算值为284.09470,实测值为284.09347;c18h16
clo[m h]
的hrms m/z计算值为283.08842,实测值为283.08734。
[0388]
(z)-1-(4-氯苯基)-4-((甲基(苯基甲基-d2)氨基)甲基)-2-苯基戊-1,4-二烯-3-酮(jn019-d2)
[0389]
从产生jn025-d的相同反应(上)中,将化合物jn019-d2分离为淡黄色蜡状物(28.8mg,71.3μmol,24%)。1h nmr(400mhz,cdcl3)δ7.43-7.38(m,2h),7.38-7.32(m,3h),7.32-7.27(m,5h),7.20(s,4h),7.02(s,1h),6.19(q,j=1.2hz,1h),6.11(q,j=1.5hz,1h),3.29(s,2h),2.06(s,3h);2h nmr(77mhz,cdcl3)δ3.46(s);
13
c nmr(126mhz,cdcl3)δ200.90,145.47,141.87,138.15,134.46,133.95,130.99,130.10,128.95,128.84,128.82,128.56,128.47,128.35,128.33,127.11,126.41,61.71(弱p,j=19.2hz),56.21,42.24;c
26h23
d2clno[m h]
的hrms m/z计算值为404.17447,实测值为404.17404。
[0390]
表征:
[0391]
[0392]
[0393]
[0394]
[0395]
[0396]
nmr)于氯仿-d中给出。2.除非另有说明,否则式是针对[m h]
的,其中m代表以其电荷中性形式存在的化合物。
[0399]
实施例1:xrpd晶体和表征
[0400]
根据以下一般方法生长所选化合物的x射线质量晶体:将化合物(约2-10mg)置于小瓶中,溶解在最少量(0.25-0.50ml)的二氯甲烷中,然后用己烷(0.50-1.0ml)稀释。通过缓慢蒸发使所得溶液浓缩,从而生成x射线质量晶体,将其留在主要含己烷的母液中,直到进一步分析。
[0401]
化合物jn032、jn110、jn034、jn097、jn117和jn103的xrpd谱分别显示在图4-9中。采集参数描述于附录a中。
[0402]
实施例2:对示例性化合物进行的生物测定
[0403]
在于其它地方描述的生化和细胞生物学测定中合成并测试jn053和jn138-jn156。[1-6]首先在细胞活力测定(mtt测定)中研究jn化合物,以测定jn化合物抑制前列腺癌细胞系的功效和特异性。
[0404]
jn化合物的生长抑制作用通过mtt测定进行评估,所述mtt测定体外评估活细胞的总数。这些实验在表达ar(ar阳性)的前列腺癌细胞系中进行以评估中靶效应,以及在ar无效(ar-null)(ar阴性)前列腺癌细胞系中进行以评估脱靶效应(即特异性)。
[0405]
表达ar的(ar-pos):
[0406]
·
lncap:全长ar
[0407]
·
lncap:全长ar的过表达
[0408]
·
22rv1:全长ar和arv7
[0409]
·
vcap:全长ar和arv7
[0410]
·
cwr22:全长ar和arv7
[0411]
ar阴性:
[0412]
·
pc3
[0413]
·
du145
[0414]
只将对表达ar的(ar阳性)前列腺癌细胞系表现出强抑制作用和对ar无效(ar阴性)前列腺癌细胞系具有最小抑制作用的那些化合物进行生化测定,以评估对ar转录活性的抑制作用。
[0415]
生化测定包括报告基因测定,以确定这些jn化合物抑制雄激素受体(ar)的转录活性的活性和特异性。报告基因测定在内源性或外源性表达全长ar和arv7的各种细胞系中进行,arv7是一种对所有临床可用的ar靶向化合物具有抗性的组成型活性剪接变体。将报告基因测定在多个重复中进行,且浓度范围很广(通常为0-10mm)。
[0416]
使用的报告基因系统包括:
[0417]
·
mmtv-萤光素酶:依赖ar的
[0418]
·
are-萤光素酶:依赖ar的
[0419]
·
gre-荧光素酶:依赖糖皮质激素受体(gr)的
[0420]
·
cre-萤光素酶:依赖creb的
[0421]
·
ap1-萤光素酶:依赖ap1(jun和fos家族)的:
[0422]
·
ar-tad-萤光素酶:依赖于ar反式激活结构域
[0423]
·
creb-tad-萤光素酶:依赖于creb反式激活结构域
[0424]
·
jun-tad-萤光素酶:依赖于c-jun反式激活结构域
[0425]
报告基因测定和mtt测定的数据概括于下表中。(图3a至图3q)。
[0426][0427]
实施例3:jn103抑制ar转录读出
[0428]
对暴露于jn103(10μm)8小时的两种去势抵抗性细胞系(lncap-ar和22rv1)进行了全转录组rna测序。通过基因集组表达分析(图10)确定的实验结果显示ar转录程序的负富集分数(nes)。结果表明ar基因标签(gene signature)的显著减少。
[0429]
实施例4:jn103选择性诱导ar的降解
[0430]
以指定的剂量和时间用jn103和放线菌酮(用于抑制翻译)处理lncap-ar细胞(图11a)。将细胞蛋白质进行针对指定蛋白质的蛋白质印迹。结果示于图11a中。对lncap-95细胞、工程改造为异位表达arδ567的hek-293细胞、pc3细胞和t47d乳腺癌细胞进行了相同的测试。从这些测试中获得的实验结果分别示于图11b至图11e中。
[0431]
测试结果表明jn103强效地诱导以下物质的时间和剂量依赖性降解:1)在lncap-ar细胞中过表达的全长ar(图11a),2)在lncap-95细胞中内源性表达的全长ar和ar-v7(组成型活性ar剪接变体)和3)在hek-293细胞中异位表达的arδ567(也是组成型活性ar变体)。重要的是,jn103不影响其它蛋白质(包括肌动蛋白或gr)的降解(图11(a)至图11(d))。另外,jn103在共表达ar、er和pr的乳腺癌细胞系中诱导ar降解,但不诱导er或pr降解(图11
(e))。
[0432]
实施例5:jn103对表达ar的癌细胞的选择性生长抑制作用。
[0433]
将du145、pc3lncap-ar(全长ar)、22rv1(全长和剪接变体ar)和vcap细胞(400个细胞/6孔板的孔)用指定的0μm(纯dmso)、2μm、4μm、6μm、8μm、10μm的jn103处理两周。用亚甲蓝染色使集落可视化。集落形成测定表明,jn103抑制去势抵抗性ar表达细胞的生长,所述ar包括lncap-ar(全长ar)、22rv1(全长和剪接变体ar)和vcap(全长和剪接变体ar)(图12)。然而,jn103对去势抵抗性、ar无效du145细胞的集落形成的影响有限,并且在高浓度下对pc3细胞集落形成只有轻微影响(图12)。
[0434]
jn103的生长抑制作用也在对20个非前列腺癌细胞系的mtt试验中进行了评估。(图13)。将细胞暴露于0μm(纯dmso)、2μm、4μm、6μm和8μm的jn103中5天,然后进行mtt测定以测定细胞活力。结果是一式四份的平均值。根据图13,jn103对表达全长ar并且生长依赖于ar表达的乳腺癌细胞系(t47d)表现出显著的生长抑制作用。
[0435]
参考文献
[0436]
1.an j,fisher m,rettig mb.vhl expression in renal cell carcinoma markedly sensitizes to bortezomib(ps-341)through a nf-κb-dependent mechanism.oncogene.24:1563-70,2005.
[0437]
2.an j and rettig mb.mechanism of von hippel-lindau protein-mediated suppression of nuclear factor kappa b(nf-κb)activity.mol cell biol.25:7546-56,2005.
[0438]
3.an j,mo n,rettig mb.egfr inhibition sensitizes renal cell carcinoma cells to bortezomib.mol can ther.6:61-9,2007.
[0439]
4.rettig,mb,heber d,an j,seeram np,rao jy,liu h,klatte t,belldegrun a,moro a,henning sm,mo d,aronson,wj,pantuck,a.pomegranate extract inhibits nf-κb activation and delays the emergence of androgen-independence in the lapc4 prostate cancer xenograft model.molecular cancer therapeutics.7:2662,2008.
[0440]
5.pantuck aj,an j,liu h,rettig mb.nf-kappa b dependent plasticity of the epithelial to mesenchymal transition induced by vhl inactivation in renal cell carcinomas.cancer research.70:752,2010.
[0441]
6.an j,liu h,magyar ce,guo y,veena ms,srivatsan es,huang j,rettig mb.hyperactivated c-jun n-terminal kinase as a therapeutic target in pvhl-deficient renal cell carcinomas.cancer research 2013 feb 15;73(4):1374-85.
[0442]
通过引用并入
[0443]
本文提到的所有公布和专利都特此通过引用整体并入,就如同每个单独的公布和专利被具体且单独地指出通过引用并入一样。在发生冲突的情况下,将以本技术(包括本文的任何定义)为准。
[0444]
等效物
[0445]
本领域技术人员将会认识到或将能够确定仅使用至多常规实验,就能实现本文所描述的化合物及其使用方法的许多等效物。此类等效物被认为在本发明的范围内并且被以
下权利要求覆盖。本领域技术人员还将认识到,本文描述的实施方案的所有组合都在本发明的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。