1.本发明涉及有机合成技术领域,具体涉及一种倍半萜类化合物的制备方法。
背景技术:
2.倍半萜类化合物含有15个碳原子、由3个异戊二烯单位构成,是自然界中数量庞大且重要的一类在天然产物中。其中,含有补身烷(drimane)骨架的萜类和混源萜类天然产物广泛存在于植物、微生物、海洋生物以及某些昆虫之中,并表现出多种多样的生物活性。例如,从坎洛树中分离出来的drimane类倍半萜成分,发现其对α4β2烟碱乙酰胆碱受体的非竞争性抑制作用高于人α3β4和α7亚型,可用于开发新的抗成瘾抗抑郁配体;从白桂皮(canella winterana)的叶子中提取的14种rimane类倍半萜,其中的8种在浮萍试验检测中具有植物毒性;含松橄榄酸浓缩到200ppm时对第二胚芽鞘和稻子皮内萌芽有很好的抑制作用。近年来,越来越多的含drimane骨架的倍半萜及复杂天然产物被发现,并表现出结构和生物活性的多样性。4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮是从烟草(nicotiana tabacum l.)中分离出来的,可能对香气有贡献。1983年,( )-drim-9(11)-en-8α-ol被shingo marumo等人从曲霉菌(aspergillus oryzae)中分离出来,它被用于烘焙和制造一些日本的饮料,如清酒、芝麻酱和烧酒。更为重要的是,这两种drimane倍半萜可以作为关键中间体,用于其他复杂萜类分子和混源萜类分子的合成,如( )-drim-9(11)-en-8α-ol被当作合成austrodoral,austrodoric acid,siphonodictyal b和liphagal等混源萜类天然产物的关键合成子。
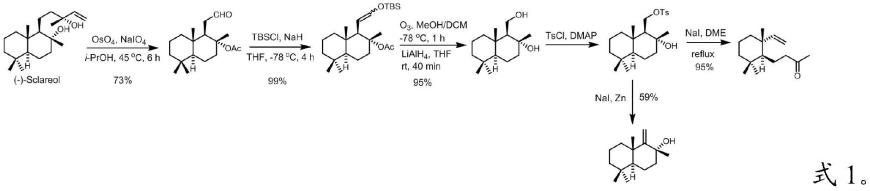
3.但目前报道的从植物中提取制备drimane倍半萜的方法普遍存在反应步骤多、反应效率低、产物异构化、贵金属使用和应用范围不广泛等缺点,大大限制了其生物活性研究,想要大量获取此类化合物需通过化学合成方法。例如,1974年,enzell,c.r等以倍半萜drimenol为初始原料经6步完成了4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮的合成(hlubucek,j.r.;aasen,a.j.;almqvist,s.o.,et al.acta chem.scand.,ser.b 1974,28(1),18)。1990年,kametani,t等以( )-wieland-miescher酮为初始原料经9步反应合成了4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮(k.shishido,y.tokunaga,n.omachi,k.hiroya,et al.chem.soc.,perkin trans.1,1990,2481-2486.),前两种方法不仅步骤长而且所使用的原料需制备得到。随后,barrero,a.f.等以香紫苏醇为初始原料经6步反应合成了4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮(barrero,a.f.;alvarez-manzaneda,e.j.;chahboun,r.,tetrahedron lett.1998,39(51),9543.( )-drim-9(11)-en-8α-ol[barrero,a.f.;manzaneda,e.a.;altarejos,j.,et al.tetrahedron 1995,51(27),7435.),反应路线如式1所示。然而,上述化学合成方法制备的drimane倍半萜的4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮的总收率为65%,( )-drim-9(11)-en-8α-ol的总收率为40%,4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮和( )-drim-9(11)-en-8α-ol的总收率均很低。
[0004]
技术实现要素:
[0005]
有鉴于此,本发明的目的在于提供一种倍半萜类化合物的制备方法,本发明提供的制备方法产物的总收率高。
[0006]
为了实现上述发明目的,本发明提供以下技术方案:
[0007]
本发明提供了一种倍半萜类化合物的制备方法,其特征在于,包括以下步骤:
[0008]
利用还原剂对香紫苏内酯进行还原反应,得到化合物1;
[0009]
将所述化合物1、碘苯二乙酸和碘混合进行碘化反应,得到化合物2;
[0010]
将所述化合物2在第一碱性试剂存在条件下进行水解反应,得到化合物3;
[0011]
将所述化合物3在第二碱性试剂存在条件下进行消除碘化氢反应,得到4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮和/或( )-drim-9(11)-en-8α-ol;
[0012][0013]
优选的,所述还原剂包括二异丙基胺基锂和/或二异丁基氢化铝;
[0014]
所述香紫苏内酯与还原剂的摩尔比为1:1~3。
[0015]
优选的,所述还原反应的温度为-78~0℃,时间为1~3h。
[0016]
优选的,所述化合物1、碘苯二乙酸和碘的摩尔比为1:1~2:1~2。
[0017]
优选的,所述碘化反应的温度为70~90℃,时间为1~3h。
[0018]
优选的,所述第一碱性试剂和第二碱性试剂独立地包括氢氧化物、碳酸盐、碱金属醇化物和含氮有机化合物;
[0019]
所述化合物2与第一碱性试剂的摩尔比为1:1~10。
[0020]
优选的,所述水解反应的时间为2~5h。
[0021]
优选的,所述化合物3与第二碱性试剂的摩尔比为1:1~10,所述消除碘化氢反应的温度为60℃,时间为6~10h,所述消除碘化氢反应得到4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮和/或( )-drim-9(11)-en-8α-ol。
[0022]
优选的,所述化合物3与第二碱性试剂的摩尔比为1:1~50,所述消除碘化氢反应的温度为25~60℃,时间为0.1~3h,所述消除碘化氢反应得到4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮。
[0023]
优选的,所述化合物3与第二碱性试剂的摩尔比为1:1~50,所述消除碘化氢反应
的温度为25~80℃,时间为3~6h,所述消除碘化氢反应得到( )-drim-9(11)-en-8α-ol。
[0024]
本发明提供了一种倍半萜类化合物的制备方法,包括以下步骤:利用还原剂对香紫苏内酯进行还原反应,得到化合物1;将所述化合物1、碘苯二乙酸和碘混合进行碘化反应,得到化合物2;将所述化合物2在第一碱性试剂存在条件下进行水解反应,得到化合物3;将所述化合物3在第二碱性试剂存在条件下进行消除碘化氢反应,得到4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮和/或( )-drim-9(11)-en-8α-ol。本发明以香紫苏内酯为初始原料,经还原、碘化、水解和消除四步反应制备得到4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮和/或( )-drim-9(11)-en-8α-ol,产物的总收率高;本发明采用的原料廉价易得,生产成本低,适宜工业化生产。而且,本发明避免使用昂贵的贵金属,进一步降低了生产成本。如实施例测试结果所示,本发明提供的制备方法,当产物为4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮和/或( )-drim-9(11)-en-8α-ol时,产物总收率分别为36.5%和27.6%;当产物为4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮时,产物总收率为62.5%;当产物为( )-drim-9(11)-en-8α-ol时,产物总收率为74.6%。
具体实施方式
[0025]
本发明提供了一种倍半萜类化合物的制备方法,包括以下步骤:
[0026]
利用还原剂对香紫苏内酯进行还原反应,得到化合物1;
[0027]
将所述化合物1、碘苯二乙酸和碘混合进行碘化反应,得到化合物2;
[0028]
将所述化合物2在第一碱性试剂存在条件下进行水解反应,得到化合物3;
[0029]
将所述化合物3在第二碱性试剂存在条件下进行消除碘化氢反应,得到4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮和/或( )-drim-9(11)-en-8α-ol;
[0030][0031]
在本发明中,若无特殊说明,所有的原料组分均为本领域技术人员熟知的市售商品。
[0032]
在本发明中,所述4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮和/或( )-drim-9(11)-en-8α-ol的反应路线如下:
[0033][0034]
本发明利用还原剂对香紫苏内酯进行还原反应,得到化合物1。
[0035]
在本发明的具体实施例中,优选将香紫苏内酯、还原剂和有机溶剂混合进行还原反应,得到化合物1。在本发明中,所述还原剂优选包括二异丙基胺基锂和/或二异丁基氢化铝。在本发明中,所述香紫苏内酯与还原剂的摩尔比为优选为1:1~3,更优选为1:1.5~2.5,进一步优选为1:2。在本发明中,所述有机溶剂优选包括二氯甲烷、四氢呋喃和甲醇中的一种或几种;本发明对于所述有机溶剂的用量没有特殊限定,能够保证所述还原反应顺序进行即可。本发明对于所述混合的方式没有特殊限定,能够将原料混合均匀即可,具体如搅拌混合。在本发明中,所述还原反应的温度优选为-78~0℃,更优选为-78~-20℃,进一步优选为-78~-50℃;所述还原反应的时间优选为1~3h更优选为1.5~2.5h,进一步优选为2h。
[0036]
完成所述还原反应后,本发明优选还包括后处理,所述后处理优选包括:在所得还原反应液中加入水终止反应,有机溶剂萃取,将所得有机相依次进行饱和氯化钠水溶液洗涤、无水硫酸钠干燥和浓缩,得到化合物1。在本发明中,所述终止反应的温度优选为室温,所述终止反应优选在搅拌条件下进行,本发明对于所述搅拌的速度没有特殊限定,能够终止反应即可;所述搅拌的时间优选为30~60min,更优选为30~50min。在本发明中,所述萃取用有机溶剂优选包括二氯甲烷或乙酸乙酯;所述有机溶剂萃取的次数优选为2~4次,更优选为3次。本发明对于所述浓缩没有特殊限定,采用本领域技术人员熟知的浓缩方式浓缩至恒重即可,具体如减压浓缩。
[0037]
得到化合物1后,本发明将所述化合物1、碘苯二乙酸和碘混合进行碘化反应,得到化合物2。
[0038]
在本发明的具体实施例中,优选将所述化合物1、碘苯二乙酸、碘和有机溶剂混合进行碘化反应,得到化合物2。在本发明中,所述化合物1、碘苯二乙酸与碘的摩尔比优选为1:1~2:1~2,更优选为1:1.2~1.8~:1.2~1.8,进一步优选为1:1.4~1.6:1.2~1.5,最优选为1:1.4:1.2。在本发明中,所述碘化反应优选在有机溶剂存在条件下进行,所述有机溶剂优选包括苯、甲苯和二甲苯中的一种或几种;本发明对于所述有机溶剂的用量没有特殊限定,能够保证所述碘化反应顺序进行即可。本发明对于所述混合的方式没有特殊限定,能够将原料混合均匀即可,具体如搅拌混合。在本发明中,所述碘化反应的温度优选为70~90℃,更优选为75~85℃,进一步优选为80℃;所述碘化反应的时间优选为1~3h,更优选为1.5~2.5h,进一步优选为2h;在本发明的具体实施例中,所述碘化反应的温度优选由金属浴提供,所述金属浴优选包括铝沙浴和/或铝丝浴。在本发明中,所述碘化反应优选在光照条件下进行,所述光照优选为金卤灯照射,所述金卤灯照射的功率优选为100~250w,更优选为150~200w。
[0039]
完成所述碘化反应后,本发明优选还包括后处理,所述后处理优选包括:将所得碘化反应液依次进行冷却至室温、预浓缩、饱和氯化钠水溶液稀释和有机溶剂萃取,将所得有机相进行饱和硫代硫酸钠水溶液洗、无水硫酸钠干燥和浓缩,得到化合物2。本发明对于所述冷却没有特殊限定,采用本领域技术人员熟知的冷却方式即可,具体如自然冷却。本发明对于预浓缩的方式没有特殊限定,采用本领域技术人员熟知的浓缩方式浓缩至恒重即可,具体如减压浓缩。在本发明中,所述饱和氯化钠稀释的倍数优选为1~5倍,更优选为2~3倍。在本发明中,所述萃取用有机溶剂优选包括乙酸乙酯或二氯甲烷;所述有机溶剂萃取的次数优选为2~4次,更优选为3次。本发明对于所述浓缩的方式没有特殊限定,采用本领域
drim-9(11)-en-8α-ol。
[0046]
在本发明中,所述化合物3与第二碱性试剂的摩尔比优选为1:1~50,更优选为1:1.5~10,进一步优选为1:2;所述消除碘化氢反应的温度优选为25~60℃,更优选为40~55℃,进一步优选为50℃,所述消除碘化氢反应的时间优选为0.1~3h,更优选为0.1~2h,进一步优选为10min,所述消除碘化氢反应得到4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮。
[0047]
在本发明中,所述化合物3与第二碱性试剂的摩尔比优选为1:1~50,更优选为1:20~50,进一步优选为1:50;所述消除碘化氢反应的温度优选为25~80℃,更优选为50~70℃,进一步优选为60℃,所述消除碘化氢反应的时间优选为3~6h,更优选为3.5~5h,进一步优选为4h,所述消除碘化氢反应得到( )-drim-9(11)-en-8α-ol。
[0048]
完成所述消除碘化氢反应后,本发明优选还包括后处理,所述后处理优选包括:将所得消除碘化氢反应液依次进行冷却至室温、预浓缩、饱和氯化铵水溶液稀释和有机溶剂萃取,将所得有机相依次进行饱和氯化钠水溶液洗涤,无水硫酸钠干燥,浓缩和柱层析分离,得到4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮和/或( )-drim-9(11)-en-8α-ol。本发明对于所述冷却没有特殊限定,采用本领域技术人员熟知的冷却方式即可,具体如自然冷却。本发明对于预浓缩的方式没有特殊限定,采用本领域技术人员熟知的浓缩方式浓缩至恒重即可,具体如真空浓缩。在本发明中,所述稀释的倍数优选为1~5倍,更优选为2~3倍。在本发明中,所述萃取用有机溶剂优选包括乙酸乙酯或二氯甲烷;所述有机溶剂萃取的次数优选为2~4次,更优选为3次。在本发明中,本发明对于浓缩的方式没有特殊限定,采用本领域技术人员熟知的浓缩方式浓缩至恒重即可,具体如真空浓缩。在本发明中,所述柱层析分离用填料优选为硅胶,所述柱层析分离用洗脱剂优选为乙酸乙酯-石油醚混合溶剂,所述洗脱剂中乙酸乙酯和石油醚的体积比优选为1:1~50,更优选为1:10~40,进一步和优选为1:20~30。
[0049]
本发明提供的制备方法,制备原料简便易得,制备条件温和,对设备的要求较低;所得产物在天然产物合成领域有广泛的应用,工艺流程附加值高,适用于大规模工业化生产。而且,本发明提供的制备方法采用价格便宜的碱性试剂作为催化剂,实现目标化合物的高效的制备;具有转化效率高、底物成本低廉、操作简单、对设备要求低、产率优良等优点。
[0050]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0051]
实施例1
[0052]
(1)在-78℃、搅拌条件下,向250ml二氯甲烷的香紫苏内酯(12.0g,50.0mmol,1.0当量)溶液中滴加二异丁基氢化铝(60.0mmol)混合均匀,保温还原反应1h,向所得还原反应液中加入水,加热到室温,搅拌30min,分别用100ml二氯甲烷萃取3次,合并有机相后用150ml饱和氯化钠水溶液洗涤,无水硫酸钠干燥,减压浓缩至恒重,得到化合物1(白色固体,12.1g,收率为96%)。
[0053]
(2)将化合物1(12.1g,48.0mmol,1.0当量)溶解于200ml苯中,加入碘苯二乙酸(21.6g,67.2mmol)和碘(14.6g,57.6mmol,1.2当量)混合均匀,在70℃(铝沙浴)中,在800r/
min、150w的金卤灯照射条件下碘代反应1h,将所得碘代反应液冷却至室温,真空浓缩,饱和氯化钠水溶液稀释2倍,分别用100ml乙酸乙酯萃取3次,合并有机层后饱和的硫代硫酸钠水溶液洗涤,无水硫酸钠干燥,减压浓缩,得到化合物2(白色固体,16.0g,收率为88%)。
[0054]
(3)在冰浴条件下,将化合物2(7.56g,20mmol,1.0当量)溶解于100ml甲醇中,加入碳酸钾(3.3g,24mmol,1.2当量)混合均匀,移除冰浴升至室温,在500r/min条件下水解反应2h,将所得水解反应液浓缩至恒重,加100ml水和100ml乙酸乙酯进行稀释,分别用100ml乙酸乙酯萃取3次,合并有机层后用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,减压浓缩,得到化合物3(白色固体,6.7g,收率为96%)。
[0055]
(4)将化合物3(70mg,0.2mmol,1.0当量)溶解于2ml甲醇中,加入碳酸钾(83mg,0.6mmol,3当量)混合均匀,然后将反应混合物在60℃(铝沙浴)、800r/min条件下消除碘化氢反应6h,将所得消除碘化氢反应液冷却至室温,真空浓缩,加入饱和氯化铵稀释5倍,分别用20ml乙酸乙酯萃取3次,合并有机层后用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,减压浓缩后柱层析分离,得到4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮(淡黄色油状,20mg,收率为45%,纯度》95%)和( )-drim-9(11)-en-8α-ol(白色固体,15mg,收率为34%,纯度为》95%);其中,柱层析分离用填料为硅胶,洗脱剂为体积比为1:10的乙酸乙酯-石油醚混合溶剂。
[0056]
实施例2
[0057]
将实施例1制备的化合物3(70mg,0.2mmol,1.0当量)溶解于10ml四氢呋喃中,加入叔丁醇钾(43.5mg,0.4mmol,2当量)混合均匀,然后将反应混合物在50℃(铝沙浴)、800r/min条件下消除碘化氢反应10min,将所得消除碘化氢反应液冷却至室温,真空浓缩,加入饱和氯化铵稀释3倍,分别用10ml乙酸乙酯萃取3次,合并有机层后用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,减压浓缩后柱层析分离,得到4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮(淡黄色油状,34mg,收率为77%,纯度》95%);其中,柱层析分离用填料为硅胶,洗脱剂为体积比为1:10的乙酸乙酯-石油醚混合溶剂。
[0058]
实施例1和实施例2制备的4-((1s,6r)-2,2,6-三甲基-6-乙烯基环己基)丁烷-2-酮的结构表征数据:
[0059]1h nmr(500mhz,cdcl3)δ5.61(dd,j=17.8,10.4hz,1h),4.90(d,j=1.1hz,1h),4.87(dd,j=5.0,1.2hz,1h),2.45(ddd,j=16.9,11.4,5.5hz,1h),2.37(ddd,j=16.8,11.4,5.4hz,1h),2.07(s,3h),1.63-1.46(m,2h),1.44-1.36(m,3h),1.34-1.09(m,4h),1.00(s,3h),0.89(s,3h),0.86(s,3h).
[0060]
13
c nmr(126mhz,cdcl3)δ209.61,151.50,110.66,52.91,47.34,42.18,41.56,40.32,34.64,33.71,29.90,21.99,21.07,18.76,17.21.
[0061]
hrms(m/z)calcd for c
15h26
o[m h]
222.1984,found 222.1975.
[0062]
实施例3
[0063]
将实施例1制备的化合物3(70mg,0.2mmol,1.0当量)溶解于10ml四氢呋喃中,加入二乙烯三胺(1ml,10mmol,50当量)混合均匀,然后将反应混合物在60℃(铝沙浴)、800r/min条件下消除碘化氢反应4h,将所得消除碘化氢反应液冷却至室温,真空浓缩,加入饱和氯化铵稀释3倍,分别用10ml乙酸乙酯萃取3次,合并有机层后用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,减压浓缩后柱层析分离,得到( )-drim-9(11)-en-8α-ol(白色固体,41mg,收率
为92%,纯度》95%);其中,柱层析分离用填料为硅胶,洗脱剂为体积比为1:10的乙酸乙酯-石油醚混合溶剂。
[0064]
实施例1和实施例3制备的( )-drim-9(11)-en-8α-ol的结构表征数据:
[0065]1hnmr(500mhz,cdcl3)δ5.22(s,1h),4.84(s,1h),2.04-1.98(m,1h),1.83-1.76(m,1h),1.73-1.62(m,2h),1.58-1.50(m,1h),1.48-1.42(m,2h),1.41(s,3h),1.40-1.31(m,3h),1.17-1.10(m,1h),1.08(s,3h),0.97(dd,j=11.5,2.5hz,1h),0.87(s,3h),0.84(s,3h).
[0066]
13
c nmr(126mhz,cdcl3)δ166.77,103.78,73.52,53.63,44.35,41.90,40.16,39.15,33.94,33.40,30.65,22.53,21.77,20.34,19.20.
[0067]
hrms(m/z)calcd for c
15h26
o[m h]
222.1984,found 222.1976.
[0068]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。