1.本发明属于新能源材料领域,具体涉及一种微米铜粉的制备方法及其应用。
背景技术:
2.hjt(异质结太阳能电池)的实验室效率可达到26%以上,厂商规模化生产的平均量产效率可达到24%,高于上一代perc(发射极和背面钝化电池),因而成为下一代太阳能电池技术。
3.然而,hjt面临的问题之一是制造成本高(0.93~0.98元/瓦),除人力成本外,其成本支出均高于perc。在成本占比中,除去硅片的成本,其中占比最终的就是低温银浆。hjt工艺因受到温度影响只能使用低温银浆,且低温银浆产品主要源于进口,价格较常规银浆要高10-20%,因此降低hjt成本一大途径就是实现低温银浆的国产化以及采用廉价金属替代部分贵重金属银,以真正降低低温银浆的成本。银包铜是在铜粉表面包覆一层银壳,可以在保证浆料电导率的同时,降低银用量,成为降低hjt的成本的有效策略。
4.银包铜的制备可分为两步:第一步是制备铜粉;第二步是在铜表面包覆银层。显然地,铜粉的形貌和结构将决定银包铜的形貌和性能,因而铜粉的制备在银包铜浆料的研发中尤为关键。
5.现阶段,在液相还原制备铜粉的过程中面临着体系浓度低、铜粉电阻率高的问题。因此,在液相还原体系中研发制备高浓度、低电阻铜粉意义重大。
技术实现要素:
6.针对现有技术的不足,本发明的目的在于提供一种微米铜粉的制备方法及其应用。不同于传统单一溶剂体系,为提高反应体系的浓度,本发明采用水和乙二醇作为混合溶剂,体系的cu
2
浓度可从0.4mol/l提高至2.0mol/l,且铜粉产率均在90%以上,铜粉粒径均在微米级别(1-10μm),适合推广应用。
7.为达到此发明目的,本发明采用以下技术方案:
8.第一方面,本发明提供一种微米铜粉的制备方法,所述制备方法包括以下步骤:
9.(1)将铜源、分散剂和溶剂混合,搅拌,得到溶液i;
10.(2)将还原剂和溶剂混合,搅拌,得到溶液ⅱ;
11.(3)将溶液i与溶液ⅱ混合,反应,得到溶液ⅲ;
12.(4)将溶液ⅲ自然沉降,烘干,研磨,得到所述微米铜粉;
13.其中,步骤(1)和步骤(2)中,所述溶剂各自独立地包括乙二醇和去离子水的组合。
14.本发明遴选出乙二醇和水混合溶剂体系,一方面可以增加反应物的溶解量,从而极大地提高体系反应浓度;另一方面,通过对乙二醇和水的比例以及体系浓度的改变,达到对铜粉形貌、粒径和电阻率的调控。尤其地,本发明所采取的制备方案所得铜粉的产率均在90%以上。
15.在本发明中,步骤(1)和步骤(2)所述溶剂中,所述乙二醇和去离子水的质量比各
自独立地为1:(0.5-2);
16.其中,“0.5-2”可以是0.5、0.7、0.9、1.1、1.3、1.5、1.7、1.9、2等。
17.优选地,步骤(1)和步骤(2)中,所述搅拌的温度各自独立地为70-90℃ (例如可以是70℃、72℃、74℃、76℃、78℃、80℃、82℃、84℃、86℃、88℃、 90℃等),时间为0.5-1h(例如可以是0.5h、0.6h、0.7h、0.8h、0.9h、1h等)。
18.在本发明中,步骤(1)中,所述铜源、分散剂和溶剂的质量比为 1:(0.8-1.5):(1.5-7.3);
19.其中,“0.8-1.5”可以是0.8、0.9、1、1.1、1.2、1.3、1.4、1.5等;
[0020]“1.5-7.3”可以是1.5、2、2.5、3、3.5、4、4.5、5、5.5、6、6.5、7、7.3 等。
[0021]
优选地,所述铜源包括五水硫酸铜、无水硫酸铜、六水硝酸铜或硝酸铜中的任意一种或至少两种的组合,优选为五水硫酸铜。
[0022]
在本发明中,步骤(1)中,所述分散剂包括聚乙烯吡咯烷酮、聚乙二醇、 d-核糖或β-环糊精中的任意一种或至少两种的组合,优选为聚乙烯吡咯烷酮和β-环糊精的组合。
[0023]
本技术采用聚乙烯吡咯烷酮和β-环糊精的组合,一方面可以增加反应物的溶解量,从而极大地提高体系反应浓度;另一方面可以降低铜粉的电阻率。
[0024]
优选地,所述聚乙烯吡咯烷酮和β-环糊精的质量比为(10-1):1;
[0025]
其中,“10-1”可以是10、9、8、7、6、5、4、3、2、1等。
[0026]
优选地,所述聚乙烯吡咯烷酮包括聚乙烯吡咯烷酮k15、聚乙烯吡咯烷酮 k30或聚乙烯吡咯烷酮k60中的任意一种或至少两种的组合,优选为聚乙烯吡咯烷酮k30。
[0027]
优选地,所述聚乙二醇包括peg400、peg1000、peg2000、peg4000或 peg10000中的任意一种或至少两种的组合。
[0028]
在本发明中,步骤(2)中,所述还原剂和溶剂的质量比为1:(2.1-10.4);
[0029]
其中,“2.1-10.4”可以是2.1、3、5、6、7、8、9、10、10.4等。
[0030]
优选地,步骤(2)中,所述还原剂包括次磷酸钠、抗坏血酸、水合肼或甲醛中的任意一种或至少两种的组合,优选为次磷酸钠和抗坏血酸的组合。
[0031]
在本发明中,还原剂为次磷酸钠和抗坏血酸的组合可以改善铜粉的电阻率。
[0032]
优选地,所述次磷酸钠和抗坏血酸的质量比为(10-1):1;
[0033]
其中,“10-1”可以是10、9、8、7、6、5、4、3、2、1等。
[0034]
在本发明中,步骤(3)中,所述溶液iii中铜源、分散剂和还原剂的质量比为(2.5-3.0):(3.0-3.5):1;
[0035]
其中,“2.5-3.0”可以是2.5、2.6、2.7、2.8、2.9、3等;
[0036]“3.0-3.5”可以是3.0、3.1、3.2、3.3、3.4、3.5等。
[0037]
优选地,步骤(3)中,所述溶液ⅲ中铜源的摩尔浓度为0.4-2mol/l(例如可以是0.4mol/l、0.6mol/l、0.8mol/l、1mol/l、1.2mol/l、1.4mol/l、1.6mol/l、 1.8mol/l、2mol/l等)。
[0038]
优选地,步骤(3)中,所述溶液ⅲ中分散剂的质量浓度为132-660g/l(例如可以是132g/l、198g/l、264g/l、330g/l、396g/l、462g/l、528g/l、600 g/l等)。
[0039]
优选地,步骤(3)中,所述溶液ⅲ中还原剂的质量浓度为38-194g/l(例如可以是38g/l、58g/l、77g/l、97g/l、116g/l、135g/l、155g/l、174g/l、 176g/l、194g/l等)。
[0040]
在本发明中,步骤(3)中,所述反应的温度为90-110℃(例如可以是90℃、 92℃、94℃、96℃、98℃、100℃、102℃、104℃、106℃、108℃、110℃等),时间为1.5-2.5h(例如可以是1.5h、1.7h、1.9h、2.1h、2.3h、2.5h等)。
[0041]
在本发明中,步骤(4)中,所述烘干的温度为45-60℃(例如可以是45℃、 47℃、49℃、51℃、53℃、55℃、57℃、59℃、60℃等),时间为5-6h(例如可以是5h、5.2h、5.4h、5.6h、5.8h、6h等)。
[0042]
优选地,步骤(4)中,所述烘干之前,还需要将溶液ⅲ自然沉降得到的沉淀依次用去离子水和无水乙醇进行清洗。
[0043]
优选地,步骤(4)中,所述微米铜粉的平均粒径为1-10μm(例如可以是1 μm、2μm、3μm、4μm、5μm、6μm、7μm、8μm、9μm、10μm等)。
[0044]
作为本发明优选地技术方案,所述的微米铜粉的制备方法包括以下步骤:
[0045]
(1)将铜源、分散剂和溶剂混合,搅拌,得到溶液i;
[0046]
其中,所述铜源包括五水硫酸铜,所述分散剂包括质量比为(10-1):1的聚乙烯吡咯烷酮k30和β-环糊精的组合,所述溶剂包括质量比为1:(0.5-2)的乙二醇和去离子水的组合;
[0047]
(2)将还原剂和溶剂混合,搅拌,得到溶液ⅱ;
[0048]
其中,所述还原剂包括质量比为(10-1):1的次磷酸钠和抗坏血酸的组合,所述溶剂包括质量比为1:(0.5-2)的乙二醇和去离子水的组合;
[0049]
(3)将溶液i与溶液ⅱ混合,反应,得到溶液ⅲ;
[0050]
其中,所述溶液ⅲ中铜源的摩尔浓度为0.4-2mol/l,所述反应的温度为 90-110℃,时间为1.5-2.5h;
[0051]
(4)将溶液ⅲ自然沉降,去除上清液,依次用去离子水和无水乙醇进行清洗沉淀,45-60℃烘干5-6h,研磨,得到所述微米铜粉;
[0052]
其中,所述微米铜粉的平均粒径为1-10μm。
[0053]
第二方面,本发明提供一种根据第一方面所述的微米铜粉的制备方法在制备银包铜浆料中的应用。
[0054]
相对于现有技术,本发明具有以下有益效果:
[0055]
(1)本发明采用乙二醇和水作为混合溶剂,不仅可以大幅度提高体系的反应浓度,增加规模化生产过程产量,还能够减少乙二醇用量,降低成产成本;
[0056]
(2)本发明提供的制备路线简单,可操作性强,所制得铜粉电阻率低,且粒径可调,为液相体系中高浓度、低电导率微米铜粉提供了普适的制备策略,适合推广应用。
附图说明
[0057]
图1为本发明在0.4-2.0m不同反应体系浓度所制得微米铜粉的sem图。
[0058]
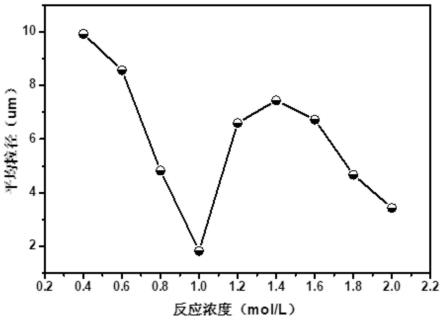
图2为铜粉粒径随反应体系浓度变化曲线图。
具体实施方式
[0059]
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0060]
实施例1
[0061]
本实施例提供一种0.4m反应浓度微米铜粉的制备方法,所述微米铜粉的制备方法包括以下步骤:
[0062]
(1)将120g(0.48mol)五水硫酸铜、144g聚乙烯吡咯烷酮k30、14.4g β-环糊精和800ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合) 混合,90℃搅拌0.8h,形成溶液i;
[0063]
(2)将42.2g次磷酸钠、4.22g抗坏血酸和400ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合)混合,90℃搅拌0.8h,形成溶液ⅱ;
[0064]
(3)将溶液i与溶液ⅱ混合均匀,100℃反应2h,得到溶液ⅲ;
[0065]
(4)将溶液ⅲ转移至烧杯,冷却至室温后,自然沉降,去除上清液,然后用去离子水清洗底部沉淀6次,无水乙醇清洗3次,放在50℃真空干燥箱中烘干5h,取出后研磨即可得到微米铜粉。
[0066]
本实施例制备的0.4m反应浓度微米铜粉的平均粒径为3.93μm。
[0067]
实施例2
[0068]
本实施例提供一种0.6m反应浓度微米铜粉的制备方法,所述微米铜粉的制备方法包括以下步骤:
[0069]
(1)将180g(0.72mol)五水硫酸铜、216g聚乙烯吡咯烷酮k30、21.6g β-环糊精和800ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合) 混合,80℃搅拌0.9h,形成溶液i;
[0070]
(2)将63.3g次磷酸钠、6.33g抗坏血酸和400ml溶剂(所述溶剂为质量比为1:1.8的乙二醇和去离子水的组合)混合,80℃搅拌0.9h,形成溶液ⅱ;
[0071]
(3)将溶液i与溶液ⅱ混合均匀,105℃反应1.9h,得到溶液ⅲ;
[0072]
(4)将溶液ⅲ转移至烧杯,冷却至室温后,自然沉降,去除上清液,然后用去离子水清洗底部沉淀5次,无水乙醇清洗3次,放在48℃真空干燥箱中烘干5.1h,取出后研磨即可得到微米铜粉。
[0073]
本实施例制备的0.6m反应浓度微米铜粉的平均粒径为8.57μm。
[0074]
实施例3
[0075]
本实施例提供一种0.8m反应浓度微米铜粉的制备方法,所述微米铜粉的制备方法包括以下步骤:
[0076]
(1)将240g(0.96mol)五水硫酸铜、288g聚乙烯吡咯烷酮k30、28.8g β-环糊精和800ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合) 混合,70℃搅拌1h,形成溶液i;
[0077]
(2)将84.4g次磷酸钠、8.44g抗坏血酸和400ml溶剂(所述溶剂为质量比为1:1.9的乙二醇和去离子水的组合)混合,70℃搅拌1h,形成溶液ⅱ;
[0078]
(3)将溶液i与溶液ⅱ混合均匀,95℃反应2.3h,得到溶液ⅲ;
[0079]
(4)将溶液ⅲ转移至烧杯,冷却至室温后,自然沉降,去除上清液,然后用去离子水清洗底部沉淀6次,无水乙醇清洗3次,放在55℃真空干燥箱中烘干5.2h,取出后研磨即可得到微米铜粉。
[0080]
本实施例制备的0.8m反应浓度微米铜粉的平均粒径为4.83μm。
[0081]
实施例4
[0082]
本实施例提供一种1.0m反应浓度微米铜粉的制备方法,所述微米铜粉的制备方法包括以下步骤:
[0083]
(1)将300g(1.2mol)五水硫酸铜、360g聚乙烯吡咯烷酮k30、36.0gβ
‑ꢀ
环糊精和800ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合) 混合,75℃搅拌0.8h,形成溶液i;
[0084]
(2)将105.5g次磷酸钠、10.55g抗坏血酸和400ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合)混合,75℃搅拌0.8h,形成溶液ⅱ;
[0085]
(3)将溶液i与溶液ⅱ混合均匀,100℃反应2h,得到溶液ⅲ;
[0086]
(4)将溶液ⅲ转移至烧杯,冷却至室温后,自然沉降,去除上清液,然后用去离子水清洗底部沉淀6次,无水乙醇清洗3次,放在50℃真空干燥箱中烘干5h,取出后研磨即可得到微米铜粉。
[0087]
本实施例制备的1.0m反应浓度微米铜粉的平均粒径为1.83μm。
[0088]
实施例5
[0089]
本实施例提供一种1.2m反应浓度微米铜粉的制备方法,所述微米铜粉的制备方法包括以下步骤:
[0090]
(1)将360g(1.44mol)五水硫酸铜、432g聚乙烯吡咯烷酮k30、43.2g β-环糊精和800ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合) 混合,85℃搅拌0.8h,形成溶液i;
[0091]
(2)将126.6g次磷酸钠、12.66g抗坏血酸和400ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合)混合,85℃搅拌0.8h,形成溶液ⅱ;
[0092]
(3)将溶液i与溶液ⅱ混合均匀,100℃反应2h,得到溶液ⅲ;
[0093]
(4)将溶液ⅲ转移至烧杯,冷却至室温后,自然沉降,去除上清液,然后用去离子水清洗底部沉淀6次,无水乙醇清洗3次,放在50℃真空干燥箱中烘干5h,取出后研磨即可得到微米铜粉。
[0094]
本实施例制备的1.2m反应浓度微米铜粉的平均粒径为6.6μm。
[0095]
实施例6
[0096]
本实施例提供一种1.4m反应浓度微米铜粉的制备方法,所述微米铜粉的制备方法包括以下步骤:
[0097]
(1)将420g(1.68mol)五水硫酸铜、504g聚乙烯吡咯烷酮k30、50.4g β-环糊精和800ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合) 混合,85℃搅拌0.8h,形成溶液i;
[0098]
(2)将147.7g次磷酸钠、14.77g抗坏血酸和400ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合)混合,85℃搅拌0.8h,形成溶液ⅱ;
[0099]
(3)将溶液i与溶液ⅱ混合均匀,100℃反应2h,得到溶液ⅲ;
[0100]
(4)将溶液ⅲ转移至烧杯,冷却至室温后,自然沉降,去除上清液,然后用去离子水清洗底部沉淀6次,无水乙醇清洗3次,放在50℃真空干燥箱中烘干5h,取出后研磨即可得到微米铜粉。
[0101]
本实施例制备的1.4m反应浓度微米铜粉的平均粒径为7.44μm。
[0102]
实施例7
[0103]
本实施例提供一种1.6m反应浓度微米铜粉的制备方法,所述微米铜粉的制备方法包括以下步骤:
[0104]
(1)将480g(1.92mol)五水硫酸铜、576g聚乙烯吡咯烷酮k30、57.6g β-环糊精和800ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合) 混合,85℃搅拌0.8h,形成溶液i;
[0105]
(2)将168.8g次磷酸钠、16.88g抗坏血酸和400ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合)混合,85℃搅拌0.8h,形成溶液ⅱ;
[0106]
(3)将溶液i与溶液ⅱ混合均匀,100℃反应2h,得到溶液ⅲ;
[0107]
(4)将溶液ⅲ转移至烧杯,冷却至室温后,自然沉降,去除上清液,然后用去离子水清洗底部沉淀6次,无水乙醇清洗3次,放在50℃真空干燥箱中烘干5h,取出后研磨即可得到微米铜粉。
[0108]
本实施例制备的1.6m反应浓度微米铜粉的平均粒径为6.72μm。
[0109]
实施例8
[0110]
本实施例提供一种1.8m反应浓度微米铜粉的制备方法,所述微米铜粉的制备方法包括以下步骤:
[0111]
(1)将540g(2.16mol)五水硫酸铜、648g聚乙烯吡咯烷酮k30、64.8g β-环糊精和800ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合) 混合,85℃搅拌0.8h,形成溶液i;
[0112]
(2)将189.9g次磷酸钠、18.99g抗坏血酸和400ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合)混合,85℃搅拌0.8h,形成溶液ⅱ;
[0113]
(3)将溶液i与溶液ⅱ混合均匀,100℃反应2h,得到溶液ⅲ;
[0114]
(4)将溶液ⅲ转移至烧杯,冷却至室温后,自然沉降,去除上清液,然后用去离子水清洗底部沉淀6次,无水乙醇清洗3次,放在50℃真空干燥箱中烘干5h,取出后研磨即可得到微米铜粉。
[0115]
本实施例制备的1.8m反应浓度微米铜粉的平均粒径为4.67μm。
[0116]
实施例9
[0117]
本实施例提供一种2.0m反应浓度微米铜粉的制备方法,所述微米铜粉的制备方法包括以下步骤:
[0118]
(1)将600g(2.4mol)五水硫酸铜、720g聚乙烯吡咯烷酮k30、72.0gβ
‑ꢀ
环糊精和800ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合) 混合,85℃搅拌0.8h,形成溶液i;
[0119]
(2)将211g次磷酸钠、21.1g抗坏血酸和400ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合)混合,85℃搅拌0.8h,形成溶液ⅱ;
[0120]
(3)将溶液i与溶液ⅱ混合均匀,100℃反应2h,得到溶液ⅲ;
[0121]
(4)将溶液ⅲ转移至烧杯,冷却至室温后,自然沉降,去除上清液,然后用去离子水清洗底部沉淀6次,无水乙醇清洗3次,放在50℃真空干燥箱中烘干5h,取出后研磨即可得到微米铜粉。
[0122]
本实施例制备的2.0m反应浓度微米铜粉的平均粒径为3.43μm。
[0123]
实施例10
[0124]
本实施例提供一种0.4m反应浓度微米铜粉的制备方法,与实施例1的区别仅在于,步骤(1)中,将聚乙烯吡咯烷酮k30替换为同等质量的聚乙烯吡咯烷酮k60,其他步骤同实施例1。
[0125]
实施例11
[0126]
本实施例提供一种0.4m反应浓度微米铜粉的制备方法,与实施例1的区别仅在于,步骤(1)中,不含有β-环糊精,将聚乙烯吡咯烷酮k30的增加至160 g,其他步骤同实施例1。
[0127]
实施例12
[0128]
本实施例提供一种0.4m反应浓度微米铜粉的制备方法,与实施例1的区别仅在于,步骤(1)中,不含有聚乙烯吡咯烷酮k30,将β-环糊精的增加至75g,其他步骤同实施例1。
[0129]
实施例13
[0130]
本实施例提供一种0.4m反应浓度微米铜粉的制备方法,与实施例1的区别仅在于,步骤(1)和步骤(2)中,溶剂为800ml,所述溶剂为质量比为1:0.4 的乙二醇和去离子水的组合,其他步骤同实施例1。
[0131]
实施例14
[0132]
本实施例提供一种0.4m反应浓度微米铜粉的制备方法,与实施例1的区别仅在于,步骤(1)和步骤(2)中,溶剂为800ml,所述溶剂为质量比为1:2.2 的乙二醇和去离子水的组合,其他步骤同实施例1。
[0133]
实施例15
[0134]
本实施例提供一种0.4m反应浓度微米铜粉的制备方法,与实施例1的区别仅在于,步骤(2)中,不含有次磷酸钠,将抗坏血酸的质量增加至180g,其他步骤同实施例1。
[0135]
实施例16
[0136]
本实施例提供一种0.4m反应浓度微米铜粉的制备方法,与实施例1的区别仅在于,步骤(2)中,不含有抗坏血酸,将次磷酸钠的质量增加至45g,其他步骤同实施例1。
[0137]
实施例17
[0138]
本实施例提供一种0.4m反应浓度微米铜粉的制备方法,与实施例1的区别仅在于,步骤(1)中,将120g(0.48mol)五水硫酸铜替换为0.48mol的六水硝酸铜,其他步骤同实施例1。
[0139]
对比例1
[0140]
本对比例提供一种0.4m反应浓度微米铜粉的制备方法,所述微米铜粉的制备方法包括以下步骤:
[0141]
(1)将120g(0.48mol)五水硫酸铜、144g聚乙烯吡咯烷酮k30、14.4g β-环糊精和1200ml溶剂(所述溶剂为质量比为1:2的乙二醇和去离子水的组合) 混合,90℃搅拌0.8h,形成溶液i;
[0142]
(2)将42.2g次磷酸钠、4.22g抗坏血酸和溶液i混合均匀,100℃反应2h,得到溶液ⅲ;
[0143]
(3)将溶液ⅲ转移至烧杯,冷却至室温后,自然沉降,去除上清液,然后用去离子水清洗底部沉淀6次,无水乙醇清洗3次,放在50℃真空干燥箱中烘干5h,取出后研磨即可得到微米铜粉。
[0144]
对比例2
[0145]
本对比例提供一种0.4m反应浓度微米铜粉的制备方法,与实施例1的区别仅在于,步骤(1)和步骤(2)中,溶剂为800ml的乙二醇,不含有去离子水,其他步骤同实施例1。
[0146]
对比例3
[0147]
本对比例提供一种0.4m反应浓度微米铜粉的制备方法,与实施例1的区别仅在于,步骤(1)和步骤(2)中,溶剂为800ml的去离子水,不含有乙二醇,其他步骤同实施例1。
[0148]
测试例1
[0149]
粒径测试
[0150]
测试样本:采用实施例1-9提供的制备方法得到的铜粉
[0151]
如图1所示,本发明在0.4-2.0m不同反应体系浓度所制得微米铜粉的sem 图。从低浓度到高浓度,本发明提供的方法均可实现微米铜粉的制备。此外,体系浓度从0.4-2.0m,铜粉形貌差别不大,均为颗粒状间杂少量片铜,铜粉的平均粒径在1.0m时最小,为2μm左右;其他浓度时,铜粉平均粒径会出现波动,但均在10μm以下。
[0152]
图2为铜粉粒径随反应体系浓度变化曲线图,从中可以看出,铜粉的平均粒径在1.0m时最小,其他浓度时,铜粉平均粒径会出现波动,但均在10μm 以下。
[0153]
测试例2
[0154]
平均电阻率测试
[0155]
测试样本:采用实施例1-17和对比例1-3提供的制备方法得到的铜粉
[0156]
测试方法:30mpa测试压力下测试铜粉的平均电阻率测试
[0157]
测试结果如下表1所示:
[0158]
表1
[0159]
[0160][0161]
由表1数据可知,采用本发明提供的制备方法制备得到的铜粉具有较低的电阻率,平均电阻率在0.00008-0.0007ω
·
cm。
[0162]
通过实施例1和实施例10-12的对比可知,分散剂的选择会影响铜粉的电阻率。
[0163]
通过实施例1、实施例13-14和对比例2-3的对比可知,只有采用质量比为 1:(0.5-2)的乙二醇和去离子水的混合溶剂,制备得到的铜粉才有较低的电阻率。
[0164]
通过实施例1和实施例15-16的对比可知,还原剂的选择会影响铜粉的电阻率。
[0165]
通过实施例1和实施例17的对比可知,将五水硫酸铜替换为同等摩尔量的六水硝酸铜会影响铜粉的电阻率。
[0166]
通过实施例1和对比例1的对比可知,直接将还原剂添加到反应体系,影响铜粉的电阻率。
[0167]
申请人声明,本发明通过上述实施例来说明本发明的工艺方法,但本发明并不局限于上述工艺步骤,即不意味着本发明必须依赖上述工艺步骤才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明所选用原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
